

Abstract
To study CD4+ T-cell suppression of AIDS virus replication, we isolated nine rhesus macaque SIVGag-specific CD4+ T-cell clones. One responding clone, Gag68, produced a typical cytotoxic CD8+ T-cell response: induction of intracellular IFN-γ, MIP-1α, MIP-1β, and CD107a degranulation. Gag68 effectively suppressed the spread of SIVmac239 in CD4+ T cells with a corresponding reduction of infected Gag68 effector cells, suggesting that CD4+ effectors need to suppress their own infection in addition to their targets to be effective. Gag68 TCR cloning and gene transfer into CD4+ T cells enabled additional experiments with this unique specificity after the original clone senesced. Our data supports the idea that CD4+ T cells can directly limit AIDS virus spread in T cells. Furthermore, Gag68 TCR transfer into CD4+ T-cell clones with differing properties holds promise to better understand the suppressive effector mechanisms used by this important component of the antiviral response using the rhesus macaque model.
Keywords: SIV, cytolytic CD4+ T cells, T-cell receptor, virus-specific, TCR transduction, virus suppression, T-cell effectors
INTRODUCTION
While much is known about the immunobiology of the CD8+ T-cell response to HIV/SIV infection, the role of CD4+ T cells as effectors that directly contribute to cell-mediated virus control remains much less studied. Classically CD4+ T cells are thought of as helper CD4+ T cells; however, they can also provide direct effector function (Marshall and Swain, 2011; Soghoian and Streeck, 2010). Circumstantial evidence for direct suppression by anti-HIV CD4+ effector T cells comes from findings that CD4+ T cells from elite controllers and vaccinees also exhibit an increased capacity to induce cytolytic responses as well as inflammatory cytokines and chemokines (Kannanganat et al., 2007a; Kannanganat et al., 2007b; Soghoian et al., 2012; Vingert et al., 2012; von Gegerfelt et al., 2010). Furthermore, HIV elite controllers have a greater bias towards Gag- and Nef- specific CD4+ T cell responses (Norris et al., 2004), which exhibit a cytotoxic response profile or cytolysis of peptide-loaded B cells (Appay et al., 2002; Nemes et al., 2010). Recent work by Johnson et al. has observed that CD4+ T cells can reduce virus levels in HIV-1-infected CD4+ T cells with a tight correlation between levels of cytotoxic T cells and HIV-1 viral load in blood over the acute-chronic infection period (Johnson et al., 2015). The authors also noted a cooperative effect between anti-HIV CD8+ and CD4+ cytolytic T cells which enhanced viral control in vitro.
In this study we sought to better understand the contribution of CD4+ T cells to SIV control by isolating SIV Gag-specific CD4+ T cells and examining their ability to act as effectors in vitro. Because one important difference between CD8+ and CD4+ effectors is that the CD4+ T-cell effectors are also targets of AIDS viruses themselves (Brenchley et al., 2006; Douek et al., 2001; Jain et al., 2015), we also examined the correlation between antiviral function and effector infection. Our results show that SIV Gag-specific CD4+ T cells can act as effectors, producing MHC class II (MHC-II)-restricted antigen-induced polyfunctional effector responses and direct suppression of SIV replication in both the CD4+ T-cell targets as well as themselves.
MATERIALS AND METHODS
Animals
All procedures were carried under protocols approved by the Animal Care and Use Committee, National Cancer Institute, National Institutes of Health (NIH). Animals used for this study were Indian rhesus macaques 86I, KTM, MK9, ZB35, EZP, ZA43, KMB, FB1, CO102, and B001. Blood draws were carried out to minimize animal discomfort. Animals were housed at the NIH animal facility in Bethesda in accordance with the Animal Welfare Act and other US federal statutes and regulations relating to animals and experiments and in accordance with the instructions of the Committee on the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council and the U.S. Public Health Service Guidelines, Guide for the Care and Use of Laboratory Animals. All animals were cared for and used humanely according to the following policies: the U.S. Public Health Service Policy on Humane Care and Use of Animals (2000); the Guide for the Care and Use of Laboratory Animals (1996); and the U.S. Government Principles for Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training (1985). All National Cancer Institute animal facilities and the animal program are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
CD4+ T-cell culture and cloning
SIV-specific CD4+ T-cell clones were isolated from PBMC drawn from 86I, a previously described SIV subunit DNA vaccinated rhesus macaque (Patel et al., 2010). Primary rhesus T cells were cultured in T-cell medium: RPMI1640 supplemented with 10% vol/vol fetal bovine serum (FBS), 2 mM L-glutamine, 100 U per ml penicillin, 100 μg per ml streptomycin and recombinant human interleukin-2 (IL-2; 50 IU/mL, NIH AIDS Reagent Repository). T cell cultures were expanded by the addition of anti-CD3 treatment (30 ng/mL; clone SP34-2; BD Biosciences, San Jose, CA), and irradiated human PBMC and Epstein-Barr virus transformed human B-cell lines (TM B-LCL) biweekly and maintained as previously described (Berger et al., 2001; Minang et al., 2009a; Riddell and Greenberg, 1990). SIVGag specific CD4+ T cells were cloned essentially as previously described (Minang et al., 2008). Briefly, the number of SIVGag-specific CD4+ T cells in 86I PBMC was amplified by stimulation with autologous PBMC pulsed with a complete coverage SIVmac239 Gag 15-mer Peptide Pool (cat # 12364: 11 amino acid overlap, NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH). After three rounds over three weeks, responding T cells were cloned by limiting dilution. The surface CD3− Jurkat derivative J.RT3-T3.5 cell line (Weiss, Wiskocil, and Stobo, 1984) was maintained in T-cell media without IL-2.
Flow cytometry
PBMC and T cells were surfaced stained with fluorochrome-conjugated mAbs: CD3 (SP34-2), CD45 (HI30), CD4 (OKT4), CD8 (SK1; BD Biosciences), NGFR (ME20.4-1.H4; Miltenyi Biotech, San Diego, CA). 1 × 106 cells were washed and stained with antibodies diluted in 100 μL staining buffer (PBS with 1% vol/vol FBS) at room temperature for 20 minutes and then washed 3 times with staining buffer. At least 100,000 events were acquired on a BD LSRII (BD Biosciences). Sorting was done using the BD FACS ARIA II (BD Biosciences). Data analysis was performed using FCS Express 4 (De Novo Software, Glendale, CA).
Intracellular cytokine staining
Peptide-pulsed autologous CD4+ T cells were used as antigen presenting cells (APC) for stimulation assays. APC were labeled with 5μM CellTrace Violet (CTV) as recommended by the manufacturer (Life Technologies, Carlsbad, CA) and pulsed with the Gag peptide for 30 m at room temperature and washed D-PBS and then resuspended in T-cell medium. 1 × 106 CD4+ T cells or T-cell clones were co-cultured with an equal amount of pulsed or unpulsed APC for 6 h with monensin (Golgi stop; BD Biosciences) and anti-human CD107a (H4A3; BD Biosciences). Cells were surface stained, fixed and permeabilized with Cytofix/Cytoperm solution (BD Biosciences) and stained for IFN-γ (#B27) and MIP1-β (# D21-1351) both from BD Biosciences.
Viral stocks
SIVmac239 of were generated by transfecting HEK293T cells with the pSIVmac239SPXFL (Genbank Accession No. AAA99261) (Kestler et al., 1990) and the Nef G2V myristylation mutant SIVmac239myr molecular clone plasmids using TransIt-293 reagent (Mirus Bio LLC, Madison, WI) as previously described (Barsov et al., 2011; Minang et al., 2008; Minang et al., 2009b).
Virus suppression assay
In vitro inhibition of virus replication was performed as previously described (Minang et al., 2008). CD4+ T cells were stimulated for 2–3 days with plate bound anti-CD3 antibody (5 μg/mL), labeled with CellTrace Violet (CTV; Life Technologies) and then infected at an MOI of 0.01 using the Viromag magnetofection procedure (Oz Biosciences, San Diego, CA). Effectors and targets were co-cultured in a 96-well U bottom plate at an effector: target (E:T) ratio of 1:1 and 10:1. Virus exposed CD4+ T cells were cultured for 7 days, with IL-2 addition every 2 to 3 days at a final concentration of 50 IU/ml. Infection spread was assessed on day 5 and 7 by flow cytometry using an FITC-conjugated anti-SIV Gag p27 mAb (Clone 55-2F12; NIH AIDS Research Reagent Program). Viral load of co-culture supernatants was measured by real-time RT-PCR as described below.
Virus quantitation
Viral RNA in supernatants and cell-associated viral DNA cell cultures was measured by real-time qRT-PCR or qPCR normalized to CCR5 gene copy numbers, respectively, as previously described (Cline et al., 2005; Lifson et al., 2001).
ELISpot
ELISpot assays were performed using the Monkey IFN-γ ELISpotPLUS kit (Mabtech, Cincinnati, OH) according to the manufacturer’s protocol. Briefly, ELISpot plates were coated with capture antibody (GZ-4) diluted in PBS (15μg/mL) overnight at 4°C. The plate was washed with PBS and blocked with T-cell medium for 30 m. CD4+ T cells were seeded at 2 × 105/well in complete RPMI 1640 media with the 5327 and 5328 Gag68 epitope peptides or SIVpeptide pools (1 μg/mL for each peptide). The plates were incubated for 18 h at 37°C in 5% CO2 and subsequently washed with PBS. Anti-IFN-γ antibody (7-B6-Biotin) diluted 1:1000 in PBS containing 0.5% (vol/vol) FBS (PBS-F) was added to the plates and incubated for 2 h at room temperature, washed 3 times in PBS-F, and incubated for 1 h at room temperature with streptavidin-horseradish peroxidase in PBS-F. Following three PBS-F washes, the plates were developed with TMB-substrate for 10–15 m and color development was stopped by washing extensively with deionized water. ELISpot plates were imaged using an AID ELISpot reader (ver 3.0 Rev.2, Autoimmun Diagnostika GmbH, Strassberg, Germany) for positive reacting wells. Epitope mapping was performed as previously described (Frahm et al., 2004).
MHC typing
MHC I and MHC II alleles expressed by macaques were genotyped by Roche/454 pyrosequencing of cDNA amplicons as described previously (Karl et al., 2014; Wiseman et al., 2013).
Retroviral vector construction
The Gag68 T-cell receptor α and β chain genes were cloned from Gag68 RNA using the SMARTer RACE cDNA Amplification kit (Takara Clontech, Mountain View, CA) according to manufacturer’s protocol with gene-specific primers and sequenced. Nucleotide sequences for the α and β variable regions are available in GenBank, (Accession #’s KP233091 and KP272132, respectively). An murine leukemia virus-based TCR expression vector was produced as previously described (Coren et al., 2015). Briefly, V α or V β region primers were used to PCR amplify their respective V regions that were then fused to our MSGV-based Rh acceptor PGK NGFR vector in a one-step golden-gate cloning procedure to make the pTCR-Gag68-NGFR transfer vector expressing both the Gag68 TCR and a truncated human low affinity nerve growth factor receptor (tNGFR). The resulting was produced by transfection into Phoenix-RD114 packaging cells (Neff et al., 2004) using TransIT-293 Reagent (Mirus Bio LLC) and harvesting 48 h post infection. Vector supernatants were clarified by filtration though a 0.45 micron filter.
TCR transduction
A non-treated tissue culture 12-well plate was coated with 25 μg/ml recombinant human fibronectin fragment (RetroNectin; Takara Clonetech). Gag68 retrovirus supernatant was added to the RetroNectin-coated plate and centrifuged 1,500 × g for 2 h at 32°C. CD4+ T cells stimulated for 48 h were transferred to the vector-loaded culture dishes (1.5 × 106 cells/well), centrifuged 1,500 × g for 1 h at 32°C and incubated at 37°C, 5% CO2 for 2 – 3 days before examining for tNGFR expression. Transductants were isolated by staining with tNGFR-PE antibody (clone C40-1457, BD Biosciences) and anti-PE paramagnetic microbead sorting (Miltenyi, Auburn, CA) according to the manufactures instructions.
Flow cytometric cytolytic assay
An adaption of the Godoy-Ramirez et al. T-cell cytolysis assay was used (Godoy-Ramirez et al., 2005). A CD4+ T-cell line from 86I was used as targets for Gag68. Target cells were labeled with CTV (life Technologies) and subsequently peptide pulsed with SIV-specific peptides (5327 and 5328; 1 μg/mL) or left untreated for 45 m at 37°C. Targets were then washed 3 times with PBS. Gag68 TCR transduced CD4+ cells were co-cultured with targets in a 96 well round bottom plate at E:T ratios from 1:1 to 10:1 in T-cell medium (4 × 105/well). Triplicate co-cultures were incubated for 18 h at 37°C, then cells were washed with PBS and stained with propidium iodide (0.5μg/mL; Sigma-Aldrich, St. Louis, MO). Lysis was measured by flow cytometry as the percent of CTV and propidium iodide double positive cells.
RESULTS
Isolation of SIV Gag-specific CD4+ T-cell clones
To isolate SIVmac239 Gag-specific CD4+ T cells, we sorted central memory (CD28+/CD95+) CD4+ T cells out of PBMC isolated from a DNA-vaccinated rhesus macaque, 86I, that had developed a primarily Gag-specific CD4+ T-cell response (Patel et al., 2010). To increase the frequency of responding cells, the sorted memory CD4+ T cells were stimulated for three rounds with autologous irradiated APC-pulsed with an SIVmac239 Gag 15-mer peptide pool, ultimately boosting the Gag response to 3% of the CD4+ T-cell population. Nine SIV Gag-specific CD4+ T-cell clones were isolated from this culture by limiting dilution cloning and selecting for cells exhibiting an induced IFN-γ response to the Gag peptide pool. However, despite initial antigen reactivity by all of the clones (Fig. 1A), only two clones, Gag6 and Gag68, remained reactive to the Gag peptide pool, the other clones senesced after two weeks of expansion. These findings are consistent with our previous experience that CD4+ T cells are relatively short-lived, thus, very difficult to study (MTT, CO unpublished data).
FIG 1.

Isolation of SIV Gag-specific CD4+ T-cell clones. Flow cytometry analysis plots of CD4+ T-cell clones derived from central memory cultures 4 h post addition of the SIV Gag peptide pool are presented: (A) intracellular IFN-γ reactivity of CD4+ T-cell clones (representative of 2 independent analyses), (B) intracellular IFN-γ and surface CD107a reactivity of Gag6 and Gag68 CD4+ T-cell clones (representative of 4 independent analyses). Analyzed samples are identified above their respective plots with parameters analyzed denoted at left and below.
Gag68 suppresses the spread of SIVmac239 replication in target CD4+ T cells
Flow cytometry analysis revealed that, while both clones had effector responses to the Gag peptide pool, Gag68 exhibited a stronger bi-functional effector response, for both degranulation as measured by the appearance of surface CD107a and induction of intracellular IFN-γ expression (Fig. 1B). In contrast, Gag6 produced mostly a monofunctional CD107a degranulation response with some bifunctional CD107a/IFN-γ staining (Fig. 1B). Interestingly, the Gag68 response was similar to our prior findings with CD8+ effector T cells that efficiently suppressed SIV replication on CD4+ target cells (Minang et al., 2008; Minang et al., 2009b) (MTT, CO, and DEO unpublished data), similar to the recent findings reported by Johnson et al. (Johnson et al., 2015).
To test whether these clones can act directly as effector T cells, we examined their ability to suppress the spread of SIVmac239 when co-cultured with autologous CTV dye-labeled target CD4+ T cells infected at a low MOI (0.01). This inoculum results in an initial low level infection that spreads throughout the target culture within 5–7 days, giving the effector T cells an opportunity to suppress the spread of virus (Minang et al., 2008). Our flow cytometry analysis revealed that 98% of the targets were infected (Gag+) at day 5 in the 1-to-1 (1:1) effector-to-target co-culture with an irrelevant CD4+ cell line as a negative effector control, a frequency similar to that of the infected target cell culture alone (compare Figs. 2A and B). In contrast, the Gag68 co-culture at the 1:1 ratio showed some suppression, 66% of the targets were infected (Fig. 2B). The suppression by Gag68 was even more apparent in parallel co-cultures at a 10:1 effector-to-target ratio with only 4% targets infected compared with 61% of the 10:1 control effector culture (Fig. 2B). Thus, Gag68 exhibits a functional effector response by suppressing the spread of SIV. Unlike the Gag68 result, Gag6 failed to suppress at either ratio (Fig. 2B), consistent with its weaker effector response (Fig. 1B).
FIG 2.

Gag68 suppression of SIVmac239 infection in CD4+ T cells. (A–C) Flow cytometry for infected CTV-labeled CD4+ T cells by surface CD4 staining and intracellular Gag staining five days post infection: Panel A, analysis of infected rhesus CD4 T-cell targets labeled by CTV is presented. Panel B, analysis of infected rhesus CD4 T-cell targets labeled by CTV co-cultured with irrelevant, Gag6 or Gag68 effector clones at an E:T ratio of 1:1, top row, or 10:1, bottom row, are presented. Panel C, analysis of infected CTV− CD4 effectors in the co-cultures are presented. Analyzed samples are identified above their respective plots with parameters denoted at left and below. Panel D, a graph of real-time RT-PCR results for SIV RNA at days 5 and 7 post co-culture is presented. Viral RNA copies relative to the irrelevant control are presented on the y-axis and the cultures indentified below the x-axis. Data representative of 2 independent experiments.
It is important to consider that CD4+ effectors are also targets for infection. Therefore, infection of the CD4+ effectors themselves can interfere with their ability to suppress virus spread due to both viral-induced cytopathogenic effects and fratricide of infected effector cells by their fellow effector T cells (Jain et al., 2015). Because this could reduce their overall effectiveness, we examined the infection status of the CD4+ effectors in the co-culture. Analysis of the CTV− effector population (Fig. 2C) showed that greater than 84% of either the irrelevant or Gag6 effectors were infected at either ratio, similar to the infection levels of the targets (Fig. 2B). In contrast, the 10:1 Gag68 co-culture contained considerably fewer infected effector cells, 6%, indicating that it was able to suppress not only the infection of the targets, but of itself. However, at the 1:1 ratio, the Gag68 cells showed a high frequency of infected effectors (Fig. 2C), apparently the result of being overwhelmed by SIV at this lower effector-to-target ratio. In fact, the frequency of infected cells in the Gag68 1:1 co-culture, 75%, was twice as high as that of their target counterparts, suggesting that the Gag68 effectors were preferentially infected by exposure to virus during the cell-to-cell interactions promoted by immune recognition of infected targets (Brenchley et al., 2006; Douek et al., 2002). Despite the relatively low levels of uninfected Gag68 effectors (10%) at the 1:1 ratio, the Gag68 cells did show some suppression. Real-time RT-PCR found that the levels of accumulated SIV RNA in supernatants from the 10:1 co-cultures at both day 5 and 7 were consistent with the flow cytometry results (Fig. 2 and data not shown): the Gag68 had considerably lower levels of SIV RNA in the supernatants than both the irrelevant and Gag6 samples. The RNA level in the day 7 Gag6 co-culture supernatants nearly doubled from that of the day 5. In contrast, the levels detected in the Gag68 samples remained nearly the same. Flow cytometry results at day 7 mirrored the RT-PCR data: Gag68 mostly controlled infection while Gag6 failed to do so. Taken together our data show that Gag68 is a potent effector clone.
Epitope mapping and effector responses of Gag68
To identify the reacting peptide(s) in the 15-mer SIV Gag pool, we screened Gag68 against a matrix of 23 Gag peptide pools for IFN-γ ELISpot reactivity. Each pool contains 12 unique Gag peptides and one redundant peptide that is shared with another pool, allowing for a single-step identification of any recognized peptide(s) by the co-reacting pools. Gag68 reacted to 2 pairs of peptide pools with the overlapping Gag peptides 465PAVDLLKNYMQLGKQ479, SIVGag(p6-1), and 469LLKNYMQLGKQQREK483, SIVGag(p6-2), (overlapping sequence underlined; data not shown), corresponding to SIVmac239 Gag amino acids 465– 483 within the p6 domain of Gag. Pulsing with either peptide produced similar induced IFN-γ responses in Gag68 cells (data not shown). Fine mapping of the overlapping epitope sequences failed to narrow down the epitope (VIA, MTT, and CO, unpublished data).
Gag68 exhibits a cytolytic-type effector response by peptide stimulation
A mixture of SIVGag(p6-1) and SIVGag(p6-2), SIVGag(p6-1/2), was used to examine the cytokine induction and CD107a degranulation profile of Gag68 by flow cytometry, 6h post-stimulation. While the results matched a Th1-type response pattern (Fig. 3A), Gag68 did not produce IL-2 and, in addition to the hallmark IFN-γ response, induced expression of other effector markers, CD107a degranulation, MIP-1α, and MIP-1β, typically associated with cytotoxic CD8+ T cells (Hersperger et al., 2010). In contrast, Gag68 expressed negligible Th2-type responses, IL-4, IL-6, and IL-10, characteristic of helper CD4+ T cells. Thus, Gag68 exhibits a cytotoxic effector cell response phenotype, consistent with our suppression data.
FIG 3.

Functional profile of SIV-Gag-specific CD4+ T-cell clone Gag68. A composite graph showing percentage of Gag68 cells producing induced cytokine intracellular cytokine responses and CD107a surface expression upon co-culture with SIVGag(p6-1/2) peptides-pulsed APCs is presented. The percent positive respondents are presented on the y-axis and the induced response is identified below the x-axis with classical Th1 and Th2 responses emphasized by brackets. Results are representative of at least 3 independent experiments.
MHC restriction of Gag68
To determine the MHC restriction of Gag68, we used a comparative restriction strategy that tested the ability of SIVGag(p6-1/2)-pulsed PBMC from a panel of 9 unrelated rhesus macaques and the original donor animal, 86I, to simulate Gag68. Gag68 produced specific IFN-γ ELISpot responses (data not shown), confirmed by IFN-γ and TNF-α flow cytometry, to peptide-pulsed PBMC from three animals (KTM, MK9, and ZB35) at levels similar that to the donor animal, 86I (Fig. 4): thus, the MHC restriction element is shared between these four animals and absent in the other five.
FIG 4.

Identification of co-reacting animals for the SIVGag(p6-1/2) peptide epitope mix by intracellular cytokine staining. Flow cytometry analysis of PBMC from different animals for IFN-γ and TNF-α intracellular responses to the presence of the SIVGag(p6-1/2) peptide mix. Analyzed samples are identified at left with peptide treatment denoted above their respective plots. Responses analyzed are denoted at left and below the plots.
To determine the MHC haplotype in common with Gag68, the MHC-I and -II loci of all ten animals were pyrosequenced (Karl et al., 2014; Wiseman et al., 2013) and correlated with the APC data, i.e., haplotypes present in 86I, KTM, MK9 and ZB35 but absent in the other animals (Table 1). All four presenting animals had the DPB1*15:01 allele associated with either DPA1*02:05 (86I, KTM, MK9) or DPA1*02:01 (ZB35) while these genotypes were absent in the nonpresenting animals. In contrast, there were no common haplotypes shared in the presenting animals for MHC-I A/B or -II DR/DQ haplotypes. It is important to note that the DPA1*02:01 and DPA1*02:05 alleles share identical alpha 1 domains and only differ by 4 amino acids in their alpha 2 domains. Thus, it is likely that the DPB1*15:01 chain can pair with either of these DPA variants and present similar peptides. Interestingly, haplotypes containing DPB1*15:01 appear to be the most frequent of all DPA/DPB combinations in Indian rhesus macaques, e.g., these have been reported to account for 23% of all DPA/DPB haplotypes at the Biomedical Primate Research Center in the Netherlands, the most thoroughly characterized breeding colony to date (Doxiadis et al., 2013).
Table 1.
MHC genotypes of rhesus macaques

|
APC function was defined by IFN-γ and TNF- α intra cellular cytokine staining as greater than 14% double positive
+, APC function; −, No APC function
ND, not determined; Gag68 presenting haplotypes are boxed and bolded
Transfer of SIV Gag-TCR confers effector responses to peptide-pulsed and infected cells
One of the difficulties in studying primary rhesus T cells is their limited life span in vitro, succumbing to senescence after about 7–12 months in culture for CD8+ T cells (Andersen et al., 2007; Minang et al., 2008). For CD4+ T cells, we observe much shorter life spans (MTT and CO unpublished data), typically 4–6 months as we found for the other 7 Gag-specific CD4+ T-cell clones in this study. To enable long-term studies of the Gag68 specificity, we cloned the Gag68 TCR α and β variable regions, and constructed a Gag68 TCR expression vector which coexpresses a tNGFR protein as a marker for transductants. A comparative search of the Gag68 variable region TCR sequences against both the human and rhesus macaque TCR gene sequences in Genbank showed no similar complementary determining region 3 (CDR3) sequences for either the α or β chain (accession #’s KP233091 and KP272132) (Fig. 5A). The V– and J–gene segment usage by the Gag68 TCR is TRAV13-1-001 and TRAJ24-001 for the α chain and TRBV12-4-0001 for the V-gene segment of the β chain. The Jβ–gene segment is not annotated in either the human or rhesus genomic sequence databases, residing at 18043948-18043091 of rhesus chromosome 3 within the β chain locus. Because we have no tetramer for the Gag68 specificity, TCR expression in the NGFR+ transductants was tested by a CD3-rescue assay where expression of TCR β-chain restores expression of CD3 on the TCR-deficient Jurkat RT3.5-T3.5-clone12 cells (Weiss, Wiskocil, and Stobo, 1984). Gag68 TCR transduction of the mutant line resulted in a clear rescue of CD3 with an intensity that strongly correlated with NGFR staining, confirming the transfer of TCR chains (Fig. 5B, data not shown).
FIG 5.

Antigen specificity and effector function conferred by transducing CD4+ T cells with the Gag68 TCR. Panel A, CDR3 sequences are presented. Panels B–D, Flow cytometry of CD4+ T-cell Gag68/tNGFR transductants is presented: Panel B, anti-CD3 and -NGFR analysis for CD3 rescue of the TCR-deficient Jurkat RT3.5-T3.5-clone12 T-cell line by Gag68 TCR transduction; Panel C, analysis of CD4 and NGFR-stained primary rhesus CD4+ T cells transduced with Gag68 TCR/tNGFR. Panel D, induced IFN- γ and MIP-1β intracellular cytokine staining analysis of the SIVGag(p6-1/2) peptide mix- pulsed Gag68 TCR-transduced primary CD4+ T-cell line. Analyzed samples are identified above their respective plots with parameters analyzed denoted at left and below.
To functionally test for TCR transfer, Gag68 TCR-transduced CD4+ T cells isolated by tNGFR immunoaffinity (Fig. 5C), were tested for effector responses to the SIVGag(p6-1/2) peptide mix by intracellular cytokine staining. Stimulation with these specific peptides induced specific effector responses, secretion of IFN-γ and MIP-1β, demonstrating that the transduced cells had been functionally reprogrammed for Gag68 specificity (Fig. 5D).
We next tested whether the Gag68 TCR, when transduced into CD4+ T cells, was capable of recognizing functional antigen by co-culturing them in SIV-infected cells. Studying MHC-II-restricted TCRs is complicated by the down-regulation of functional MHC-II levels on SIV infected cells by viral proteins such as Nef (Schindler et al., 2004; Schindler et al., 2003; Stumptner-Cuvelette et al., 2001). Nevertheless, SIVmac239-infected target CD4+ T cells in two cell lines isolated from 86I PBMC, CD4+ 1 and CD4+ 2, induced IFN-γ responses in co-culture with Gag68, similar to those of peptide-pulsed CD4+ 1 and CD4+2 targets (Fig 6). To examine Gag68 recognition without the confounding influence of Nef, we infected cells with our previously described SIVmac239 Nef myristylation mutant, G2V, that expresses a nonfunctional protein, Nef/G2V (Barsov et al., 2011; Minang et al., 2009b; Schindler et al., 2004). The IFN-γ response to SIVmac239 Nef/G2V-infected targets by Gag68 TCR transduced CD4+ T cells was higher than that to wild type SIVmac239 in both CD4+ target cell lines, suggesting higher levels of DP contribute to better recognition of SIV-infected CD4+ T cells by the Gag68 TCR. Together these data demonstrate that our Gag68-transduced CD4+ T cells while capable of recognizing wild-type SIV-infected CD4+ targets, are potentially limited by the downregulation of viral antigen presentation from Nef blocking MHC-II peptide loading presentation (Schindler et al., 2003).
FIG 6.

Recognition of SIV-infected CD4+ target T cells by 86I CD4+ T-cell Gag68 TCR-transductants. Flow cytometry plots for induced IFN-γ intracellular cytokine production by primary rhesus Gag68 TCR-transduced CD4+ T cells in response to co-culture with SIVGag(p6-1/2) peptide mix-pulsed, SIVmac239−, or SIVmac239 Nef/G2V-infected CD4+ T cell targets, either the CD4+ 1 or CD4+-2 bulk 86I T-cell lines, are presented. Gating on the effectors is displayed with CD4 staining on the x-axis and IFN-γ on the y-axis and the treatment of the co-cultured targets identified above each column of plots. Data are representative of two independent experiments.
Suppression of virus spread by Gag68 CD4+ transductants
Next, we tested whether the Gag68 TCR transductants could confer virus suppression to 86I CD4+ T cells. NGFR+-sorted Gag68 TCR transductants (Fig. 7A) were used in our SIV suppression assay. Flow cytometry analysis of the co-cultures at day 5 revealed that Gag68 TCR-transduced 86I CD4+ T cells effectively suppressed the spread of SIV in the target CD4+ T cells compared to the targets alone, 7% versus 72% Gag+ respectively (Fig 7A and B). This suppression was specific as I86 CD4+ T cells transduced with the mismatching A0*1-restricted CM9-6 SIV Gag specific TCR failed to suppress virus spread in the Mamu A0*4/0*4 86I targets, 70% Gag+. Similar to the native Gag68 clone, effective suppression was correlated with a corresponding control of infection in the CD4+ effectors themselves, 0.3% in Gag68 transductants versus 83% in the MHC-mismatched control (Fig7C). Thus, Gag68 TCR transduction transfers functional suppression of virus in both targets and the transductants.
FIG 7.

Gag68 TCR transductant suppression of SIVmac239 infection in CD4+ T cells. Panel A, Histograms from an NGFR flow cytometry analysis of 86I CD4+ T cells after transduction with the Gag68 TCR and paramagnetic sorting for the tNGFR coexpressed marker are presented. (B–D) Flow cytometry for infected CTV-labeled CD4+ T cells by surface CD4 staining and intracellular Gag staining five days post infection: Panel B, analysis of infected rhesus CD4 T-cell targets labeled by CTV is presented. Panel C, analyses of infected rhesus CD4 T-cell targets labeled by CTV co-cultured with either MHC-I mismatched CM9-6 TCR or Gag68 TCR 86I CD4+ transductant effector lines at an E:T ratio of 10:1 are presented. Panel C, analyses of infected CTV− CD4 effectors in the co-cultures are presented. Analyzed samples are identified above their respective plots with parameters denoted at left and below. Suppression data are representative of two independent experiments.
Cytolysis of peptide-pulsed targets by Gag68 transductants
To determine whether Gag68 TCR transfer could effect a lytic response, 86I CD4+ T cells were transduced with Gag68 TCR and examined for their ability to lyse CTV-labeled, SIVGag(p6-1/2) peptide-loaded CD4+ target cells by flow cytometry for CTV and propidium iodide staining. The results showed that the Gag68 transductants lysed the peptide-loaded CD4+ targets to levels observed by other groups (Sacha et al., 2009) while cells without peptide showed little lysis (Fig. 8), demonstrating that the Gag68 TCR enabled an antigen-specific lytic response in CD4+ T cells.
FIG 8.
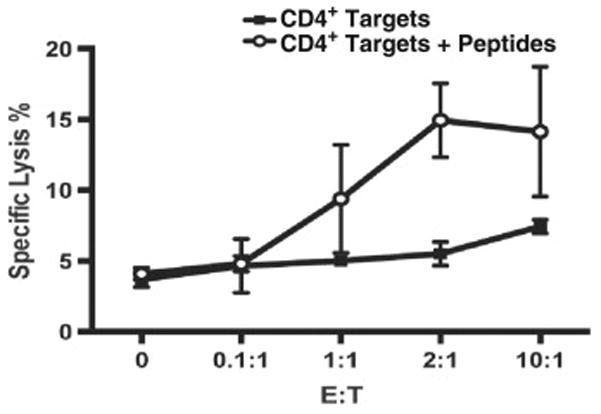
Gag68 cytolysis. Graph of results from CTV/PI flow cytometry analyses of SIVGag(p6-1/2) peptide mix-pulsed target CD4+ T-cell lysis by Gag68 TCR transduced CD4+ T cells is presented. Specific lysis was calculated as the percent of lysed, i.e. CTV+/PI+ cells in the presence of effectors minus the baseline lysis observed in the target cell only control and is presented on the y-axis and with the effector to target ratio (E:T) indicated on the x-axis. Error bars are the standard deviations of two independent triplicate assays.
DISCUSSION
Here, we present a detailed examination of an SIV Gag-specific MHC-II-restricted rhesus macaque CD4+ T-cell clone which can exhibit a direct effector function, acting alone to suppress the spread of SIV replication in CD4+ T cells. This finding is consistent with that of Zheng et al. who identified two Nef–specific cytotoxic CD4+ T-cell clones that could suppress the production of HIV-1 in primary CD4+ T-cell targets in vitro (Zheng et al., 2009). The exact mechanism(s) for viral suppression by the Gag68 CD4+ T-cell clone is not known. CD8+ cytotoxic T cells can directly suppress AIDS viruses through three mechanisms, cell killing by release of perforin/granzyme B or engaging the cell surface death receptor Fas on targets by FasL present on effectors, or by interfering with viral replication by the release of β-chemokines. Interestingly, Gag68 did produce a polyfunctional response to antigen stimulation characteristic of the suppressive cytotoxic CD8+ T cells that we have studied, expressing cytotoxic and chemokine effector markers (Barsov et al., 2011; Minang et al., 2008; Minang et al., 2009b) (MTT, VIA, and CO, unpublished data). These results are consistent with the primary HIV/CD4+ T-cell line data of Johnson et al. (Johnson et al., 2015), suggesting that CD8+ and CD4+ effector T cells share a common suppressive mechanism. Our Gag68 results, combined with those of Zheng et al. and Johnson et al., point to the potential of AIDS virus-specific CD4+ T cells to act directly as effectors to control AIDS-virus replication in CD4+ T cells and not simply helpers in the antiviral response.
Our findings differ from those of Sacha et al., who isolated and characterized five SIV Gag-specific MHC-II-restricted CD4+ T-cell clones that vigorously recognized and suppressed SIV infection in macrophages yet failed to suppress infection in CD4+ T-cell targets (Sacha et al., 2009). One difference between our two studies is the source of the effector CD4+ T cells, from vaccinated versus CD8-depleted infected rhesus macaques. Since a loss or the lack of CD8+ T cells in vivo has been correlated with an increase in cytotoxic CD4+ T cells (Soghoian and Streeck, 2010; Zajac et al., 1996; Zhou et al., 2012), it is possible that the CD4+ T cells induced by vaccination and CD8 depletion have different properties. Indeed, differences in antigen priming, e.g. vaccination versus infection, are correlated with qualitative differences in effector responses in influenza models (Mueller et al., 2010; Nagaoka et al., 2014). Alternatively, and more provocatively, because HIV and SIV-specific T cells are preferentially infected in vivo (Brenchley et al., 2006; Douek et al., 2002), the strongest effector CD4+ T cells might be eliminated early in infection, selecting against stronger responding cells in vivo while sparing those with weaker responses. Finally, it is important to note that our suppression assay fundamentally differs from that of Sacha et al. who synchronously infects a large percentage of the target cells with an MOI of 1 which are then analyzed for loss of infected cells after 24 h. Thus, this suppression assay mostly relies on cytolysis of targets. In contrast, our suppression assay measures the ability of an effector CD4+ T-cell clone to reduce virus spread among the virus susceptible CD4+ target T cells by infecting at a relatively low MOI, approximately 0.01, to generate a initially low level spreading infection that peaks at 5–7 day, providing opportunities for the effectors to employ both cytolytic and noncytolytic mechanisms over time (Demers, Reuter, and Betts, 2013). Nevertheless, all three reports of suppressing CD4+ T-cell clones, Sacha et al. (Sacha et al., 2009), Zheng et al. (Zheng et al., 2009), and ours, as well as those of T-cell lines by Johnson et al. (Johnson et al., 2015), strongly support the direct involvement of CD4+ T cells as effectors in limiting viral spread in both macrophages and CD4+ T cells.
While both the Gag68 and Gag6 clones exhibited bifunctional effector responses, only Gag68, displaying a better peptide response than Gag6, effectively suppressed SIV in our assay. However, even though effector responses to presented peptide are informative, they may not always correlate with suppression ((Minang et al., 2009b), MTT unpublished data).
Unlike the prior studies (Johnson et al., 2015; Sacha et al., 2009; Zheng et al., 2009), we also examined the infection status of the effectors themselves. One striking difference between the two clones is the reduced levels of infected Gag68 cells compared to those in the Gag6 samples, highlighting the importance of CD4+ effectors reducing their own infection with its associated cytopathogenicity for efficient virus suppression in the targets (Jain et al., 2015). Similarly, the reduced Gag68 suppression in the 1:1 effector-to-target co-culture compared to its 10:1 counterpart was accompanied by an extensive amount of effector infection versus the 10:1 Gag68 co-culture. Therefore, failure of suppression is strongly linked to the extent of effector cell infection, reflecting a saturable ability of the antiviral effectors to limit the spread infection in both the targets and, more importantly themselves. The need for robust infection suppression is especially important as virus-specific CD4+ T cells are more susceptible to infection and elimination in vivo (Brenchley et al., 2006; Douek et al., 2002) and in vitro (SJ, MTT, DEO, manuscript in preparation). Our data together with clinical data describing preferential infection of virus-specific T cells suggest that the CD4+ effector response is dampened during infection in vivo and possibly eliminated in high viremic settings.
Initially, Gag68 was one of 9 Gag-specific CD4+ T cells. However, soon after initial culturing, 7 of the clones became anergic, failing to respond to presented antigen. In our studies we have routinely found that CD4+ T-cell clones have much shorter useful lifespans than similarly cultured CD8+ T-cell clones (MTT unpublished data). This represents a severe limitation to the study of CD4+ T cell specificities which we overcome here with our retroviral TCR transfer strategy (Barsov et al., 2011), enabling additional study of the Gag68 TCR properties long after the original clone died. Transfer of Gag68 to CD4+ T cells conferred antigen-specific virus suppression and cytolytic activity to them. Thus, the availability of an effective MHC-II-restricted TCR now allows for extended studies of the effector functions of CD4+ T cells by engineering SIV-specific CD4+ T cells from naïve animals without a vaccine/infection source bias. Considering the relatively high prevalence (23%) of the DPA1*02:05/01/DPB1*15:01MHC-II haplotypes (Doxiadis et al., 2013), nearly half of an Indian rhesus macaque population should be suitable for additional Gag68 transduction experiments, especially adoptive transfer studies. Combining the power of adoptive transfer and retroviral transduction of this capable MHC-II-restricted TCR opens the possibility to engineer SIV-specific CD4+ T-cell populations with various phenotypes (e.g., central memory, effector memory, naive), cytokine secretion profiles, and CD107a expression to test questions such as the contribution of the various T-cell effector functions and phenotypes to direct antiviral effector function both in vitro and vivo. Specifically, transducing CD4+ T-cell clones expressing various effector molecules with the Gag68 TCR will allow for experiments that examine the mechanism(s) of AIDS virus suppression used by MHC-II-restricted effector T cells.
The existence of Gag68 supports the idea that CD4+ T cells assist antiviral responses directly by contributing to the effector response against infected T cells, the primary targets of infection in SIV/HIV. Our results not only confirm the ability of an MHC-II- restricted T-cell specificity to recognize antigen presented by infected CD4+ T cells and suppress viral replication both in their targets and themselves, but also provide a means to study the lesser understood CD4+ T-cell contribution to AIDS virus control.
Highlights.
The rhesus macaque MHC class II-restricted CD4+ T-cell clone, Gag68, possesses a strong ability to suppress SIV infection in CD4+ T cells.
Gag68 was able to suppress its own infection as well as that of the CD4+ T-cell target.
Effective suppression by CD4+ T-cell clones required control of their own infection.
In vivo, CD4+ T cells likely are limited by infection when recognizing their infected targets.
A Gag68 TCR transfer vector was created for T-cell engineering studies with CD4+ T cells and a suppressive MHC class II-restricted TCR.
Acknowledgments
We thank the Kelli Oswald and Rebecca Shoemaker for PCR viral load analysis, Donald Johnson, Adam Wiles, Rodney Wiles, and Vicky Coalter for tissue processing and sample support and Cynthia Matthews of the Blood Services Section, Department of Transfusion Medicine, NIH for human leukopacks. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Human rIL-2 from Dr. Maurice Gately, Hoffmann - La Roche Inc; SIVmac p27 Hybridoma (55-2F12) from Dr Niels Pedersen; SIVmac239 Gag Peptide Pool. MHC genotyping efforts were supported by the WNPRC Base Grant from the National Center for Research Resources (P51 RR000167) and Office of Research Infrastructure Programs (P51 OD011106) of the National Institutes of Health. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen H, Barsov EV, Trivett MT, Trubey CM, Giavedoni LD, Lifson JD, Ott DE, Ohlen C. Transduction with human telomerase reverse transcriptase immortalizes a rhesus macaque CD8+ T cell clone with maintenance of surface marker phenotype and function. AIDS research and human retroviruses. 2007;23(3):456–465. doi: 10.1089/aid.2006.0194. [DOI] [PubMed] [Google Scholar]
- Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, Easterbrook P, Grey P, Smith D, McMichael AJ, Cooper DA, Rowland-Jones SL, Kelleher AD. Characterization of CD4(+) CTLs ex vivo. J Immunol. 2002;168(11):5954–5958. doi: 10.4049/jimmunol.168.11.5954. [DOI] [PubMed] [Google Scholar]
- Barsov EV, Trivett MT, Minang JT, Sun H, Ohlen C, Ott DE. Transduction of SIV-Specific TCR Genes into Rhesus Macaque CD8 T Cells Conveys the Ability to Suppress SIV Replication. PLoS ONE. 2011;6(8):e23703. doi: 10.1371/journal.pone.0023703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C, Huang ML, Gough M, Greenberg PD, Riddell SR, Kiem HP. Nonmyeloablative Immunosuppressive Regimen Prolongs In Vivo Persistence of Gene-Modified Autologous T Cells in a Nonhuman Primate Model. Journal of Virology. 2001;75(2):799–808. doi: 10.1128/JVI.75.2.799-808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Ruff LE, Casazza JP, Koup RA, Price DA, Douek DC. Preferential infection shortens the life span of human immunodeficiency virus-specific CD4+ T cells in vivo. J Virol. 2006;80(14):6801–6809. doi: 10.1128/JVI.00070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline AN, Bess JW, Piatak M, Lifson JD. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. Journal of Medical Primatology. 2005;34(5–6):303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- Coren LV, Jain S, Trivett MT, Ohlen C, Ott DE. Production of retroviral constructs for effective transfer and expression of T-cell receptor genes using Golden Gate cloning. Biotechniques. 2015;58(3):135–139. doi: 10.2144/000114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers KR, Reuter MA, Betts MR. CD8(+) T-cell effector function and transcriptional regulation during HIV pathogenesis. Immunol Rev. 2013;254(1):190–206. doi: 10.1111/imr.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, Betts MR, Hill BJ, Little SJ, Lempicki R, Metcalf JA, Casazza J, Yoder C, Adelsberger JW, Stevens RA, Baseler MW, Keiser P, Richman DD, Davey RT, Koup RA. Evidence for increased T cell turnover and decreased thymic output in HIV infection. J Immunol. 2001;167(11):6663–8. doi: 10.4049/jimmunol.167.11.6663. [DOI] [PubMed] [Google Scholar]
- Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, Koup RA. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417(6884):95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- Doxiadis GG, de Groot N, Otting N, de Vos-Rouweler AJ, Bolijn MJ, Heijmans CM, de Groot NG, van der Wiel MK, Remarque EJ, Vangenot C, Nunes JM, Sanchez-Mazas A, Bontrop RE. Haplotype diversity generated by ancient recombination-like events in the MHC of Indian rhesus macaques. Immunogenetics. 2013;65(8):569–84. doi: 10.1007/s00251-013-0707-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm N, Korber BT, Adams CM, Szinger JJ, Draenert R, Addo MM, Feeney ME, Yusim K, Sango K, Brown NV, SenGupta D, Piechocka-Trocha A, Simonis T, Marincola FM, Wurcel AG, Stone DR, Russell CJ, Adolf P, Cohen D, Roach T, StJohn A, Khatri A, Davis K, Mullins J, Goulder PJ, Walker BD, Brander C. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J Virol. 2004;78(5):2187–200. doi: 10.1128/JVI.78.5.2187-2200.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy-Ramirez K, Makitalo B, Thorstensson R, Sandstrom E, Biberfeld G, Gaines H. A novel assay for assessment of HIV-specific cytotoxicity by multiparameter flow cytometry. Cytometry A. 2005;68(2):71–80. doi: 10.1002/cyto.a.20189. [DOI] [PubMed] [Google Scholar]
- Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. 2010;6(5):e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Trivett MT, Ayala VI, Ohlen C, Ott DE. African Green Monkey TRIM5alpha Restriction in SIV-Specific Rhesus Macaque Effector CD4 T Cells Enhances Their Survival and Antiviral Function. J Virol. 2015 doi: 10.1128/JVI.03598-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Eller M, Teigler JE, Maloveste SM, Schultz BT, Soghoian DZ, Lu R, Oster AF, Chenine AL, Alter G, Dittmer U, Marovich M, Robb ML, Michael NL, Bolton D, Streeck H. Cooperativity of HIV-Specific Cytolytic CD4 T Cells and CD8 T Cells in Control of HIV Viremia. J Virol. 2015;89(15):7494–7505. doi: 10.1128/JVI.00438-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannanganat S, Ibegbu C, Chennareddi L, Robinson HL, Amara RR. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. Journal of Virology. 2007a;81(16):8468–8476. doi: 10.1128/JVI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannanganat S, Kapogiannis BG, Ibegbu C, Chennareddi L, Goepfert P, Robinson HL, Lennox J, Amara RR. Human immunodeficiency virus type 1 controllers but not noncontrollers maintain CD4 T cells coexpressing three cytokines. J Virol. 2007b;81(21):12071–6. doi: 10.1128/JVI.01261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl JA, Heimbruch KE, Vriezen CE, Mironczuk CJ, Dudley DM, Wiseman RW, O’Connor DH. Survey of major histocompatibility complex class II diversity in pig-tailed macaques. Immunogenetics. 2014;66(11):613–623. doi: 10.1007/s00251-014-0797-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestler H, Kodama T, Ringler D, Marthas M, Pedersen N, Lackner A, Regier D, Sehgal P, Daniel M, King N, et al. Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science. 1990;248(4959):1109–1112. doi: 10.1126/science.2160735. [DOI] [PubMed] [Google Scholar]
- Lifson JD, Rossio JL, Piatak M, Jr, Parks T, Li L, Kiser R, Coalter V, Fisher B, Flynn BM, Czajak S, Hirsch VM, Reimann KA, Schmitz JE, Ghrayeb J, Bischofberger N, Nowak MA, Desrosiers RC, Wodarz D. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J Virol. 2001;75(21):10187–10199. doi: 10.1128/JVI.75.21.10187-10199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NB, Swain SL. Cytotoxic CD4 T cells in antiviral immunity. J Biomed Biotechnol. 2011;2011:954602. doi: 10.1155/2011/954602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minang JT, Barsov EV, Yuan F, Trivett MT, Piatak M, Lifson JD, Ott DE, Ohlen C. Efficient inhibition of SIV replication in rhesus CD4+ T-cell clones by autologous immortalized SIV-specific CD8+ T-cell clones. Virology. 2008;372(2):430–441. doi: 10.1016/j.virol.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Minang JT, Trivett MT, Bolton DL, Trubey CM, Estes JD, Li Y, Smedley J, Pung R, Rosati M, Jalah R, Pavlakis GN, Felber BK, Piatak M, Roederer M, Lifson JD, Ott DE, Ohlen C. Distribution, Persistence, and Efficacy of Adoptively Transferred Central and Effector Memory-Derived Autologous Simian Immunodeficiency Virus-Specific CD8+ T Cell Clones in Rhesus Macaques during Acute Infection. The Journal of Immunology. 2009a;184(1):315–326. doi: 10.4049/jimmunol.0902410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minang JT, Trivett MT, Coren LV, Barsov EV, Piatak M, Jr, Ott DE, Ohlen C. Nef-mediated MHC class I down-regulation unmasks clonal differences in virus suppression by SIV-specific CD8(+) T cells independent of IFN-gamma and CD107a responses. Virology. 2009b;391(1):130–9. doi: 10.1016/j.virol.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Langley WA, Carnero E, Garcia-Sastre A, Ahmed R. Immunization with live attenuated influenza viruses that express altered NS1 proteins results in potent and protective memory CD8+ T-cell responses. J Virol. 2010;84(4):1847–1855. doi: 10.1128/JVI.01317-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka M, Hatta Y, Kawaoka Y, Malherbe LP. Antigen signal strength during priming determines effector CD4 T cell function and antigen sensitivity during influenza virus challenge. J Immunol. 2014;193(6):2812–2820. doi: 10.4049/jimmunol.1401358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff T, Peterson LJ, Morris JC, Thompson J, Zhang X, Horn PA, Thomasson BM, Kiem HP. Efficient gene transfer to hematopoietic repopulating cells using concentrated RD114-pseudotype vectors produced by human packaging cells. Mol Ther. 2004;9(2):157–9. doi: 10.1016/j.ymthe.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Nemes E, Bertoncelli L, Lugli E, Pinti M, Nasi M, Manzini L, Manzini S, Prati F, Borghi V, Cossarizza A, Mussini C. Cytotoxic granule release dominates gag-specific CD4+ T-cell response in different phases of HIV infection. AIDS. 2010;24(7):947–957. doi: 10.1097/QAD.0b013e328337b144. [DOI] [PubMed] [Google Scholar]
- Norris PJ, Moffett HF, Yang OO, Kaufmann DE, Clark MJ, Addo MM, Rosenberg ES. Beyond help: direct effector functions of human immunodeficiency virus type 1-specific CD4(+) T cells. J Virol. 2004;78(16):8844–51. doi: 10.1128/JVI.78.16.8844-8851.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Valentin A, Kulkarni V, Rosati M, Bergamaschi C, Jalah R, Alicea C, Minang JT, Trivett MT, Ohlen C, Zhao J, Robert-Guroff M, Khan AS, Draghia-Akli R, Felber BK, Pavlakis GN. Long-lasting humoral and cellular immune responses and mucosal dissemination after intramuscular DNA immunization. Vaccine. 2010;28(30):4827–36. doi: 10.1016/j.vaccine.2010.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128(2):189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- Sacha JB, Giraldo-Vela JP, Buechler MB, Martins MA, Maness NJ, Chung C, Wallace LT, Leon EJ, Friedrich TC, Wilson NA, Hiraoka A, Watkins DI. Gag- and Nef-specific CD4+ T cells recognize and inhibit SIV replication in infected macrophages early after infection. Proc Natl Acad Sci U S A. 2009;106(24):9791–9796. doi: 10.1073/pnas.0813106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler M, Munch J, Brenner M, Stahl-Hennig C, Skowronski J, Kirchhoff F. Comprehensive analysis of nef functions selected in simian immunodeficiency virus-infected macaques. J Virol. 2004;78(19):10588–10597. doi: 10.1128/JVI.78.19.10588-10597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler M, Wurfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Munch J, Kirchhoff F. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol. 2003;77(19):10548–10556. doi: 10.1128/JVI.77.19.10548-10556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, Ranasinghe S, Lindqvist M, Davis I, Lane K, Rychert J, Rosenberg ES, Piechocka-Trocha A, Brass AL, Brenchley JM, Walker BD, Streeck H. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci Transl Med. 2012;4(123):123ra25. doi: 10.1126/scitranslmed.3003165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soghoian DZ, Streeck H. Cytolytic CD4(+) T cells in viral immunity. Expert Rev Vaccines. 2010;9(12):1453–1463. doi: 10.1586/erv.10.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumptner-Cuvelette P, Morchoisne S, Dugast M, Le Gall S, Raposo G, Schwartz O, Benaroch P. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci U S A. 2001;98(21):12144–12149. doi: 10.1073/pnas.221256498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingert B, Benati D, Lambotte O, de Truchis P, Slama L, Jeannin P, Galperin M, Perez-Patrigeon S, Boufassa F, Kwok WW, Lemaitre F, Delfraissy JF, Theze J, Chakrabarti LA. HIV controllers maintain a population of highly efficient Th1 effector cells in contrast to patients treated in the long term. J Virol. 2012;86(19):10661–74. doi: 10.1128/JVI.00056-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gegerfelt A, Valentin A, Alicea C, Van Rompay KK, Marthas ML, Montefiori DC, Pavlakis GN, Felber BK. Emergence of simian immunodeficiency virus-specific cytotoxic CD4+ T cells and increased humoral responses correlate with control of rebounding viremia in CD8-depleted macaques infected with Rev-independent live-attenuated simian immunodeficiency virus. J Immunol. 2010;185(6):3348–3358. doi: 10.4049/jimmunol.1000572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A, Wiskocil RL, Stobo JD. The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J Immunol. 1984;133(1):123–128. [PubMed] [Google Scholar]
- Wiseman RW, Karl JA, Bohn PS, Nimityongskul FA, Starrett GJ, O’Connor DH. Haplessly hoping: macaque major histocompatibility complex made easy. IILAR J. 2013;54(2):196–210. doi: 10.1093/ilar/ilt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Quinn DG, Cohen PL, Frelinger JA. Fas-dependent CD4+ cytotoxic T-cell-mediated pathogenesis during virus infection. Proc Natl Acad Sci U S A. 1996;93(25):14730–14735. doi: 10.1073/pnas.93.25.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Fujiwara M, Ueno T, Oka S, Takiguchi M. Strong ability of Nef-specific CD4+ cytotoxic T cells to suppress human immunodeficiency virus type 1 (HIV-1) replication in HIV-1-infected CD4+ T cells and macrophages. J Virol. 2009;83(15):7668–77. doi: 10.1128/JVI.00513-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Ramachandran S, Mann M, Popkin DL. Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: past, present and future. Viruses. 2012;4(11):2650–69. doi: 10.3390/v4112650. [DOI] [PMC free article] [PubMed] [Google Scholar]