

Summary
Studies of long-lived individuals have revealed few genetic mechanisms for protection against age-associated disease. Therefore, we pursued genome sequencing of a related phenotype – healthy aging – to understand the genetics of disease-free aging without medical intervention. In contrast with studies of exceptional longevity, usually focused on centenarians, healthy aging is not associated with known longevity variants but is associated with reduced genetic susceptibility to Alzheimer and coronary artery disease. Additionally, healthy aging is not associated with a decreased rate of rare pathogenic variants, potentially indicating the presence of disease-resistance factors. In keeping with this possibility, we identify suggestive common and rare variant genetic associations implying that protection against cognitive decline is a genetic component of healthy aging. These findings, based on a relatively small cohort, require independent replication. Overall, our results suggest healthy aging is an overlapping but distinct phenotype from exceptional longevity that may be enriched with disease-protective genetic factors.
eTOC Blurb
The genomic characterization of humans that age without developing diseases suggests that healthy aging is a distinct phenotype from exceptional longevity, and that it may be enriched with disease-protective genetic factors, such as resistance against cognitive decline.
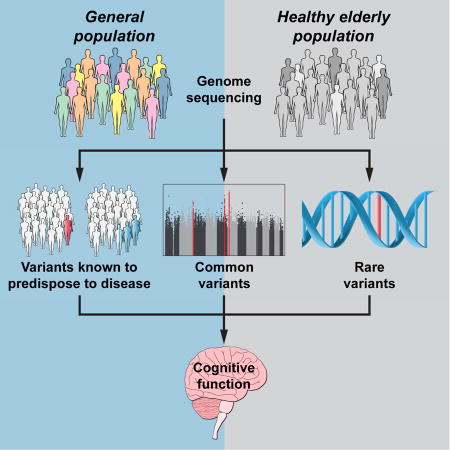
Introduction
Age-associated diseases account for two-thirds of human deaths globally and 90% of all deaths in industrialized nations. Management of age-related disease is a major source of disability and health care costs, and thus management of age-associated disease is a major driver of medical research and health policy (Goldman et al., 2013). Age-standardized death rates for the leading causes of death have declined substantially over the past half century due to improved understanding of behavioral risks and more effective medical interventions (Ma et al., 2015). However, as average life expectancy improves, a shift in increased death due to frailty and chronic degenerative disease has been observed (Lozano et al., 2012). Thus, overall longevity is likely due to the avoidance of multiple vulnerabilities such as death due to infectious disease and severe early onset genetic diseases, mid-life death due to chronic and potentially sporadic diseases, and late-life death due to degenerative diseases and frailty – each of which is differentially influenced by the interplay of genetics and environment. Moreover, technologic progress with pharmacotherapy for cancer and with implantable devices, such as defibrillators and prosthetic joints, can promote longevity as a function of medical interventions per se.
Age at death in adulthood has a moderate genetic component overall, with a heritability of approximately 25% (Murabito et al., 2012). Heritability of longevity increases with age, with a negligible genetic contribution to survival up to approximately 60 years of age, after which an increasing genetic component to survival is observed (Brooks-Wilson, 2013; Christensen et al., 2006). Most genetic studies of aging have focused on long-lived individuals, typically defined as centenarians 100 years or older, who may have had exceptional survival due to medical interventions (Murabito et al., 2012). A number of genetic associations with exceptional longevity have been made (Atzmon et al., 2006; Bojesen and Nordestgaard, 2008; Hurme et al., 2005; Kuningas et al., 2007; Melzer et al., 2007; Pawlikowska et al., 2009; Sanders et al., 2010; Suh et al., 2008; Willcox et al., 2008), with only markers at APOE and FOXO3A being well replicated (Murabito et al., 2012). Overall, the results of genetic and epidemiological longevity studies suggest aging is a complex trait, and that achievement of exceptional longevity may not best capture the genetics of resistance to or delay of age-associated disease (Christensen et al., 2006).
In this light, we pursued a genomic study of an alternate but related aging phenotype – healthy aging – in order to expose its potential to uncover genetic factors for protection against age-associated disease. It is important to differentiate longevity from our healthy aging phenotype, which as we have defined it for our healthy aging cohort (Wellderly), attempts to understand the genetics of disease-free aging in humans, without medical interventions. Towards this end, we performed whole genome sequencing (WGS) of the Wellderly and compared their genetic characteristics to an ethnicity-matched population control. Our findings suggest that healthy aging is associated with a disease-protective genetic profile that overlaps with but differs from that observed in exceptional longevity cohorts. These findings include no enrichment of true longevity variants, a lower genetic risk from common susceptibility alleles for Alzheimer and coronary artery disease, and no decrease in the rate of rare pathogenic variants. We identify suggestive common and rare variant genetic associations which implicate genetic protection against cognitive decline in healthy aging. Our data is made available for the discovery of additional disease protective genetic factors by the research community.
Results
Wellderly Cohort Definition and Demographics
The Wellderly phenotype is defined as individuals who are >80 years old with no chronic diseases and who are not taking chronic medications. Individuals with any of the following phenotypes were excluded from enrollment: autoimmune disease, blood clots, cancer (except basal and squamous cell carcinoma), type I or II diabetes, dementia, myocardial infarction, renal failure, and stroke. The enrollment procedure is described in Experimental Methods, and the specific ICD code, CPT code, lab value, and medication status exclusion criteria are provided in Supplemental Text.
To date, a total of 1,354 Wellderly individuals ranging from 80 to 105 years old have enrolled in our study. Demographic characteristics of these Wellderly individuals were compared with the characteristics of the general US population of 70+ or 80+ year old individuals (depending upon availability) utilizing data from the US 2010 Census data or the National Health and Nutrition Examination Survey (Table 1). The Wellderly cohort individuals differ from these age-matched controls in the following ways: 1) they are comprised of a slightly but significantly higher proportion of male individuals overall, 2) they contain a small but significantly elevated rate of male smokers, 3) they exercise significantly more frequently, 4) they are leaner on average but not significantly so due to a wide weight distribution (Figure 1C), and 5) they have attained a significantly higher level of education relative to the general population. The majority of individuals were enrolled between the age of 80 and 85 with similar overall age distributions for female and male Wellderly individuals (Figure 1A and 1B).
Table 1.
Wellderly Demographic Data
| Characteristic | Wellderly Cohort | U.S. Population | ITMI Cohort |
|---|---|---|---|
| Median Age | 84.2 yrs (±9.3 yrs) | 70+yrsa or 80+yrsb | 33.3 yrs |
|
| |||
| Gendera | Male = 39.3% (±1.3%) | Male = 36.3% | Male = 52.7% |
| Female = 60.7% (±1.3%) | Female = 63.7% | Female = 47.3% | |
|
| |||
| Median Heightb | Male = 69″ (±5.0″) | Male = 68″ | Male = N/A |
| Female = 63″ (±5.7″) | Female = 63″ | Female = 66″ | |
|
| |||
| Median Weightb | Male = 168lbs (±47lbs) | Male = 190lbs | Male = N/A |
| Female = 132lbs (±45lbs) | Female = 160lbs | Female = 142lbs | |
|
| |||
| Ever Smokerb | Male = 61% (±2.6%) | Male = 54% | Male = 31.5% |
| Female = 42% (±2.6%) | Female = 43% | Female = 26.2% | |
|
| |||
| Exerciseb | 66.8% (±2.5%) | 44.1% | N/A |
|
| |||
| Educationb | |||
| No high school | 0.5% (±0.4%) | 24.6% | 8.0% |
| High school | 16.8% (±2.0%) | 56.2% | 17.2% |
| Bachelor’s | 55.0% (±2.7%) | 11.9% | 38.7% |
| Advanced | 27.7% (±2.4%) | 7.3% | 36.1% |
U.S. Population 80+ years,
U.S. population 70+ years. 95% confidence interval in parenthesis.
Figure 1. Wellderly Demographic Characteristics.
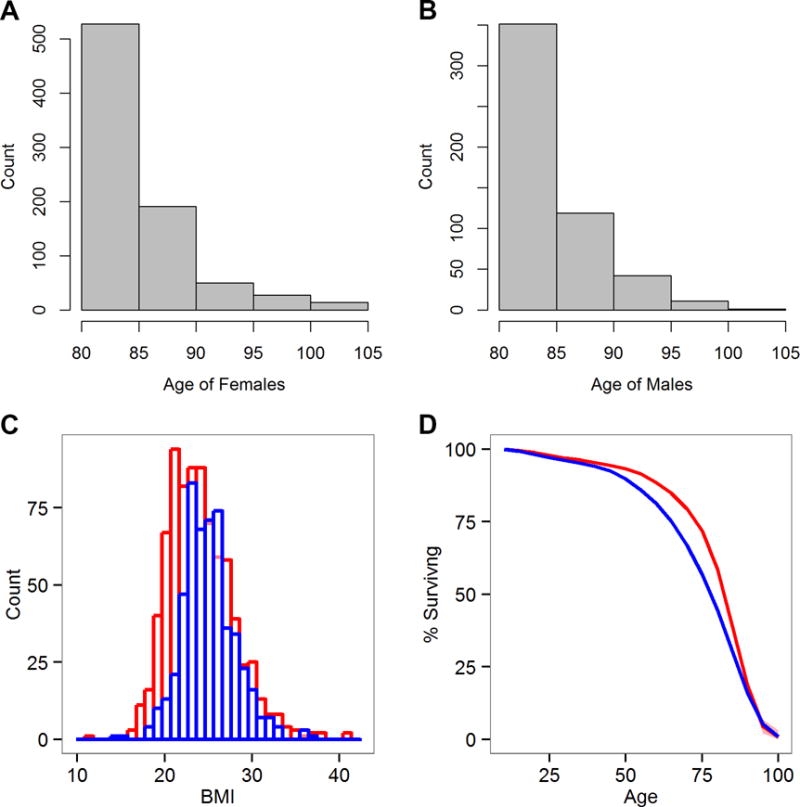
A. The age distribution of Wellderly female individuals. B. The age distribution of male Wellderly individuals. C. The BMI distribution of Wellderly individuals for males (blue) and females (red). D. Survival curves for the Wellderly siblings (red) and the expected survival of a 1920s birth cohort (blue). A significant difference in survival was observed (log-rank test p-value = 3.67 × 10−8) in middle-age (40–79 years) but not overall longevity. 95% confidence interval is provided for the Wellderly siblings in red shading (only visible at the end of the survival curve).
Healthy Aging vs. Longevity
It is possible that the Wellderly phenotype simply represents the tail end of a stochastic age-related disease incidence process – in which the Wellderly individuals are simply “lucky” and by chance do not develop disease. If this were true, one would expect that the siblings of Wellderly individuals should live no longer than the average individual, and that no shared factor, whether genetic or environmental, should have an influence on Wellderly sibling survival.
To address this possibility, we performed survival analysis comparing the US Social Security 1920s birth cohort life table vs. Wellderly siblings – the majority being deceased individuals who were obviously genetically similar to the Wellderly individuals. Survival analysis was initiated at 10 years of age to eliminate a significant excess of childhood death in the 1920s birth cohort life table (Bell and Miller, 2005) relative to that reported by the Wellderly individuals. Survival analysis demonstrated significant increased survival (log-rank test p-value = 3.67 × 10−8) in Wellderly siblings relative to the 1920s birth cohort (Figure 1D). Inspection of the survival curve reveals a strong survival advantage in middle age to middle old age (40–79 years), a limited survival advantage from 80–90 yrs of age, and no survival advantage after 90+ yrs of age. 83% of Wellderly siblings survive to age 70 yrs whereas 75% of the 1920s birth cohort survives to 70 yrs. This suggests Wellderly individuals avoid or delay the onset of middle-age onset diseases through some shared factor. This analysis cannot differentiate between genetic, environmental, or behavioral characteristics shared by Wellderly individuals and their siblings, but indicates the phenotype is not simply due to chance.
Genome Sequencing and Variant Filtration
To determine whether genetic factors underlie the Wellderly phenotype, WGS of 600 Wellderly individuals was performed using the Complete Genomics platform (Complete Genomics Inc., Mountain View, CA) (see Experimental Methods). These genomes were compared to 1,507 adult members of the Inova Translational Medicine Institute (ITMI) pre-term birth cohort also sequenced on the Complete Genomics platform (Bodian et al., 2014). Individuals from these cohorts were filtered to retain only individuals of 95+% European ancestry and a maximum relatedness of 12.5% (see Experimental Methods). After cohort filtration, 511 Wellderly individuals and 686 ITMI individuals were carried forward to downstream analyses. Variants were combined within and across cohorts and filtered stringently for downstream statistical analyses (see Experimental Methods). Genomic variant filtration resulted in reduction of the initial ~57 million total raw variants to 24,205,551 variants after filtration. Supplemental Table S1 displays the number of variants removed by each filtration step. The count of variants by type and coding impact is presented in Supplemental Table S2. Our variant filtration strategy was reinforced by observing no evidence of genomic inflation in the genome-wide association results (see below – λ = 0.98) (Reich and Goldstein, 2001).
Genetic Longevity and Disease Risk
First, we explored whether previously reported longevity variants may underlie the Wellderly phenotype (Atzmon et al., 2006; Bojesen and Nordestgaard, 2008; Hurme et al., 2005; Kuningas et al., 2007; Melzer et al., 2007; Pawlikowska et al., 2009; Sanders et al., 2010; Suh et al., 2008; Willcox et al., 2008). No deviation in allele frequency of longevity variants was observed between the Wellderly cohort and the ITMI cohort or the 1000 Genomes European individuals (Sudmant et al., 2015) (Table 2). No trend in allele frequency deviations implying even minimal enrichment of longevity variants was observed, suggesting the Wellderly healthy aging phenotype is likely distinct from traditionally defined longevity.
Table 2.
Allele Frequencies of Known Longevity Variants
| Variant | Gene | Longevity Allele | Wellderly AF | ITMI AF | p-value | 1000G CEU AF |
|---|---|---|---|---|---|---|
| rs2802292 | FOXO3A | G | 0.41 | 0.38 | 0.059 | 0.43 |
| rs1935949 | FOXO3A | A | 0.31 | 0.30 | 0.431 | 0.35 |
| rs3758391 | SIRT1 | T | 0.32 | 0.31 | 0.586 | 0.33 |
| rs5882 | CETP | G | 0.33 | 0.33 | 0.726 | 0.33 |
| rs1042522 | TP53 | C | 0.74 | 0.74 | 0.677 | 0.72 |
| rs1800795 | IL6 | C | 0.40 | 0.40 | 0.761 | 0.42 |
| rs2811712 | CDKN2A | G | 0.11 | 0.10 | 0.727 | 0.10 |
| rs34516635 | IGF1R | A | 0.01 | 0.01 | 0.394 | 0.00 |
| rs2542052 | APOC3 | C | 0.61 | 0.59 | 0.433 | 0.61 |
| rs3803304 | AKT1 | C | 0.73 | 0.74 | 0.511 | 0.73 |
Comparison of allele frequency for known longevity variants. Frequency of allele associated with longer lifespan is displayed.
Next, we considered whether the Wellderly have decreased genetic risk of common disease by calculating genetic risk scores for the Wellderly and ITMI individuals for the top 5 leading causes of death with a genetic component – heart disease, cancer, stroke, Alzheimer disease, and diabetes (CDC Vital Statistics) – using genetic loci identified from the latest large genome-wide association studies and/or meta-analyses (Al-Tassan et al., 2015; Consortium et al., 2013; Kilarski et al., 2014; Lambert et al., 2013; Michailidou et al., 2015; Replication et al., 2014; Seshadri et al., 2010; Timofeeva et al., 2012; Wolpin et al., 2014; Zheng et al., 2008) (Table 3). The 5 most common and deadly cancer types were considered; breast, colon, lung, pancreatic, and prostate – comprising over 75% of cancer deaths in the USA. We observed no difference in cancer, stroke, or type 2 diabetes genetic risk. On the other hand, the Wellderly have a significantly lower genetic risk for Alzheimer disease (p-value = 9.84 × 10−4) and coronary artery disease (p-value = 2.54 × 10−3). Individual marker level differences are presented in Supplemental Table S3.
Table 3.
Genetic Risk For Leading Causes of Death with a Component.
| Wellderly Genetic Risk | ITMI Genetic Risk | Wellderly Odds Ratio | p-value | |
|---|---|---|---|---|
| Alzheimer disease | 0.0075 (±0.0418) | 0.1060 (±0.0409) | 0.906 (±0.051) | 0.009 |
| Breast Cancer | 0.596 (±0.045) | 0.600 (±0.045) | 0.996 (±0.064) | 1.0 |
| Colorectal Cancer | 1.11 (±0.030) | 1.12 (±0.03) | 0.992 (±0.032) | 1.0 |
| Coronary Artery Disease | 8.27 (±0.035) | 8.35 (±0.040) | 0.922 (±0.047) | 0.023 |
| Lung Cancer | 0.130 (±0.027) | 0.142 (±0.022) | 0.988 (±0.034) | 1.0 |
| Pancreatic Cancer | 0.364 (±0.033) | 0.387 (±0.026) | 0.977 (±0.040) | 1.0 |
| Prostate Cancer | 4.80 (±0.100) | 4.75 (±0.090) | 1.050 (±0.131) | 1.0 |
| Stroke | 2.04 (±0.030) | 2.09 (±0.030) | 0.958 (±0.043) | 0.66 |
| Type 2 Diabetes | 6.40 (±0.050) | 6.39 (±0.04) | 1.004 (±0.0580) | 1.0 |
Mean genetic risk score is presented with 95% confidence interval in parenthesis. Wellderly odds ratio is the overall odds ratio of Wellderly individuals for the development of each disease relative to the general population – also with 95% confidence interval in parenthesis. p-values corrected for multiple testing are reported. See also Supplemental Table S3
The difference in Alzheimer disease genetic risk is strongly influenced by APOE-ε4 marker (rs2075650; p-value = 7.02 × 10−4), though a trend for marginal differences in allele frequency, in the direction consistent with decreased Alzheimer risk, can be observed for the majority of the 18 Alzheimer disease risk variants (Supplemental Table S3). The next most significant single marker (rs11218343; p-value = 2.96 × 10−2) was also the next strongest genetic risk marker for Alzheimer disease in the published GWAS meta-analysis – at the SORL1 locus – suggesting the magnitude of the deviation in allele frequency between the Wellderly and ITMI cohort is related to the strength of the genetic risk locus in mediating disease (correlation between −log10 single marker p-value and genetic risk weight = 0.89, p-value <0.0001).
Similarly, 83 of the 141 previously identified markers for coronary artery disease variants displayed allele frequency differences consistent with decreased genetic risk for coronary artery disease in the Wellderly (p-value = 0.043). The correlation between deviation in allele frequency between the Wellderly and ITMI cohort and the strength of the genetic risk locus was again significant, though not as strong as that observed for Alzheimer disease (r = 0.15, p-value = 0.04). The strongest differences were observed at APOE (rs2075650; p-value =7.02 × 10−4), PECAM1 (rs2070783; p-value = 1.26 × 10−2), SMG6 (rs2281727; p-value = 1.35 × 10−2), and SCARB1 (rs11057841; p-value = 1.69 × 10−2).
Correspondingly, the single strongest risk markers for Alzheimer disease (APOE above), and coronary artery disease (9p21 rs1333049; p-value =3.57 × 10−2) (Wellcome Trust Case Control, 2007), demonstrated deviations in allele frequency between the Wellderly and ITMI cohorts, whereas no significant difference was observed for the strongest risk markers for diabetes or cancer: FTO (rs9939609; p-value = 0.809) (Wellcome Trust Case Control, 2007), TCF7L2 (rs7903146; p-value = 0.728) (Wellcome Trust Case Control, 2007), 8q24 (rs6983267; p-value = 0.421) (Wokolorczyk et al., 2008).
Common Variant Genome-Wide Association Study
Next, we globally considered whether any common variants (minor allele frequency > 5%) were associated with the Wellderly phenotype. We performed a WGS-based genome-wide association study (GWAS) using a logistic model with principal component correction (Figure 2A) to account for any remaining population stratification after restriction to individuals with 95+% European ancestry – though inspection of the principal component plots demonstrates the cohorts are well balanced (Figure 2A). The quantile-quantile (QQ) plot of association results demonstrates no genomic inflation (λ = 0.98) (Figure 2B). No associations were observed at the genome-wide significance level (p-value < 5 × 10−8, Figure 2C), though a number of interesting marginally significant peaks were observed. The full list of top hits is provided in Supplemental Table S4.
Figure 2. Common Variant Association Results.
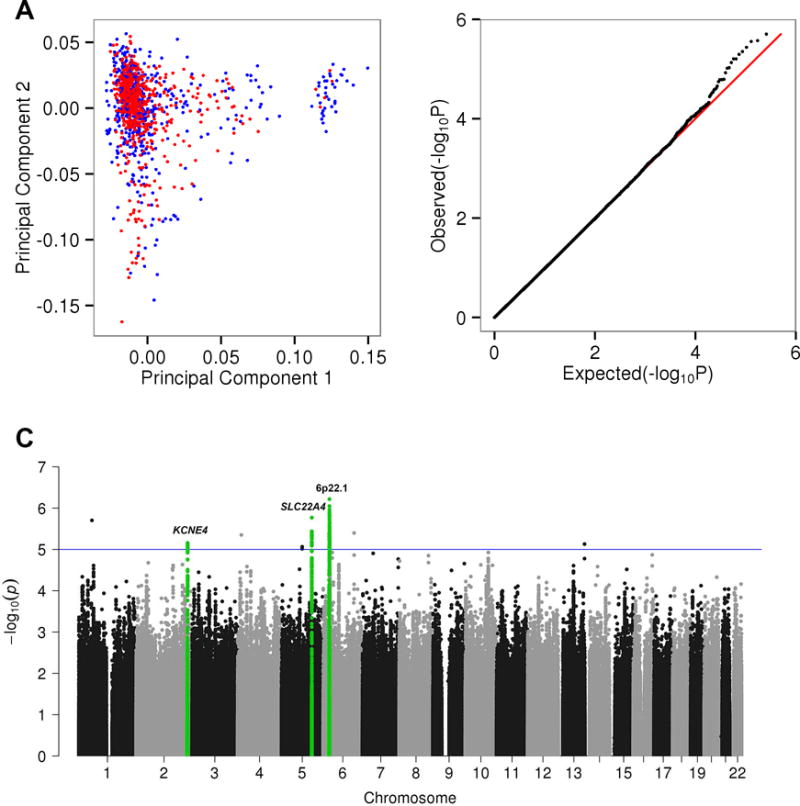
A. Principal component plot of the Wellderly (red) and ITMI (blue) cohort based on filtered and LD pruned (R2 > 0.5) common variants (allele frequency > 5%). B. QQ plot of expected −log10(P-values) (x-axis) vs. observed −log10(P-values) (y-axis) (one black point per variant). Expected vs. expected −log10(P-values) (red line) is along the diagonal. The λ value for this QQ plot is 0.98 – no genomic inflation observed. C. Manhattan plot of the unbiased GWAS results. Each point represents a single SNP P-value determined by a logistic model adjusted for the first ten principal components. Points are organized by chromosome and chromosomal coordinate (x-axis) and −log10(P-values) (y-axis). The blue line indicates P-values < 10−5. Green highlighted SNPs correspond to the top most significant regions, plotted in Figure 3. See also Supplemental Table S4.
The top region is a large linkage block at MHC locus 6p22.1 containing lead SNPs rs13217620 (chr6:27,653,120) (p-value = 6.1 × 10−7) (Figure 3A) and rs41266839 (chr6:26,409,890) (p-value = 4.1 × 10−6) (Figure 3B). This region contains a handful of SNPs that have been replicated and confidently associated with cognitive traits including rs1056667; associated with cognitive performance (Rietveld et al., 2014; Rietveld et al., 2013), rs13194053; associated with schizophrenia and bipolar disorder (International Schizophrenia et al., 2009; Shi et al., 2009). The two lead SNPs (rs13217620 and rs41266839) from our study are in perfect disequilibrium (D′ = 1.0) with the aforementioned cognitive trait associated SNPs (rs1056667 and rs13194053 respectively) (Figure 3A and 3B).
Figure 3. Regional Association Plots.
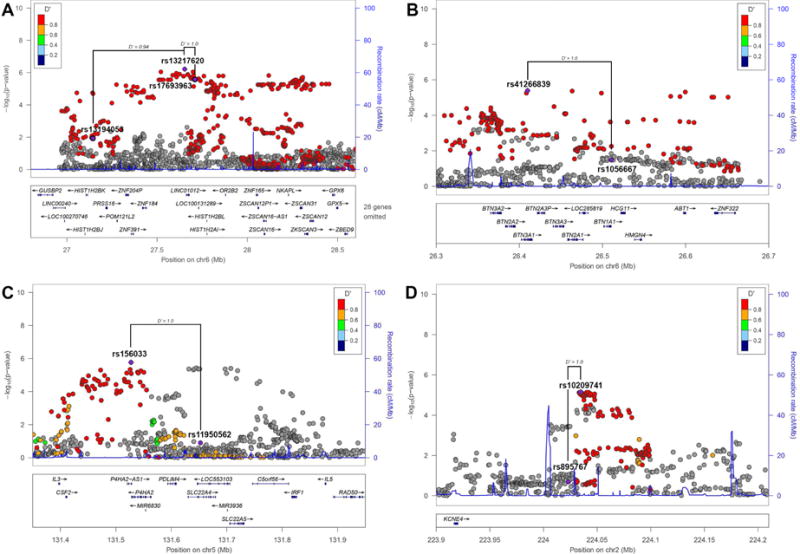
Regional plots for the top 3 most significant regions for GWAS comparing the Wellderly and ITMI cohorts. For all plots, each point represents a SNP, where the x-axis represents the position of the SNP and the y-axis (right) the −log10 p-value of the genome-wide association results. Each point is color coded with the D′ value as calculated within the Wellderly and ITMI cohorts. The D′ value within the 1000 Genomes European individuals is indicated by connecting lines. Recombination rate is also plotted at each genomic position with the rate indicated on the y-axis (left) A. Lead SNP rs13217620 (chr6:27,653,120) (p-value = 6.1 × 10−7) is highlighted along with SNPs previously associated with schizophrenia (rs13194053 and rs17693963). B. Lead SNP rs41266839 (chr6:26,409,890) (p-value = 4.1 × 10−6) is highlighted along with rs1056667, previously associated with cognitive performance. C. Lead SNP rs156033 (p-value = 1.7 × 10−6) is highlighted along with rs11950562, previously associated with isovalerylcarnitine levels. D. Lead SNP rs10209741 (p-value = 7.0 × 10−6) is highlighted along with rs895767, previously marginally associated with cognitive decline.
The second sub-significant region included lead SNP rs156033 (p-value = 1.7 × 10−6), in the 5q31.1 region, which contains SLC22A4 and multiple variants (rs7727544, rs419291, rs11950562, rs273914, rs272889, rs272869, rs274567) strongly associated with carnitine, and carnitine-related metabolite levels (Shin et al., 2014) (Figure 3C). Again, our lead SNP displays strong disequilibrium with all of these previously associated SNPs, especially rs11950562 (D′ = 1.0) (Figure 3C), which was specifically associated with isovalerylcarnitine levels (p-value = 2 × 10−41) (Shin et al., 2014).
Finally, our third sub-significant region included lead SNP rs10209741 (p-value = 7.0 × 10−6) in the 2q36.1 region containing KCNE4, and nearby (11.4kb) rs895767, which was previously associated with cognitive decline (Zhang and Pierce, 2014) (Figure 3C). Again, rs10209741 and rs895767 are in perfect disequilibrium (D′= 1.0).
While no individual locus is genome-wide significant, the observation that at least two of three of our top GWAS hits are associated with cognitive function is unlikely to be due to chance. With the cautious assumption that ~100 loci have been previously associated with cognitive function via GWAS, the probability that two of our three top GWAS hits land amongst these ~100 cognitive function loci by chance is p-value = 3.0 × 10−8. This observation is also consistent with the finding that the Wellderly have a lower overall genetic risk for Alzheimer disease and cognitive decline (above), a lower APOE-ε4 rate (rs429358 p-value = 7.6 × 10−5), and a greatly reduced frequency of the Alzheimer disease-associated rare coding variant rs145999145 in PLD3 as previously reported (Cruchaga et al., 2014).
High Penetrance Pathogenic Variants
Next, we explored the contribution of rare monogenic disease variants to the Wellderly phenotype. We considered monogenic disorders that are the most prevalent and relevant to the Wellderly phenotype – hereditary dementia, cancer, and common monogenic diseases as defined by the American College of Medical Genetics secondary findings (Green et al., 2013). No difference in the rate of rare pathogenic variants in the Wellderly vs. ITMI cohorts was observed for any gene set (chi-square p-values 1.0, 0.39, 0.80 respectively) (Figure 4A), nor at the individual gene level (Supplemental Table S5). The full list of genes per disease category is presented in Supplemental Text.
Figure 4. Rare Variant Burden.
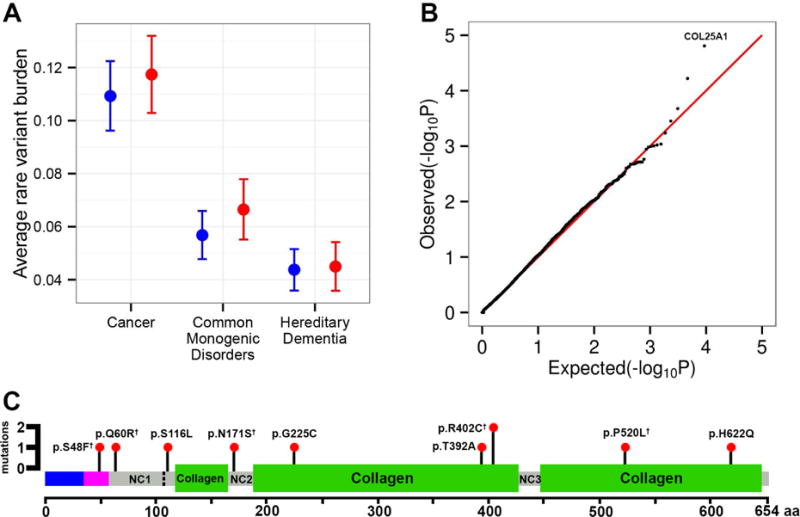
A. The rate of rare pathogenic cancer, hereditary dementia and common monogenic disease (secondary finding) variants is displayed for the Wellderly (red) and ITMI (blue) cohorts with standard error bars. No difference is observed between the cohorts. See also Supplemental Table S5. B. QQ plot of expected −log10(P-values) (x-axis) vs. observed −log10(P-values) (y-axis) (one black point per gene). Expected vs. expected −log10(P-values) (red line) is along the diagonal. The λ value for this QQ plot is 0.99 – no genomic inflation observed. The point corresponding to the top gene, COL25A1 is labeled. See also, Supplemental Table S6. C. The location, impact, and count of COL25A1 coding variants are indicated along the COL25A1 protein. The COL25A1 protein and domains is displayed horizontally from the N-terminal to C-terminal amino acid (x-axis). The blue region indicates the cytoplasmic region of COL25A1, the maroon region indicates the transmembrane domain, green regions are collagen domains, and grey regions are intervening non-collagen stretches. The cleavage site for secretion of COL25A1 is indicated by the vertical dashed line. COL25A1 interacts with the amyloid beta protein at NC2. Each observed mutation position is indicated by a black pin with red head. The height of each pin corresponds to the number of alleles observed in the Wellderly cohort (all variants observed in a single individual except p.R402C). The coding impact of each variant is indicated next to each pin. Super-script daggers indicate variants that have been previously observed in the Exome Aggregation Consortium database. See also Supplemental Figure S1 and Supplemental Table S7.
Rare Variant Genome-Wide Association Study
Finally, we considered whether rare coding variants in any gene (minor allele frequency < 1%) were associated with the Wellderly phenotype using the SKAT-O method (see Experimental Procedures). The QQ plot of gene-level association results demonstrates no genomic inflation (genomic inflation factor = 0.99) (Figure 4B). No genome-wide significant associations (p-value < 4.8 × 10−6 for Bonferroni correction of 10,447 individual gene tests) were observed. However, the top hit, COL25A1 (p-value = 1.56 × 10−5), is of major interest. The full list of top hits is provided in Supplemental Table S6.
COL25A1 contained 9 ultra-rare coding variants carried by 10 distinct Wellderly individuals, where 8 variants were observed as singletons and one variant was observed in two distinct individuals (Figure 4C). No COL25A1 variants were observed in any ITMI individual (variants provided in Supplemental Table S7, Sanger validation traces displayed in Figure S1). Five of these nine variants have been previously observed in the Exome Aggregation Consortium Database. COL25A1 is a brain-specific, secreted collagenous protein that associates with amyloid plaques (Soderberg et al., 2005). The distribution of the observed coding mutations relative to the various functional domains and elements of COL25A1 is displayed in Figure 4C. No clustering in any particular region of COL25A1 was observed, though many of the mutations result in highly non-conservative amino acid substitutions (p.S48F, p.G225C, p.R402C, p.P520L), or are nearby critical regions for plaque association (p.S116L being nearby the cleavage site at amino acid 113 and p.N171S being in the NC2 domain which interacts with Aβ).
Discussion
We identified no major, singular contributor to healthy aging. Instead, healthy aging appears to demonstrate characteristics similar to other complex polygenic phenotypes. Further, our results suggest that healthy aging is a genetically overlapping but divergent phenotype from exceptional longevity, and that the healthy aging phenotype is potentially enriched for heritable components of both reduced risk of, as well as resistance to, age-associated disease.
First, we observe minimal and non-significant enrichment of the previously identified FOXO3A longevity allele in the Wellderly (Willcox et al., 2008). In contrast, we observe a significant depletion of APOE-ε4 alleles, which is associated with longevity but considered to be ‘frailty’ allele for late onset disease rather than a true longevity allele (Christensen et al., 2006; Gerdes et al., 2000).
Second, and again in contrast to exceptional longevity cohorts, where no overall reduction in common disease genetic risk is generally observed (Beekman et al., 2010; Fortney et al., 2015; Sebastiani et al., 2012), we observe a significant decrease in genetic risk for Alzheimer disease and coronary artery disease genetic risk in the Wellderly. Importantly, these results reflect an overall decrease in genetic risk scores, a result not observed in exceptional longevity cohorts (Beekman et al., 2010; Sebastiani et al., 2012), possibly due to the indirect and/or a mixed relationship between individual genetic disease risk loci and exceptional longevity (as discussed by (Fortney et al., 2015)) vs. the potentially more direct relationship between aging in the absence of disease and overall genetic disease risk.
On the other hand, no difference in genetic risk is observed for type 2 diabetes genetic risk and cancer. Some of these findings (type 2 diabetes, colon and lung cancer) can be explained by the fact that risk for these diseases are strongly modulated by behavioral and environmental risk factors. However, we also observed no difference in genetic risk for more heritable cancer types (breast and prostate especially) neither via common variant risk nor rare pathogenic variant burden. The lack of a difference in rare pathogenic variant burden, for cancer as well as hereditary dementia and other common monogenic disorders, could be indicative disease resistance via behavioral or other genetic characteristics.
In fact, while no individually genome-wide significant associations were observed, both our rare and common variant genetic associations implicate protection against cognitive decline as a potential molecular mechanisms governing healthy aging and longevity – an area where genetic associations have been notoriously difficult to identify despite reasonably high heritability estimates. Perhaps most interestingly, mutation of COL25A1 may disrupt Alzheimer disease pathogenesis due to the critical role soluble COL25A1 plays in regulating the buildup of Aβ fibrils (Kakuyama et al., 2005; Osada et al., 2005). This finding provides potential genetic support for the targeting of COL25A1 for the prevention or treatment of Alzheimer disease.
This work represents one of the first combined rare and common variant genome-wide association studies utilizing whole genome sequencing data, and highlights some of the opportunities and challenges associated with WGS-based case-control analysis. For complex polygenic phenotypes, achieving genome-wide significance is improbable without much larger sample sizes, which will take years to accrue for rare phenotypes such as healthy aging. It should be re-emphasized that the Wellderly phenotype is a narrowly defined phenotype likely with fewer eligible individuals than those who would be eligible for an exceptional longevity cohort. Thus, we acknowledge that, due to the relatively small size of our cohort, independent replication is needed to confirm our preliminary findings. In the meantime, it is important to discern patterns that hint at underlying molecular and physiological pathways that may inform follow-up studies. While the patterns in our unbiased analysis highlight the role of genetic resistance to cognitive decline in healthy aging, due to the global absence of disease in the cohort, we suggest that the Wellderly are likely to be enriched with other genetic factors for disease resistance. These factors will likely be gleaned only via more detailed analyses of the Wellderly dataset via interaction with individual genetic risk factors, or as a result of more detailed analyses of genes of interest. Moreover, the Wellderly dataset can act as independent replication and validation of candidate disease protective genetic factors identified in other cohorts. The identification of genetic factors protective against disease is of paramount importance to understanding disease biology and a proven means for drug target identification. Thus, we make the Wellderly genomic data available broadly for further mining by the research community.
Experimental Procedures
Healthy Elderly Active Longevity Cohort Study (IRB-13-6142) was approved by the Scripps Institutional Review Board in July 2007.
Data Availability
Aggregate unfiltered annotated Wellderly variants and their allele and genotype frequencies are available via Scripps Translational Science Institute Variant Browser (http://genomics.scripps.edu/browser). Variant presence is also queryable via a Global Alliance for Genomics and Health Beacon (http://genomics.scripps.edu/browser/ga4gh). Individual level variant data are available from Complete Genomics Inc. under terms determined by Complete Genomics Inc., from Scripps Genomic Medicine for scientific collaboration with not for profit entities, and will be deposited in dbGAP under similar data use restrictions.
Wellderly Cohort Recruitment
The Wellderly cohort (1,354 individuals) was collected over the course of eight years. Individuals with no chronic diseases and not taking chronic medications were screened for enrollment. Specific inclusion/exclusion criteria are provided in Supplemental Text. The study and inclusion criteria were advertised to the public, for example, via the Scripps newsletters, radio announcements, newspaper articles in various cities around the country and in senior community newsletters. Participants volunteered and were self-referred. Research staff did not recruit individuals who had not already contacted the study staff expressing interest in participating. Potential study participants were then interviewed in order to assess health history according to the inclusion criteria.
ITMI Cohort
Individuals were enrolled in the ITMI research study titled “Molecular Study of Pre-term Birth,” as described in (Bodian et al., 2014). The sub-cohort for this study consists of adults who consented to research use of their genomic and clinical data. Demographic data were obtained from self-report questionnaires. Of the individuals with European ancestry, 68.1% (467) were parents of a neonate born full term, and 31.9% (219) had a preterm infant. They range in age from 20–44 years. Collection of peripheral blood and sample processing for WGS were previously described (Bodian et al., 2014).
Survival Analysis
The number of living and deceased (with age of death) Wellderly siblings was determined by self-report. The number of at risk for death Wellderly siblings, starting at age 10 (3,024 individuals total), and the number of deaths per 5-year interval was calculated based on these self-report data. Survival analysis was conducted by determining the expected number of deaths in a cohort of 3,024 individuals born in the 1920s, under the assumption of equal male and female individuals at 10 years of age, and calculating the expected number of deaths per 5-year interval based on the conditional probability of death (qx) as supplied in the Social Security Actuarial Study No. 120 (Bell and Miller, 2005) for the 1920s birth cohort. The significance of the difference in survival between Wellderly siblings and an equal-sized population born in the 1920s was then calculated via the log-rank test for survival analysis.
Genome Sequencing and Cohort Filtration
WGS of both the Wellderly cohort and ITMI cohort was performed by Complete Genomics Inc., Mountain View, CA – via their standard WGS service. Variant calling was performed via cgatools v2.0.1 through v2.0.4. A mean whole genome coverage of 56X per Wellderly individual and 55X per ITMI individual was achieved (Figure S2A and Figure S2B). An average of 98.8% and 98.9% of the protein coding portion of the genome was covered by >10 reads in the Wellderly and ITMI cohorts respectively (Figure S2C and Figure S2D).
Genetic ancestry estimation was defined via the ADMIXTURE algorithm (Alexander and Lange, 2011) using ~16,000 ancestry informative markers and a reference panel of 83 populations around the world (Libiger and Schork, 2012). Individuals of 95+% European ancestry were retained for downstream association analyses. Related individuals were identified via pairwise identity by descent estimation in PLINK (Purcell et al., 2007).
Variant Filtrations
Variants were combined across the Wellderly and ITMI cohorts via GenomeComb and filtered using metrics based on optimized features selected based on concordance between monozygotic twins (Reumers et al., 2012) and refined based on observed GWAS genomic inflation. These filters excluded variants that were: 1) variants labeled as VQLOW in all individuals, 2) variants clustered in >10% of individuals from either cohort, 3) variants with >10% missingness in either cohort, 4) variants with median coverage <10 or >100 in either cohort, 5) variants in simple repeats, homopolymer repeats ≥ 6bp, segmental duplications, microsatellite repeats, or low complexity repeats, 5) variants out of Hardy-Weinberg Equilibrium (p-value < 1 × 10−5), and 6) variants in non-unique 36mers (Derrien et al., 2012). VQLOW genotypes were set to missing. For common variant analyses, an additional filter for variants <5% minor allele frequency in either cohort was applied. For rare variant analyses, an additional filter for variants >1% minor allele frequency in either cohort was applied, as well as a minimum allele depth filter, where if 20% of individuals with a rare variant alternate allele call had a minimum alternate allele depth of <25% of total reads or fewer than 3 supporting reads – the variant was considered a false positive and removed.
Genetic Risk Scores
Weighted genetic risk scores based on known susceptibility markers for the top 5 causes of death with a genetic component were calculated in both cohorts. Markers and corresponding weights used to construct the genetic risk scores included markers identified in the latest large-scale meta-analyses (when possible) of: Alzheimer disease replicated markers in Table 2 of (Lambert et al., 2013) and APOE risk markers (Seshadri et al., 2010); breast cancer markers in Table 1 and Supplementary Table 3a of (Michailidou et al., 2015); colorectal cancer markers in Supplementary Table 3 of (Al-Tassan et al., 2015); coronary artery disease markers in Supplementary Table 9 of (Consortium et al., 2013); lung cancer markers in Supplemental Table S2 of (Timofeeva et al., 2012) that were presented in Figure 2A–G, using the fixed effect model for weights; pancreatic cancer markers in Supplemental Table 4 of (Wolpin et al., 2014), using stage 1 odds ratios for weights; prostate cancer markers in Table 3 of (Zheng et al., 2008); stroke markers in Table e-2 of (Kilarski et al., 2014) with the All_IS phenotype and type 2 diabetes markers in Supplemental Table 3 and Supplemental Table 6 of (Replication et al., 2014), using the European weight estimates. Markers were coded additively, and the logarithms of the reported odds ratios were used as weights. All markers were pruned by pairwise linkage disequilibrium (R2 > 0.8) prior to constructing the genetic risk score. The full list of these markers, corresponding weights, allele frequencies in each of our cohorts, and our single marker association statistics are available in Supplemental Table S3.
Variant Annotations and Pathogenicity
Variant annotation was performed by Cypher Genomics Inc. as described by the SG-ADVISER annotation tool (Pham et al., 2015). High confidence known pathogenic variants are defined as any variant whose allele frequency is no greater than 0.5% in all 1000 Genomes (Sudmant et al., 2015), NHLBI Exome Sequencing Project (Fu et al., 2013), and Exome Aggregation Consortium (http://exac.broadinstitute.org) populations and catalogued as pathogenic or likely pathogenic in ClinVar (Landrum et al., 2014) without any benign or likely benign assertions. Secondary finding variants were determined as defined by (Green et al., 2013). Analyses of rare pathogenic variants considered variant counts on the cohort level only; individual pathogenic variants and actionability at the individual level were not examined.
Genome-Wide Association Studies
Common variants, filtered as described above, were subject to a standard GWAS analysis via PLINK (Purcell et al., 2007) using a logistic model and corrected for the first ten principal components. The QQ plot was generated by pruning association results at a linkage disequilibrium threshold of 0.50. Chromosome positions are provided in GrCh37/hg19 coordinates. Regional plots were generated with LocusZoom with modifications (Pruim et al., 2010). Pairwise linkage disequilibrium from 1000 Genomes (Sudmant et al., 2015) European individuals was calculated with SNAP (Johnson et al., 2008). For rare variants, burden testing was performed with SKAT-O on all rare coding exonic variants (Lee et al., 2012) adjusted for the first two principal components. A gene must have had 5 or more rare variants (as recommended by the SKAT authors) to be included in testing – 9,343 genes satisfied this requirement. The QQ plot was generated as one point per gene.
Supplementary Material
Highlights.
Healthy aging is complex polygenic trait related but distinct from longevity.
Healthy aging is associated with decreased genetic risk for select diseases.
Healthy aging is potentially linked to protection against cognitive decline.
Genome data is made available for further analysis
Acknowledgments
Complete Genomics, Inc. produced and provided all Wellderly WGS data. We would like to thank Wayne Pfeiffer (San Diego Supercomputer Center) for helpful comments on the manuscript and Chunlei Wu, Jiwen Xin, Jackie Atkins and Quang Tran for IT support. This work is supported by Scripps Genomic Medicine, an NIH-NCATS Clinical and Translational Science Award (CTSA; 5 UL1 RR025774) to STSI, the Inova Health System, and a generous gift of the Odeen family to ITMI. Further support is from the Gary and Mary West Foundation, Lavin Family Foundation, NIH-U01 HG006476 and U54GM114833 (AT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
G.A.E., D.L.B., and M.R. performed data curation, developed software, and performed formal analysis and visualization. B.M. developed software for data curation. E.R.S. conceived of the Wellderly sibling survival analysis. A.A.S and S.E.T. obtained study resources. N.E.W conceived of the genetic risk score and provided formal statistical analysis and supervision. E.T. and J.E.N. conceived of research goals, reviewed and edited the manuscript, and acquired funding. A.T. conceived of research goals, wrote the original draft and edits, executed formal analysis and visualization, and provided project supervision, administration, and acquisition of funding.
References
- Al-Tassan NA, et al. A new GWAS and meta-analysis with 1000Genomes imputation identifies novel risk variants for colorectal cancer. Scientific reports. 2015;5:10442. doi: 10.1038/srep10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC bioinformatics. 2011;12:246. doi: 10.1186/1471-2105-12-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzmon G, et al. Lipoprotein genotype and conserved pathway for exceptional longevity in humans. PLoS biology. 2006;4:e113. doi: 10.1371/journal.pbio.0040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekman M, et al. Genome-wide association study (GWAS)-identified disease risk alleles do not compromise human longevity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18046–18049. doi: 10.1073/pnas.1003540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell FC, Miller ML. (Social Security Actuarial Study No 120).Life Tables for the United States Social Security Area 1900–2100. 2005 [Google Scholar]
- Bodian DL, et al. Germline variation in cancer-susceptibility genes in a healthy, ancestrally diverse cohort: implications for individual genome sequencing. PloS one. 2014;9:e94554. doi: 10.1371/journal.pone.0094554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojesen SE, Nordestgaard BG. The common germline Arg72Pro polymorphism of p53 and increased longevity in humans. Cell Cycle. 2008;7:158–163. doi: 10.4161/cc.7.2.5249. [DOI] [PubMed] [Google Scholar]
- Brooks-Wilson AR. Genetics of healthy aging and longevity. Human genetics. 2013;132:1323–1338. doi: 10.1007/s00439-013-1342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen K, et al. The quest for genetic determinants of human longevity: challenges and insights. Nature reviews Genetics. 2006;7:436–448. doi: 10.1038/nrg1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium CAD, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature genetics. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505:550–554. doi: 10.1038/nature12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, et al. Fast computation and applications of genome mappability. PloS one. 2012;7:e30377. doi: 10.1371/journal.pone.0030377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortney K, et al. Genome-Wide Scan Informed by Age-Related Disease Identifies Loci for Exceptional Human Longevity. PLoS genetics. 2015;11:e1005728. doi: 10.1371/journal.pgen.1005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes LU, et al. Estimation of apolipoprotein E genotype-specific relative mortality risks from the distribution of genotypes in centenarians and middle-aged men: apolipoprotein E gene is a “frailty gene,” not a “longevity gene”. Genet Epidemiol. 2000;19:202–210. doi: 10.1002/1098-2272(200010)19:3<202::AID-GEPI2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Goldman DP, et al. Substantial health and economic returns from delayed aging may warrant a new focus for medical research. Health Aff (Millwood) 2013;32:1698–1705. doi: 10.1377/hlthaff.2013.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in medicine: official journal of the American College of Medical Genetics. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurme M, et al. Interleukin-6 -174G/C polymorphism and longevity: a follow-up study. Mechanisms of ageing and development. 2005;126:417–418. doi: 10.1016/j.mad.2004.10.001. [DOI] [PubMed] [Google Scholar]
- International Schizophrenia, C et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuyama H, et al. CLAC binds to aggregated Abeta and Abeta fragments, and attenuates fibril elongation. Biochemistry. 2005;44:15602–15609. doi: 10.1021/bi051263e. [DOI] [PubMed] [Google Scholar]
- Kilarski LL, et al. Meta-analysis in more than 17,900 cases of ischemic stroke reveals a novel association at 12q24.12. Neurology. 2014;83:678–685. doi: 10.1212/WNL.0000000000000707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuningas M, et al. SIRT1 gene, age-related diseases, and mortality: the Leiden 85-plus study. The journals of gerontology Series A, Biological sciences and medical sciences. 2007;62:960–965. doi: 10.1093/gerona/62.9.960. [DOI] [PubMed] [Google Scholar]
- Lambert JC, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature genetics. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic acids research. 2014;42:D980–985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. American journal of human genetics. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libiger O, Schork NJ. A Method for Inferring an Individual’s Genetic Ancestry and Degree of Admixture Associated with Six Major Continental Populations. Frontiers in genetics. 2012;3:322. doi: 10.3389/fgene.2012.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, et al. Temporal Trends in Mortality in the United States, 1969–2013. Jama. 2015;314:1731–1739. doi: 10.1001/jama.2015.12319. [DOI] [PubMed] [Google Scholar]
- Melzer D, et al. A common variant of the p16(INK4a) genetic region is associated with physical function in older people. Mechanisms of ageing and development. 2007;128:370–377. doi: 10.1016/j.mad.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K, et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nature genetics. 2015;47:373–380. doi: 10.1038/ng.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murabito JM, et al. The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long-lived individuals. The journals of gerontology Series A, Biological sciences and medical sciences. 2012;67:470–479. doi: 10.1093/gerona/gls089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada Y, et al. CLAC binds to amyloid beta peptides through the positively charged amino acid cluster within the collagenous domain 1 and inhibits formation of amyloid fibrils. J Biol Chem. 2005;280:8596–8605. doi: 10.1074/jbc.M413340200. [DOI] [PubMed] [Google Scholar]
- Pawlikowska L, et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging cell. 2009;8:460–472. doi: 10.1111/j.1474-9726.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham PH, et al. Scripps Genome ADVISER: Annotation and Distributed Variant Interpretation SERver. PLoS One. 2015;10:e0116815. doi: 10.1371/journal.pone.0116815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Goldstein DB. Detecting association in a case-control study while correcting for population stratification. Genet Epidemiol. 2001;20:4–16. doi: 10.1002/1098-2272(200101)20:1<4::AID-GEPI2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Replication DIG, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nature genetics. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reumers J, et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nature biotechnology. 2012;30:61–68. doi: 10.1038/nbt.2053. [DOI] [PubMed] [Google Scholar]
- Rietveld CA, et al. Common genetic variants associated with cognitive performance identified using the proxy-phenotype method. Proc Natl Acad Sci U S A. 2014;111:13790–13794. doi: 10.1073/pnas.1404623111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietveld CA, et al. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science. 2013;340:1467–1471. doi: 10.1126/science.1235488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AE, et al. Association of a functional polymorphism in the cholesteryl ester transfer protein (CETP) gene with memory decline and incidence of dementia. Jama. 2010;303:150–158. doi: 10.1001/jama.2009.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani P, et al. Genetic signatures of exceptional longevity in humans. PloS one. 2012;7:e29848. doi: 10.1371/journal.pone.0029848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SY, et al. An atlas of genetic influences on human blood metabolites. Nature genetics. 2014;46:543–550. doi: 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg L, et al. Characterization of the Alzheimer’s disease-associated CLAC protein and identification of an amyloid beta-peptide-binding site. J Biol Chem. 2005;280:1007–1015. doi: 10.1074/jbc.M403628200. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh Y, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeeva MN, et al. Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Human molecular genetics. 2012;21:4980–4995. doi: 10.1093/hmg/dds334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control, C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, et al. FOXO3A genotype is strongly associated with human longevity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wokolorczyk D, et al. A range of cancers is associated with the rs6983267 marker on chromosome 8. Cancer research. 2008;68:9982–9986. doi: 10.1158/0008-5472.CAN-08-1838. [DOI] [PubMed] [Google Scholar]
- Wolpin BM, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nature genetics. 2014;46:994–1000. doi: 10.1038/ng.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Pierce BL. Genetic susceptibility to accelerated cognitive decline in the US Health and Retirement Study. Neurobiol Aging. 2014;35:1512 e1511–1518. doi: 10.1016/j.neurobiolaging.2013.12.021. [DOI] [PubMed] [Google Scholar]
- Zheng SL, et al. Cumulative association of five genetic variants with prostate cancer. The New England journal of medicine. 2008;358:910–919. doi: 10.1056/NEJMoa075819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Aggregate unfiltered annotated Wellderly variants and their allele and genotype frequencies are available via Scripps Translational Science Institute Variant Browser (http://genomics.scripps.edu/browser). Variant presence is also queryable via a Global Alliance for Genomics and Health Beacon (http://genomics.scripps.edu/browser/ga4gh). Individual level variant data are available from Complete Genomics Inc. under terms determined by Complete Genomics Inc., from Scripps Genomic Medicine for scientific collaboration with not for profit entities, and will be deposited in dbGAP under similar data use restrictions.