

Abstract
The human Epstein-Barr virus (EBV) evades the immune system by entering a transcriptionally latent phase in B cells. EBV in tumor cells express distinct patterns of genes referred to as latency types. Viruses in tumor cells also display varying levels of lytic transcription resulting from spontaneous reactivation out of latency. We measured this dynamic range of lytic transcription with RNA deep sequencing and observed no correlation with EBV latency types among genetically different viruses, but type I cell lines reveal more spontaneous reactivation than isogenic type III cultures. We further determined that latency type and spontaneous reactivation levels predict the relative amount of induced reactivation generated by cytotoxic chemotherapy drugs. Our work has potential implications for personalizing medicine against EBV-transformed malignancies. Identifying latency type or measuring spontaneous reactivation may provide predictive power in treatment contexts where viral production should be either avoided or coerced.
Keywords: Epstein-Barr virus, transcriptome, latency, reactivation, RNA-seq
1. Introduction
The human Epstein-Barr virus (EBV) generates a global health burden. This lymphotrophic DNA herpesvirus evades the immune system by entering a latent phase in B cells that persists for life. Compared to lytic replication, during which about a hundred genes are expressed to amplify virus, latency restricts transcription to approximately a dozen or fewer genes [1]. EBV can immortalize B cells in vitro [2]. EBV is also associated with Burkitt lymphoma, Hodgkin lymphoma, nasopharyngeal carcinoma, gastric carcinoma, and other malignancies in vivo [1]. The requirement of specific latent genes for in vitro transformation strongly argues that viral infection contributes to oncogenesis in vivo [3]. Indeed, persistent infection correlates with ~1% of all cancer worldwide [4]. Detailed analysis of viral transcription during latency forms the foundational context for understanding how EBV interacts with cancer cells.
Latent forms of EBV express different sets of genes partially dependent on the developmental state of the cancer cell prior to immortalization [1]. While many exceptions to the rule exist, transcriptional programs generally stereotype to one of a few patterns. Type I latency limits expression to EBNA1, the gene encoding the DNA-binding protein responsible for latent replication and segregation, the EBER non-coding RNAs, and the BamHI A rightward transcript (BART) RNAs. Type III latency further includes other EBNA isoforms and messages for the three LMP products. BHRF1 is expressed under certain conditions [5, 6].
Improving upon previous genomic technologies, RNA deep sequencing (RNA-seq) methods have illuminated novel details of viral transcription [7–12]. Increased sensitivity identified new transcripts and splice variants. Examination of Burkitt lymphoma cell lines uncovered both latent and lytic gene expression [7, 8, 11]. Lymphoblastoid cell lines (LCLs) display a wide range of spontaneous reactivation [9]. We asked if the dynamic range of lytic transcription in cell culture lines correlated with different EBV latency types. We further use measurement of spontaneous reactivation by RNA-seq to predict the induction response to cytotoxic chemotherapy drugs. Our work may have implications for personalizing medicine against EBV-transformed malignancies.
2. Material and methods
2.1. Cell Culture
We maintained Burkitt lymphoma cell lines at 37 °C with 5% CO2 (v/v) in RPMI-1640 media containing 25 mM HEPES and 10% (v/v) fetal bovine serum. We maintained LCLs similarly except with 15% (v/v) fetal bovine serum. MutuI [13] cells grew under standard conditions [14]. Raji [15] (CCL-86) and Daudi [16] (CCL-213) cell lines were obtained from ATCC (Manassas, VA). The GM12878 [17] (GM12878) cell line was obtained from the Coriell Institute for Medical Research (Camden, NJ). Jeffery T. Sample (Pennsylvania State University) provided the KemI and KemIII [18] cell lines, Andrew I. Bell (University of Birmingham) provided the RaeI [19] cell line, and Bill Sugden (University of Wisconsin, Madison) provided the 721 LCL [20]. We generated MutuIII by prolonged passaging of MutuI in cell culture [13].
2.2. RNA-Seq
We isolated RNA from 4 × 106 log phase cells homogenized with a QIAshredder spin column (Qiagen). Total RNA was purified by silica-based membrane affinity as packaged in the RNeasy Mini Kit (Qiagen). Preparations included the optional DNAse treatment step. Single primer isothermal linear amplification to cDNA [21] was achieved using the Ovation RNA-Seq System V2 (NuGEN) and 20 ng of RNA. 3 μg of cDNA was then sheared in a 40 μL volume using a Covaris S2 Focused-ultrasonicator. We prepared deep sequencing libraries by adaptor-mediated amplification [22] as packaged in either the Encore NGS Library System I (NuGEN) or Ovation Ultralow Library System V2 (NuGEN).
Each library was sequenced on a HiSeq (Illumina). 50 bp reads were mapped using Bowtie [23] to an index containing both the human hg19 and EBV reference [GenBank ID: NC_007605.1] genomes. Parameters allowed for up to two mismatches and only considered reads that mapped to a unique sequence. The number of hits at each base was counted and then normalized per million mapped reads. RNA-seq profiling for every cell line was performed with two or three biological replicates and yielded ~30–100 million mapped sequences each experiment with reproducible transcriptome profiles.
2.3 Viral Reactivation
We induced lytic replication of EBV with chemicals [14] and measured reactivation by staining for the immediate-early lytic transactivator BZLF1 using the paraformaldehyde-methanol method [24] with a BZ1 antibody (Santa Cruz Biotechnology) and goat anti-mouse IgG-FITC (Santa Cruz Biotechnology). Cells were treated for three days with 100 μM bendamustine (Sigma-Aldrich or Millipore), 1 μg/mL gemcitabine (Sigma-Aldrich), or 20 nM romidepsin (Selleck Chemicals).
3. Results
3.1. Dynamic Range of Spontaneous Reactivation
We began by asking what a strictly latent EBV transcriptome looks like as measured by RNA-seq. To do so we examined the Raji [15] and Daudi [16] cell lines, which adopt latency type III and I, respectively. Both express very few viral gene products either spontaneously or in response to stimuli: these lines contain little early antigen under basal conditions, chemical treatment only weakly increases early antigen production, and chemical treatment cannot generate viral capsid antigen production [25, 26]. By the above metrics, Raji cells are generally more latent than Daudi cells. Mechanistically, Raji genomes have a deletion in the BALF2 gene necessary for progression through lytic replication [27]. Daudi genomes have deletions in other genes [28], although the source of the lytic replication defect is less understood. Both Raji and Daudi transcriptomes predominantly consist of signals from latent genes (Figure 1). Identified Raji transcripts include EBNA2, BHRF1, and LMP1. Other overlapping EBNA exons are also expressed. In Daudi cells, the strongest signal is the BHRF1 message. Our results resemble analysis performed by others with the same lines [11]. Signals from lytic gene expression generally peak at ~0–2 counts per million mapped reads. These profiles act as a baseline for defining a latent transcriptome.
Fig. 1.

Deep sequencing of EBV transcription in Raji and Daudi cell lines. The X axis denotes nucleotide position and the Y axis denotes the number of counts per million mapped reads. RNA signals with unambiguously assignable annotations are marked. BHRF1 and the BARTs are latent transcripts labeled blue. One representative transcriptome from independent replicates is shown.
Cell lines with the same latency type display a broad range of spontaneous reactivation. We measured the transcriptomes of MutuI, KemI, and RaeI cells as a sampling of type I latency lines (Figure 2). In agreement with previously published data [7], we detected widespread lytic transcription in the MutuI line. BHLF1 displays the strongest signal. Initiation of lytic DNA replication requires transcription of this gene [29] and its expression level correlates with spontaneous reactivation [9]. Other predominant signals correspond to lytic transcripts as well. The KemI expression profile is similar: BHLF1 displays the strongest signal amidst many lytic transcripts. The RaeI line, however, contrasts significantly and does not reveal lytic transcription on the same scale. The only notable signals consist of BHRF1 and the BARTs. Trace levels of lytic gene expression resemble amounts observed with Raji and Daudi cells. RaeI transcriptomes therefore also reflect the more latent end of the dynamic range of spontaneous reactivation.
Fig. 2.

Deep sequencing of EBV transcription in latency I and latency III cell lines. The X axis denotes nucleotide position and the Y axis denotes the number of counts per million mapped reads. RNA signals with unambiguously assignable annotations are marked. BHRF1, Cp, EBNA2, and LMP1 are latent transcripts labeled blue; all others are lytic transcripts labeled red. One representative transcriptome from independent replicates is shown.
Type III latency lines display variability of spontaneous reactivation similar to type I lines (Figure 2). The GM12878 LCL reveals a pattern consisting of the strong BHLF1 signal and other lytic genes. Significant levels of latent transcripts, such as LMP1 and the 5′ end of the EBNA message from the C promoter, are also observed. The 721 LCL, KemIII, and MutuIII lines, however, predominantly express latent transcripts such as EBNA messages, BHRF1, and LMP1. We were reluctant to apply quantitative metrics previously used to compare lyticness, such as percentage of viral compared to human reads, or strength of the BHLF1 signal, to rank the lines we studied. While those approaches work when comparing transcriptomes with similar profiles and the same sequence [9], different levels of latent gene expression between viruses with different sequences confound analysis. We find that visual inspection, qualitatively comparing signals from latent and lytic genes, proves informative. Because of the large variability in levels of spontaneous reactivation within each latency type, bulk comparison of type I and type III lines does not illuminate any general trend between latency type and spontaneous reactivation levels.
3.2. Latency Type Predicts Spontaneous Reactivation
Cell lines with type I latency undergo higher levels of spontaneous lytic transcription as measured by RNA-seq compared to isogenic type III cultures. Genetic differences in both the host and virus confound general comparisons between latency types using a hodgepodge of cell lines. We therefore chose two sets of isogenic pairs for detailed study. We derived the MutuIII line from MutuI cells in culture [13]. While the MutuI transcriptome mostly consists of signals from lytic genes, MutuIII predominantly reveals latent transcripts (Figure 2). The KemI and KemIII lines were both obtained from the same tumor and yet adopt different latency programs [18]. The same relative difference holds as observed with the Mutu cell lines: the KemI transcriptome is more lytic than the KemIII transcriptome (Figure 2). The dynamic range of spontaneous reactivation between different lines of the same latency program is vast (Figure 2), therefore a randomly selected type I line may yield either more or less lytic transcription when compared to a non-isogenic type III line. When comparing isogenic pairs with RNA-seq profiling, however, type I lines reveal more spontaneous reactivation than the matched type III line.
3.3. Spontaneous Reactivation Predicts Induction Levels
Perturbation with cytotoxic chemotherapy agents induces greater lytic reactivation in type I latency lines with higher spontaneous reactivation levels compared to isogenic type III cultures. We suspected that the biochemical mechanism allowing for more spontaneous reactivation would also permit greater lytic induction upon chemical treatment. DNA damaging agents and chromatin modifiers such as the alkylator bendamustine [14], the nucleoside analog gemcitabine [30], and the histone deacetylase inhibitor romidepsin [31] disrupt EBV latency. Incubation of MutuI cells with bendamustine, gemcitabine, and romidepsin readily induces lytic reactivation as measured by increased expression of the BZLF1 immediate early protein (Figure 3). Although we detect BZLF1 in less than 1% of untreated cells, ~20–40% of treated cells stain positively by flow cytometry. Parallel treatment of MutuIII cells yields much less or no reactivation. Only 0–2% of both treated and untreated cells stain positively. A similar, though less stark, contrast is seen between Kem lines. Incubation of KemI cells with romidepsin moderately increases lytic reactivation from 0% in untreated cells to 6% in treated cells. KemIII cells yield no significant increase between 1–2% of positively staining cells (Figure 4). We unfortunately could not obtain parallel data with bendamustine and gemcitabine because both Kem lines were resistant to induction by those chemicals. The trend observed for latency reversing agents with which we have data, however, is consistent: type I lines yield greater amounts of induced reactivation compared to the isogenic type III line upon drug treatment. Because isogenic type I cell lines display both more spontaneous reactivation and more induced reactivation when compared to matched type III lines, the levels of lytic transcription under spontaneous and induced conditions correlate. Relative spontaneous reactivation measured with RNA-seq therefore also predicts relative amounts of induced reactivation in isogenic lines.
Fig. 3.

EBV reactivation in MutuI and MutuIII cells induced by bendamustine, gemcitabine, and romidepsin. BZLF1 expression was measured by flow cytometry. Error bars represent the standard deviation of n = 4 replicates.
Fig. 4.
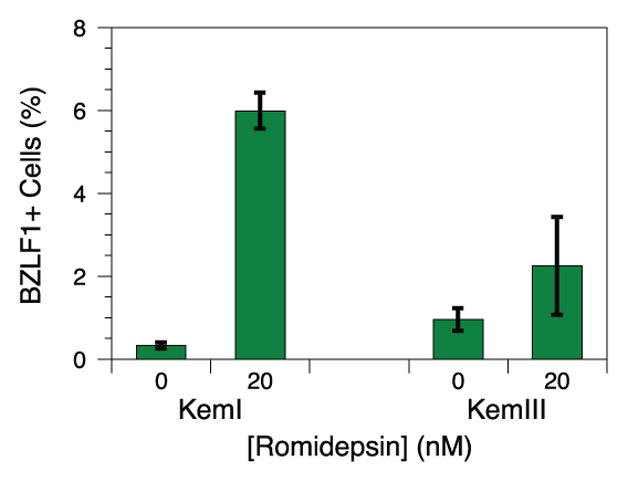
EBV reactivation in KemI and KemIII cells induced by romidepsin. BZLF1 expression was measured by flow cytometry. Error bars represent the standard deviation of n = 4 replicates.
4. Discussion
The biochemical basis of spontaneous reactivation in EBV lends the system to prediction of input/output behavior. Expression of the immediate early gene BZLF1 initiates a transcriptional cascade that results in the activation of lytic genes to assemble new viral particles [32]. Small amounts of abortive replication, an incomplete cascade, are detectable by RNA-seq of LCLs [9]. Different biochemical events generate stochastic gene expression [33]. We do not formally know if spontaneous lytic transcription results from intrinsic noise, such as gene-, network-, and cell-level variability that activates the BZLF1 promoter, or from environmental stimuli triggering signaling events sensed by EBV under physiological conditions. Levels of spontaneous reactivation in LCLs oscillate periodically [34]. Because cyclical processes suggest the presence of a changing stimulus, we suspect that spontaneous lytic transcription results from signaling events. If true, then under equal environmental conditions for two cell lines, greater levels of spontaneous reactivation in one line may reflect greater responsiveness of that EBV transcriptional network to the current stimulus. That greater responsiveness may be leveraged upon treatment with small molecules known to reactive virus. We sought to test, and argue that we have verified, this hypothesis.
The ability to predict the extent of induced reactivation in EBV-containing tumor cells may inform clinical strategies to cure cancer. Lytic induction therapy attempts to generate replicating virus to serve as targets for immune responses or antiviral drugs [35]. Unfortunately, we currently know of few molecular markers to predict the efficacy of latency-disrupting agents. The viral LMP1 protein suppresses reactivation by anti-IgM and phorbol esters [36]. Our work advances predictive power in two practical ways. First, comparing latency types focuses on a frequently observed distinction between viruses in tumors; rarely does the transcriptional difference between two isolates consist only of LMP1 expression. Indeed, both type I and type III latency occur in lymphomas with similar presentation [12]. Second, we examine cytotoxic chemotherapy drugs that are currently used to treat patients with EBV-associated malignancies in the clinic. Although our analysis only predicts differences in isogenic backgrounds, suggesting that other factors confound comparisons, we identified regulated molecular signatures of reactivation potential that add to our understanding of this complex transition from latency. Knowledge of potential lytic replication benefits multiple contexts. In some cases, reactivation should be avoided to reduce adverse effects caused by increased viral load. In other cases, reactivation would be desired to execute lytic induction therapy. Profiling viral transcription may provide predictive power that could yield tangible improvements in treatment decisions.
Supplementary Material
Highlights.
EBV latency type predicts spontaneous reactivation levels.
EBV latency type predicts induced reactivation generated by drugs.
Spontaneous reactivation levels predict induced reactivation generated by drugs.
Acknowledgments
We thank Jeffery T. Sample (Pennsylvania State University), Andrew I. Bell (University of Birmingham), and Bill Sugden (University of Wisconsin, Madison) for providing cell lines. We are grateful to the Gladstone Genomics Core and the UCSF Center for Advanced Technology for use of shared equipment. Flow cytometry experiments were made possible with help from the University of California San Francisco-Gladstone Center for AIDS Research (CFAR), an NIH-funded program (P30 AI027763). Our work was supported by funding from the University of California, San Francisco and a subcontract of NIH grant P30 CA082103.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nature reviews Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Pattengale PK, Smith RW, Gerber P. Selective transformation of B lymphocytes by E.B. virus. Lancet. 1973;2:93–94. doi: 10.1016/s0140-6736(73)93286-8. [DOI] [PubMed] [Google Scholar]
- 3.Young LS, Murray PG. Epstein-Barr virus and oncogenesis: from latent genes to tumours. Oncogene. 2003;22:5108–5121. doi: 10.1038/sj.onc.1206556. [DOI] [PubMed] [Google Scholar]
- 4.Parkin DM. The global health burden of infection-associated cancers in the year 2002. International journal of cancer Journal international du cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 5.Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS pathogens. 2009;5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xing L, Kieff E. Epstein-Barr virus BHRF1 micro- and stable RNAs during latency III and after induction of replication. Journal of virology. 2007;81:9967–9975. doi: 10.1128/JVI.02244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Z, Xu G, Deng N, Taylor C, Zhu D, Flemington EK. Quantitative and qualitative RNA-Seq-based evaluation of Epstein-Barr virus transcription in type I latency Burkitt’s lymphoma cells. Journal of virology. 2010;84:13053–13058. doi: 10.1128/JVI.01521-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Concha M, Wang X, Cao S, Baddoo M, Fewell C, Lin Z, Hulme W, Hedges D, McBride J, Flemington EK. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. Journal of virology. 2012;86:1458–1467. doi: 10.1128/JVI.06537-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell host & microbe. 2012;12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moss WN, Steitz JA. Genome-wide analyses of Epstein-Barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC genomics. 2013;14:543. doi: 10.1186/1471-2164-14-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao S, Strong MJ, Wang X, Moss WN, Concha M, Lin Z, O’Grady T, Baddoo M, Fewell C, Renne R, Flemington EK. High-throughput RNA sequencing-based virome analysis of 50 lymphoma cell lines from the Cancer Cell Line Encyclopedia project. Journal of virology. 2015;89:713–729. doi: 10.1128/JVI.02570-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arvey A, Ojesina AI, Pedamallu CS, Ballon G, Jung J, Duke F, Leoncini L, De Falco G, Bressman E, Tam W, Chadburn A, Meyerson M, Cesarman E. The tumor virus landscape of AIDS-related lymphomas. Blood. 2015;125:e14–e22. doi: 10.1182/blood-2014-11-599951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregory CD, Rowe M, Rickinson AB. Different Epstein-Barr virus-B cell interactions in phenotypically distinct clones of a Burkitt’s lymphoma cell line. The Journal of general virology. 1990;71(Pt 7):1481–1495. doi: 10.1099/0022-1317-71-7-1481. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez SG, Miranda JL. Bendamustine reactivates latent Epstein-Barr virus. Leuk Lymphoma. 2015:1–3. doi: 10.3109/10428194.2015.1079317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pulvertaft JV. Cytology of Burkitt’s Tumour (African Lymphoma) Lancet. 1964;1:238–240. doi: 10.1016/s0140-6736(64)92345-1. [DOI] [PubMed] [Google Scholar]
- 16.Klein E, Klein G, Nadkarni JS, Nadkarni JJ, Wigzell H, Clifford P. Surface IgM-kappa specificity on a Burkitt lymphoma cell in vivo and in derived culture lines. Cancer research. 1968;28:1300–1310. [PubMed] [Google Scholar]
- 17.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshioka M, Crum MM, Sample JT. Autorepression of Epstein-Barr virus nuclear antigen 1 expression by inhibition of pre-mRNA processing. Journal of virology. 2008;82:1679–1687. doi: 10.1128/JVI.02142-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly G, Bell A, Rickinson A. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med. 2002;8:1098–1104. doi: 10.1038/nm758. [DOI] [PubMed] [Google Scholar]
- 20.Kavathas P, Bach FH, DeMars R. Gamma ray-induced loss of expression of HLA and glyoxalase I alleles in lymphoblastoid cells. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:4251–4255. doi: 10.1073/pnas.77.7.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurn N, Chen P, Heath JD, Kopf-Sill A, Stephens KM, Wang S. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clinical chemistry. 2005;51:1973–1981. doi: 10.1373/clinchem.2005.053694. [DOI] [PubMed] [Google Scholar]
- 22.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 23.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imbert-Marcille BM, Coste-Burel M, Robillard N, Foucaud-Gamen J, Billaudel S, Drouet E. Sequential use of paraformaldehyde and methanol as optimal conditions for the direct quantification of ZEBRA and rta antigens by flow cytometry. Clin Diagn Lab Immunol. 2000;7:206–211. doi: 10.1128/cdli.7.2.206-211.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.zur Hausen H, O’Neill FJ, Freese UK, Hecker E. Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature. 1978;272:373–375. doi: 10.1038/272373a0. [DOI] [PubMed] [Google Scholar]
- 26.Luka J, Kallin B, Klein G. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology. 1979;94:228–231. doi: 10.1016/0042-6822(79)90455-0. [DOI] [PubMed] [Google Scholar]
- 27.Decaussin G, Leclerc V, Ooka T. The lytic cycle of Epstein-Barr virus in the nonproducer Raji line can be rescued by the expression of a 135-kilodalton protein encoded by the BALF2 open reading frame. Journal of virology. 1995;69:7309–7314. doi: 10.1128/jvi.69.11.7309-7314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones MD, Foster L, Sheedy T, Griffin BE. The EB virus genome in Daudi Burkitt’s lymphoma cells has a deletion similar to that observed in a non-transforming strain (P3HR-1) of the virus. The EMBO journal. 1984;3:813–821. doi: 10.1002/j.1460-2075.1984.tb01890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rennekamp AJ, Lieberman PM. Initiation of Epstein-Barr virus lytic replication requires transcription and the formation of a stable RNA-DNA hybrid molecule at OriLyt. Journal of virology. 2011;85:2837–2850. doi: 10.1128/JVI.02175-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng WH, Hong G, Delecluse HJ, Kenney SC. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. Journal of virology. 2004;78:1893–1902. doi: 10.1128/JVI.78.4.1893-1902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hui KF, Cheung AK, Choi CK, Yeung PL, Middeldorp JM, Lung ML, Tsao SW, Chiang AK. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. International journal of cancer Journal international du cancer. 2016;138:125–136. doi: 10.1002/ijc.29698. [DOI] [PubMed] [Google Scholar]
- 32.Countryman J, Miller G. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:4085–4089. doi: 10.1073/pnas.82.12.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 34.Davies ML, Xu S, Lyons-Weiler J, Rosendorff A, Webber SA, Wasil LR, Metes D, Rowe DT. Cellular factors associated with latency and spontaneous Epstein-Barr virus reactivation in B-lymphoblastoid cell lines. Virology. 2010;400:53–67. doi: 10.1016/j.virol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Israel BF, Kenney SC. Virally targeted therapies for EBV-associated malignancies. Oncogene. 2003;22:5122–5130. doi: 10.1038/sj.onc.1206548. [DOI] [PubMed] [Google Scholar]
- 36.Adler B, Schaadt E, Kempkes B, Zimber-Strobl U, Baier B, Bornkamm GW. Control of Epstein-Barr virus reactivation by activated CD40 and viral latent membrane protein 1. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:437–442. doi: 10.1073/pnas.221439999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.