

Abstract
The number of validated biomarkers of tobacco smoke exposure is limited, and none exist for tobacco-related cancer. Additional biomarkers for smoke, effects on cellular systems in vivo are needed to improve early detection of lung cancer, and to assist the Food and Drug Administration in regulating exposures to tobacco products. We assessed the effects of smoking on the gene expression using human cell cultures and blood from a cross-sectional study. We profiled global transcriptional changes in cultured smokers’ peripheral blood mononuclear cells (PBMCs) treated with cigarette smoke condensate (CSC) in vitro (n = 7) and from well-characterized smokers’ blood (n = 36). ANOVA with adjustment for covariates and Pearson correlation were used for statistical analysis in this study. CSC in vitro altered the expression of 1 178 genes (177 genes with > 1.5-fold-change) at P < 0.05. In vivo, PBMCs of heavy and light smokers differed for 614 genes (29 with > 1.5-fold-change) at P < 0.05 (309 remaining significant after adjustment for age, race, and gender). Forty-one genes were persistently altered both in vitro and in vivo, 22 having the same expression pattern reported for non-small cell lung cancer. Our data provides evidence that persistent alterations of gene expression in vitro and in vivo may relate to carcinogenic effects of cigarette smoke, and the identified genes may serve as potential biomarkers for cancer. The use of an in vitro model to corroborate results from human studies provides a novel way to understand human exposure and effect.
Keywords: cigarette smoke, gene expression, peripheral blood mononuclear cells
INTRODUCTION
Tobacco smoking is a major cause of worldwide mortality and morbidity, and specifically it is causally related to cancer of the lung and other malignancies, chronic obstructive pulmonary disease, and heart disease [1]. A deeper understanding of smoking-related cancer mechanisms and new biomarkers of exposure and risk are needed to improve early detection methods, for example, for lung cancer screening. Separately, the recent authority to the Food and Drug Administration (FDA) over tobacco products allows the FDA to mandate product performance standards governing smoke exposure to toxic constituents and also requires the FDA to evaluate manufacture’s health claims for newly modified-tobacco products of purported reduced exposure. To do this, new methods are needed to compare different tobacco products in the laboratory, and new biomarkers are needed to assess differential effects in smokers using modified-tobacco products. Currently there are only a few biomarkers of exposure, and even fewer validated biomarkers of lung cancer risk [2,3].
There are only a few validated biomarkers of tobacco smoke exposure, such as cotinine, and none that have been validated as markers for disease risk such as lung cancer, although some may be [4]. Cotinine is a well-established biomarker for tobacco exposure, but only for recent exposures because of its short half-life (16 h). Numerous other non-invasive biomarkers are in development indicating a more direct biological effect, such as DNA methylation, immune, inflammation, untargeted metabolomics, mitochondrial mutations, oxidative damage, and others [4–13]. For this study, we are focusing on altered gene expression as an alternative biomarker for smoke exposure, which will provide complementary information to other biomarkers as well as explore new carcinogenic pathways related to smoking.
Cigarette smoke condensate (CSC) is a complex mixture of thousands of chemicals, many of which have been implicated as toxins and carcinogens [14]. Studies of gene expression related to cigarette smoke reveal complex relationships for pathogenetic pathways of disease; for example, there are differential gene expression profiles in airway epithelial cells [15,16] and blood cells [17–19]. Exposure of CSC in vitro causes inflammation in human monocytes [20,21], oxidative stress in cardiomyocytes [22], DNA damage in HPV-transform cervical cells [23,24], and extracellular matrix degradation [25]. However, almost all these previous studies compared smoking to non-smoking exposure; none have assessed gene expression as a function of dose. Importantly, as the human studies were conducted using cross-sectional study designs that do not establish direct relationships for smoking and gene expression, results may be confounded by smokers’ lifestyle or demographics. In order to establish better evidence for the relationship of smoke to altered gene expression in vitro, one strategy is to use experimental conditions in vitro and determine if the experimental results can be extrapolated to human experience. Thus, the goals of this study were to evaluate novel gene expression profiles and pathways affected by cigarette smoking, and to identify potential biomarkers for cigarette smoke exposure and harm. By using a corroborative in vitro and in vivo experimental approach, we hypothesized that persistent alterations of gene expression could be identified by systematic transcriptome analysis and there would be a CSC dose-response relationship for gene expression.
MATERIALS AND METHODS
Ethics Statement
The study was approved by the Institutional Review Board of Georgetown University Medical Center and The Ohio State University. Each participant provided a written informed consent for study participation.
Study Population and Sample Collection
Subjects were recruited through local print advertising, and screened for eligibility. Only healthy smokers without current infections, medication use or illnesses that can affect the immune system were included in this study. All smokers had to have been smoking for at least five years and had a stable smoking pattern for the prior 6 months (cigarette brand and cigarettes per day, and no smoking-cessation attempts). Following eligibility screening, current smokers completed an extensive interview and provided a blood specimen. Subjects were classified as light smokers if they smoked fewer than 12 cigarettes per day, as heavy smokers if they smoked more than 23 cigarettes per day, and as moderate smokers if they smoked in between, based on tertiles.
Blood samples from each subject were collected into BD Vacutainer® CPT™ tubes (BD Biosciences, Laurel, MD), and peripheral blood mononuclear cells (PBMCs) were segregated according to the manufacturer’s instructions. All blood samples were delivered and processed within 2 h of phlebotomy. From the company’s documentation, lymphocytes account for a relatively fixed majority (85.9 ± 4.3%) of these mononuclear cells when isolated according to their methods. The mononuclear fractions were placed in TRIzol reagent and kept at −80°C until RNA isolation (uncultured).
Preparation of Cigarette Smoke Condensate (CSC)
CSC was prepared by smoking 2R4F low tar reference cigarettes (University of Kentucky, Lexington, KY) on a GCSM 2072i rotary smoking machine (CH Technologies, Westwood, NJ) using FTC conditions (35 cc puffs of 2 s duration every 60 s). The CSC was collected on Cambridge filter pads and then extracted with dimethyl sulfoxide (DMSO). The extracts were frozen at −80°C until use.
In vitro Treatment of Cultured PBMCs
Freshly isolated PBMCs from five light and three moderate smokers were cultured in RPMI 1640 medium (Biosource, Rockville, MD), supplemented with 1.5% phytohemagglutinin (PHA, GIBCO), 1 000 U/ml heparin, 2 mM L-glutamine, and 15% fetal bovine serum for 72 h. The cultures were then exposed to the CSC at a final concentration of 40 μg/ml (CSC treated), which was previously determined to retain > 90% cell survival (data not shown). Because smokers are chronically exposed to cigarette smoke, we chose to apply an exposure period of 18 h [17], rather than a pulse treatment (4–6 h) [17,26]. A CSC dose of 40 μg/ml was chosen based on studies that showed no cytotoxic effects on human PBMCs, but were able to modulate at gene expression level [17,27]. In this study, an increase in CYP1A1 expression was used as one indicator of the effect of CSC on the exposed cells, given its well established relationship to smoking [28–30]. Cultured PBMCs were exposed to CSC, and the mRNA expression patterns were compared to unexposed (DMSO treated) cultured PBMCs.
Total RNA Isolation
Total RNA was isolated directly from uncultured and cultured PBMCs using the TRIzol reagent method according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). RNA samples then underwent an additional clean-up step using RNeasy Micro Kit (Qiagen, Valencia, CA). Total RNA quality was assessed by visualization of the ethidium bromide-stained 28S and 18S rRNA bands and the 260/280 ratios of absorbance reading.
Gene Expression Microarray
The gene expression microarray was carried out using Affymetrix HG-U133A Genechips containing 22,283 human transcripts (Affymetrix, Santa Clara, CA) at the Genomics Core Facility of Georgetown University. Gene expression data are available through GSE12587.
Quality Control
We chose QC criteria as follows: spike-in hybridization controls (BioB, BioC, BioD and CreX), 3′/5′ glyceraldehydes-3-phosphate dehydrogenase (GAPDH) and β-actin signal ratios were to be less than three, percent present call was to be within 35–65% range and scale factor should be within threefold of one another. Two samples (one sample was treated with DMSO, and the other one was treated with CSC) from 16 cultured PBMCs did not meet the criteria and were omitted from the analysis, which was therefore carried out on seven sets of the paired samples including CSC treated and DMSO treated matched samples. Six samples from uncultured PBMCs did not pass QC and we subsequently carried out analyses on nine samples originating from heavy smokers, 10 samples from moderate smokers and 11 samples from light smokers. Besides the quality control filters for individual microarray, Pearson correlation coefficients were computed for pairs of samples from the same group (across all genes) to allow assessment of the reproducibility and variation of biological replicate samples. Assessment of global gene expression levels among different individuals within the CSC treated group, DMSO treated group, light smoker group and heavy smoker group (data not shown) revealed highly significant correlations with R2 values of more than 0.96. This indicates that there were no outlier samples that needed to be rejected from the analysis, and that all samples from the same group yielded similar overall expression values. A total of 12,501 probe sets passed our filtering and were retained for all further analyses.
Expression Analysis and Statistical Methods
Raw microarray data were normalized using the Robust Multichip Average (RMA) algorithm in the Expression Console (Affymetrix). A differential gene expression analysis was carried out in Transcriptome Analysis Console (Affymetrix) on log2 expression value from the RMA output.
To examine the effect of CSC treatment on the gene expression of human PBMCs, a paired-sample ANOVA test was conducted to test for differential expression between CSC-treated and DMSO treated (control) PBMCs. To examine the effect of smoking on smoker’s PBMCs, an unpaired-sample ANOVA test was used to test for differential expression between heavy smokers and light smokers. To quantify how well a given gene’s expression level correlates with the daily number of cigarettes the subject smoked, Pearson’s correlation coefficients were calculated. Fold change was determined by comparing the normalized biweight average gene expression values between two groups. A change in gene expression is deemed to be significant if the P value is less than 0.05. To account for multiple testing, we controlled the false-discovery rate (FDR) by calculating adjusted P value based on raw P value and listed differential genes according to FDR P values [31]. Two dimensional hierarchical clustering of all smokers using the genes that were differentially expressed between heavy smokers and light smokers were carried out. Prior to clustering, each gene was normalized across all samples to have a mean center. Clustering was performed using Pearson correlation and complete linkage clustering with MeV software program (http://www.tm4.org) [32].
Because of the differences in age, gender, and race between heavy and light smokers, we performed an analysis to test the effect of smoking status (heavy vs. light) on gene expression while controlling for the effects of age, race and gender. Four-way ANOVA was performed with Partek Genomics Suite 6.6 (St. Louis, MO).
Gene Ontology (GO) Analysis
To investigate associations of the differentially expressed genes with biological pathways and networks, Cytoscape v3.0.2 software (http://cytoscape.org) [33] with ClueGO plug-in v2.0.8 (http://www.ici.upmc.fr/cluego/) [34] were used. GO is a bioinformatics platform that provides common vocabularies and classifications to allow genes from different organisms to be compared based on the annotations. ClueGO is a tool to analyze and visualize functionally grouped annotation networks using GO biological process, GO molecular function, and GO cellular component (http://www.geneontology.org/) as ontologies and pathways [35]. The size of the nodes reflects the statistical significance of the terms and color of the nodes reflects the same group. The degree of connectivity between terms (edge) was calculated using κ score. The default setting of the program and a κ score of 0.5 was used. The leading group term was based on the highest significance of the group. The network integrates only the positive κ score term associations and was automatically generated using the organic layout algorithm supported by Cytoscape. A right-sided hypergeometric test was performed to enrich GO-terms. FDR for multiple testing defined the significance (P value < 0.001) [31]. After performing a ClueGO functional analysis, genes associated to term was visualized together on the network. The name of the gene was colored in red. Terms and their genes shared the color. If a gene was found in two or more terms, it had two or more colors. Ingenuity pathway analysis (IPA) software was used to organize the differentially expressed genes into pathways of interacting genes. The identified genes were mapped to canonical pathways in Ingenuity knowledge base and then ranked by significance analysis.
Quantitative RT-PCR Confirmation in Independent Samples
Genes for qRT-PCR were selected from the micro-array data based on the analysis reported herein. Primer sequences were purchased from Applied Biosystems. RNA (500 ng) was reversely transcribed into cDNA using the SuperScript III Reverse Transcriptase (Invitrogen). RT-PCR was performed by using 2X TaqMan Gene Expression Master Mix (Applied Biosystems, Grand Island, NY) on a StepOne Plus PCR system apparatus (Applied Biosystems). Levels of the GAPDH transcript were also measured as internal control. The reactions were performed in triplicate. Relative expression levels were defined as 2−ΔCt. Primer information and conditions of amplification are available in Supplementary Table S1.
RESULTS
Study Population
Demographic data on the 36 smokers are presented in Table 1. Among these subjects, using tertiles, 14 were classified as light smokers (<13 cigs/day), 13 were classified as moderate smokers (15–20 cigs/day), and nine were heavy smokers (> 23 cigs/day) according to the self-reported numbers of cigarettes smoked during the 24 h before blood draw. The mean ages for these groups (47, 47, and 50, respectively), and BMI (28.0, 27.6, and 29.5, respectively) were not statistically different. The numbers of years smoked (18.9, 30.2, and 32.2, respectively) and pack-years (8.7, 28.6, and 57.1, respectively) were statistically different between heavy and light smokers. There were no statistically significant differences in proportion by gender and race.
Table 1.
Clinical Demographics of Study Subjects
| Smoking status
|
|
|||
|---|---|---|---|---|
| Light (L) | Moderate | Heavy (H) | P-value H versus L | |
| Variable | 14 | 13 | 9 | — |
| Sex (Male/Female) | 6/8 | 10/3 | 5/4 | ns |
| Age (yrs) | 46.71 ± 2.68 | 47.43 ± 2.39 | 50.44 ± 3.46 | ns |
| Race (White/Black) | 6/8 | 7/6 | 4/5 | ns |
| BMI | 27.98 ± 0.93 | 27.61 ± 1.29 | 29.46 ± 1.33 | ns |
| Cigarettes per day (last 24 h) | 8.71 ± 0.67 | 18.69 ± 0.60 | 34.11 ± 3.13 | <0.0001 |
| Years of smoking | 18.93 ± 3.33 | 30.15 ± 3.56 | 32.22 ± 4.06 | 0.02 |
| Pack-years | 8.729 ± 1.77 | 28.55 ± 3.67 | 57.11 ± 11.45 | <0.0001 |
Mean (±SEM) is shown for continuous variables.
ns, indicates nonsignificant; —, not applicable.
Pack-years = Divide the number of cigarettes per day by 20, then multiply by the number of years smoked.
Differential Gene Expression After Exposure to CSC in Vitro
A total of 1 178 genes were found to be differentially expressed in seven paired CSC and DMSO-treated cultured PBMCs (Figure 1A) (P < 0.05). Among them, 11 genes had a FDR < 0.1 (ARPC2, CYP1A1, HSD11B1, KIAA0355, NQO1, PIR, PRUNE, SCPEP1, TIPARP, TXNRD1, and WDR19). Very few differentially expressed genes pass multiple testing, which might be due to the small sample size in this study. Of the 1 178 genes, 55% (647) were up-regulated in response to CSC treatment whereas 45% (531) were down-regulated. Among them, there were 177 genes with P < 0.05, and fold change greater than 1.5 (Supplementary Table S2). The most significantly elevated genes included the genes that code for xenobiotic functions, such as phase I enzymes cytochrome P450 1A1 (CYP1A1, 8.9-fold), CYP1B1 (5.43-fold), NAD(P)H dehydrogenase, quinone 1 (NQO1) (3.75-fold), and thioredoxin reductase 1 (TXNRD1, 2.31-fold). Genes that code for iron-binding nuclear protein (PIR, 5.3-fold), FTL (2.06-fold) and FTH1 (1.67-fold) were also up-regulated. The genes encoding cytokines and chemokines were among the most down-regulated genes, such as CCL8 (7.06-fold), CXCL13 (3.5-fold), IL21 (2.8-fold), and IL18RAP (1.5-fold).
Figure 1.
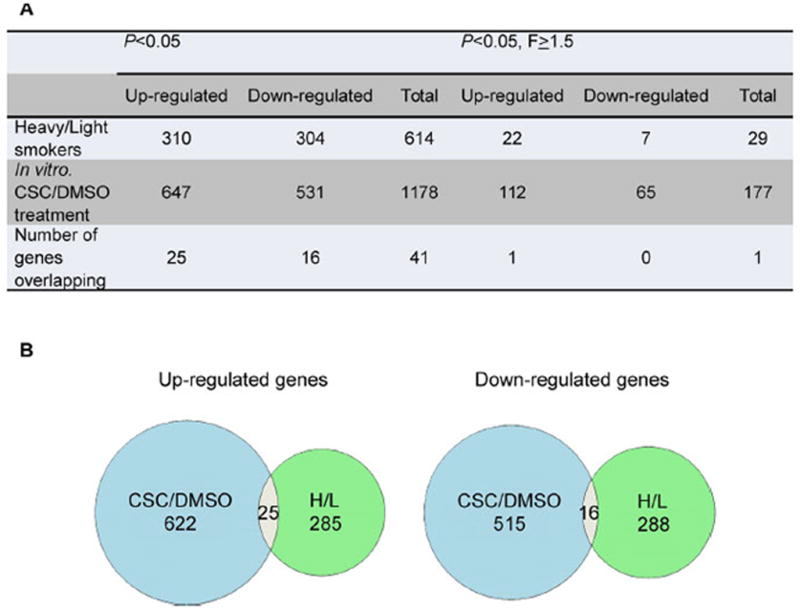
Number of genes differentiating heavy smokers from light smokers (H/L) and CSC treatment from DMSO treatment (CSC/DMSO) in all subjects. Venn diagram showing 41 persistent alternated genes (25 up-regulated and 16 down-regulated) identified in separate analyses of the two different data sets, CSC/DMSO and H/L.
In addition, differentially expressed genes involved in carcinogenesis were also identified (Supplementary Table S2), including up-regulation of oncogenes and down-regulation of tumor suppressor genes. Nine putative oncogenes significantly increased after CSC treatment, such as EREG (1.49-fold), JUN (1.75-fold), MAFF (2.06-fold), MAFG (1.3-fold), RAB38 (1.73-fold), SERPINI1 (1.55-fold), SERPINB2 (5.24-fold), SKIL (1.19-fold), and TOM1 (1.53-fold). Several putative tumor suppressor genes were slightly but significantly decreased by CSC treatment, such as APC (1.13-fold), BRCA2 (1.35-fold), CDKN2D (1.28-fold), SFN (1.25-fold), and TP63 (1.1-fold).
Class Identification of Genes Responsive to CSC Treatment in Vitro
We investigated biological interactions among the genes responsive to CSC treatment using the Cytoscape with ClueGO plug-in. Analysis of all the 1 178 genes identified 36 significant enrichment in a Benjamini-Hochberg analysis (corrected term P value < 0.001). Significant terms are visualized in Figure 2A. These most significant networks identified for CSC treatment invitroare listed in Supplementary Table S3.
Figure 2.

Gene ontology analysis of 1 178 genes associated with CSC/DMSO (A) and 614 genes associated with H/L (B). All genes were analyzed using GlueGO to identify significant biological processes (P <0.001 after FDR correction). Different GO groups are colored with different colors. The detailed list of significant GO terms is shown in Supplementary Table S3, and S5.
Genes Differentially Expressed Between Heavy and Light Smokers
The PBMCs expression profiles from blood were compared between 11 light smokers and 9 heavy smokers. A total of 614 genes were found to be differentially expressed in the PBMCs of heavy smokers as compared with those of light smokers (Figure 1A) (P <0.05). Because the analysis of the differential gene expression in heavy and light smokers was informed by an a priori hypothesis based on the in vitro study, we provided P values that are unadjusted for multiple comparison. Of the 614 genes, 50.5% (310) were up-regulated in heavy smokers versus light smokers, whereas 49.5% (304) were down-regulated. Among them, there were 29 genes with P <0.05, and fold change greater than 1.5 (Figure 1A). The most significantly (P <0.01) up-regulated genes were IGHD (2.3-fold), CD79A (1.6-fold), IGHM (1.63-fold), BLK (1.57-fold), VPREB3 (1.56-fold), and BCL11A (1.51-fold). Genes that were cigarette-smoke responsive in vivo with the down regulation were HIST1H2BH (1.74-fold), HIS-T1H2AC (1.71-fold), TMEM70 (1.7-fold), PDGFC (1.65-fold), SDPR (1.65-fold), PTPN12 (1.61-fold), and C12orf7 (1.59-fold) (Supplementary Table S4).
These 614 genes fell into various categories of biological functions or pathways, such as DNA damage (PPP1R15A) and stress-related signaling genes (APOE, MTF1, and TNF). Functional analysis using Cytoscape with GlueGO plug-in identified 10 significant biological networks (corrected term P value <0.001) (Figure 2B). They included genes involved in immune system, metabolic process, and chromatin remodeling (Supplementary Table S5).
A linear correlation for cigarettes per day and gene expression was assessed for those 614 altered genes. It was found that 78 genes had a statistically significant (P <0.01, FDR <0.1) linear correlation (Table 2). Thirty one genes were positively correlated with the number of cigarette smoked whereas 47 genes were negative correlated with smoking status. To assess how other variables may confound gene expression, a general linear model was used to explore the relationship between gene expression and smoking level adjusted for age, race, and gender. Among the above 614 genes significantly differentiated between heavy and light smokers, 309 genes remained statistically significant after adjustment (P ≤ 0.05) (Supplementary Table S6).
Table 2.
Genes Correlated With Cigarettes Per Day (cpd) Among Smokers
| Symbol | r-value | P-value | FDR |
|---|---|---|---|
| C1RL | 0.6635 | 0.0001 | 0.0208 |
| ERMAP | −0.6620 | 0.0001 | 0.0208 |
| HLF | −0.6218 | 0.0002 | 0.0341 |
| EPHA1 | −0.6190 | 0.0003 | 0.0341 |
| DCAF7 | 0.6175 | 0.0003 | 0.0341 |
| CBX5 | 0.6087 | 0.0004 | 0.0366 |
| FOLH1 | −0.5952 | 0.0005 | 0.0457 |
| IGHD | 0.5844 | 0.0007 | 0.0534 |
| IL23A | −0.5698 | 0.0010 | 0.0635 |
| CRYBA4 | −0.5690 | 0.0010 | 0.0635 |
| SYK | 0.5592 | 0.0013 | 0.0707 |
| C12orf43 | 0.5545 | 0.0015 | 0.0707 |
| TCF21 | −0.5480 | 0.0017 | 0.0707 |
| GLT8D2 | −0.5475 | 0.0017 | 0.0707 |
| RABL2A | 0.5459 | 0.0018 | 0.0707 |
| WEE1 | −0.5441 | 0.0019 | 0.0707 |
| TCTN2 | −0.5424 | 0.0020 | 0.0707 |
| FA2H | −0.5334 | 0.0024 | 0.0730 |
| COMMD4 | 0.5330 | 0.0024 | 0.0730 |
| IRF3 | 0.5326 | 0.0024 | 0.0730 |
| PRR16 | 0.5303 | 0.0026 | 0.0730 |
| USP33 | −0.5280 | 0.0027 | 0.0730 |
| DBH | −0.5273 | 0.0028 | 0.0730 |
| NCAM1 | −0.5228 | 0.0030 | 0.0730 |
| TGOLN2 | 0.5207 | 0.0032 | 0.0730 |
| NAPG | −0.5195 | 0.0033 | 0.0730 |
| CSHL1 | −0.5180 | 0.0034 | 0.0730 |
| AIP | 0.5151 | 0.0036 | 0.0730 |
| KAZALD1 | −0.5150 | 0.0036 | 0.0730 |
| KLK3 | −0.5132 | 0.0037 | 0.0730 |
| PI4KB | 0.5123 | 0.0038 | 0.0730 |
| PGK2 | −0.5094 | 0.0040 | 0.0730 |
| RIMS2 | −0.5081 | 0.0042 | 0.0730 |
| MAP7 | −0.5068 | 0.0043 | 0.0730 |
| SPIB | 0.5062 | 0.0043 | 0.0730 |
| TMPRSS5 | −0.5057 | 0.0044 | 0.0730 |
| VPREB3 | 0.5053 | 0.0044 | 0.0730 |
| EHBP1L1 | 0.5027 | 0.0046 | 0.0746 |
| B4GALNT1 | −0.4986 | 0.0050 | 0.0746 |
| GJA9 | −0.4949 | 0.0054 | 0.0746 |
| ADAMTS13 | −0.4933 | 0.0056 | 0.0746 |
| TXN2 | 0.4932 | 0.0056 | 0.0746 |
| SIPA1 | 0.4908 | 0.0059 | 0.0746 |
| ELMO2 | 0.4887 | 0.0061 | 0.0746 |
| STC2 | −0.4875 | 0.0063 | 0.0746 |
| HOXD12 | −0.4868 | 0.0064 | 0.0746 |
| RPL36 | 0.4842 | 0.0067 | 0.0746 |
| C17orf80 | −0.4839 | 0.0067 | 0.0746 |
| SARDH | −0.4838 | 0.0068 | 0.0746 |
| BCL11A | 0.4833 | 0.0068 | 0.0746 |
| PRSS2 | −0.4822 | 0.0070 | 0.0746 |
| CD79A | 0.4820 | 0.0070 | 0.0746 |
| CAPN9 | −0.4814 | 0.0071 | 0.0746 |
| POLD4 | 0.4807 | 0.0072 | 0.0746 |
| HRH1 | −0.4804 | 0.0072 | 0.0746 |
| PRUNE2 | −0.4789 | 0.0074 | 0.0746 |
| PDGFC | −0.4788 | 0.0074 | 0.0746 |
| LPPR3 | −0.4780 | 0.0076 | 0.0746 |
| DPEP1 | −0.4771 | 0.0077 | 0.0746 |
| BAG6 | 0.4766 | 0.0077 | 0.0746 |
| UMOD | −0.4759 | 0.0079 | 0.0746 |
| ATG13 | 0.4757 | 0.0079 | 0.0746 |
| QKI | 0.4756 | 0.0079 | 0.0746 |
| VRK3 | 0.4756 | 0.0079 | 0.0746 |
| NAALAD2 | −0.4749 | 0.0080 | 0.0746 |
| MTFR1 | 0.4730 | 0.0083 | 0.0746 |
| DICER1 | −0.4729 | 0.0083 | 0.0746 |
| KCNK7 | −0.4728 | 0.0083 | 0.0746 |
| LOC93432 | −0.4725 | 0.0084 | 0.0746 |
| MED24 | 0.4710 | 0.0086 | 0.0755 |
| RBMXL2 | −0.4699 | 0.0088 | 0.0760 |
| SLC6A14 | −0.4682 | 0.0091 | 0.0772 |
| DST | −0.4675 | 0.0092 | 0.0772 |
| MAPK1IP1L | −0.4660 | 0.0095 | 0.0773 |
| NAA60 | 0.4655 | 0.0095 | 0.0773 |
| ITGB4 | −0.4645 | 0.0097 | 0.0773 |
| MIR4784 | 0.4641 | 0.0098 | 0.0773 |
| HSD3B1 | −0.4631 | 0.0099 | 0.0773 |
Hierarchical Clustering
To identify the relationship among samples and the underlying patterns of gene expression, two-way hierarchical cluster analysis was performed on the samples and the subset of 614 genes showing the significant expression with smoking. The samples separated into two clusters, with nine (82%) of the light smokers clustering into one cluster, and nine (100%) of the heavy smokers clustering together (Figure 3A). The expression of a subset of genes in two light smokers was similar to that of heavy smokers. The result demonstrated inter-individual variability within the heavy and light smoker groups, and revealed subgroups of individuals with similar patterns of expression, distinct from the other subgroup. The genes also separated into two main clusters; the top cluster consisting of genes up-regulated in heavy smokers, the bottom cluster containing down-regulated genes (Figure 3A).
Figure 3.
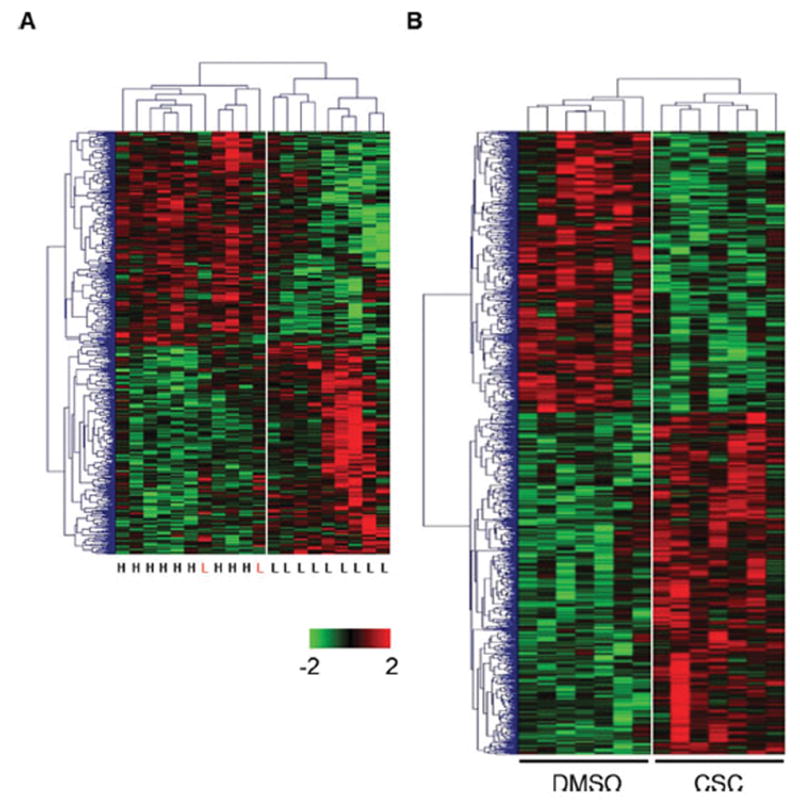
Hierarchical clustering of significant differentially expressed genes in in vitro and in vivo. (A) 310 up-regulated and 304 down-regulated genes in heavy (labeled with H) versus light (labeled with L) smokers. (B) 647 up-regulated and 531 down-regulated genes in CSC versus DMSO treated PBMCs. The dendrogram on the top shows correlations between subjects, and on the left shows correlations between genes. Red, up-regulation; green, down-regulation.
Two-way hierarchical cluster analysis was also performed on the samples and the subset of 1 178 genes showing the significant expression with CSC treatment. The samples separated into two clusters, with seven (100%) of the CSC treated PBMCs clustering into one cluster, and seven (100%) of the DMSO treated PBMCs clustering together (Figure 3B).
Overlap of Differentially Expressed Genes in Vitro and in Vivo
To select transcripts consistently changed upon cigarette smoke, we combined the two lists of 614 and 1 178 significant genes, obtained in the in vivo and in vitro analysis, respectively. We identified a set of 41 genes comprised of 25 up-regulated and 16 down-regulated genes (Figure 1B, and Table 3). They included lipid metabolic process genes such as GM2A, IDS, and LDLR; double-strand break repair genes such as RAD54L, TP53BP1; and transcriptional regulation genes such as GON4L, HLF, MTF1, PPP1R15A, and ZNF301. One gene, PPP1R15A, had an absolute fold change higher than 1.5 both in vivo and in vitro.
Table 3.
Gene Expression Signature of Cigarette Smoke
| Probe set | Gene symbol | Description | Heavy versus Light smoker
|
CSC versus DMSO treatment
|
|||
|---|---|---|---|---|---|---|---|
| Fold change | P-value | Fold change | P-value | ||||
| Up-regulated genes | |||||||
| 1 | 204567_s_at | ABCG1 | ATP-binding cassette, sub-family G (WHITE), member 1 | 1.23 | 0.009608 | 2.04 | 0.005918 |
| 2 | 214736_s_at | ADD1 | adducin 1 (alpha) | 1.18 | 0.03279 | 1.2 | 0.036888 |
| 3 | 218004_at | BSDC1 | BSD domain containing 1 | 1.18 | 0.041418 | 1.34 | 0.002537 |
| 4 | 211034_s_at | C12orf51 | chromosome 12 open reading frame 51 | 1.1 | 0.014476 | 1.24 | 0.045051 |
| 5 | 218089_at | C20orf4 | chromosome 20 open reading frame 4 | 1.12 | 0.046835 | 1.16 | 0.021807 |
| 6 | 201253_s_at | CDIPT | CDP-diacylglycerol–inositol 3-phosphatidyltransferase | 1.19 | 0.038556 | 1.4 | 0.000901 |
| 7 | 55093_at | CHPF2 | chondroitin polymerizing factor 2 | 1.16 | 0.049118 | 1.53 | 0.005841 |
| 8 | 211928_at | DYNC1H1 | dynein, cytoplasmic 1, heavy chain 1 | 1.23 | 0.01802 | 1.36 | 0.047912 |
| 9 | 217788_s_at | GALNT2 | UDP-N-acetyl-alpha-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase 2 (GalNAc-T2) | 1.14 | 0.037033 | 1.3 | 0.029329 |
| 10 | 212737_at | GM2A | GM2 ganglioside activator | 1.32 | 0.021234 | 1.42 | 0.027955 |
| 11 | 201056_at | GOLGB1 | golgin B1 | 1.14 | 0.01177 | 1.52 | 0.002311 |
| 12 | 218873_at | GON4L | gon-4-like (C. elegans) | 1.24 | 0.002474 | 1.15 | 0.046341 |
| 13 | 212221_x_at | IDS | iduronate 2-sulfatase | 1.18 | 0.013825 | 1.89 | 0.000865 |
| 14 | 210111_s_at | KLHDC10 | kelch domain containing 10 | 1.13 | 0.037398 | 1.36 | 0.019973 |
| 15 | 205323_s_at | MTF1 | metal-regulatory transcription factor 1 | 1.22 | 0.030284 | 1.2 | 0.039075 |
| 16 | 37028_at | PPP1R15A | protein phosphatase 1, regulatory subunit 15A | 1.63 | 0.033035 | 2.22 | 0.039873 |
| 17 | 213313_at | RABGAP1 | RAB GTPase activating protein 1 | 1.13 | 0.04469 | 1.33 | 0.002135 |
| 18 | 204675_at | SRD5A1 | steroid-5-alpha-reductase, alpha polypeptide 1 (3-oxo-5 alpha-steroid delta 4-dehydrogenase alpha 1) | 1.2 | 0.037698 | 1.2 | 0.033153 |
| 19 | 221752_at | SSH1 | slingshot homolog 1 (Drosophila) | 1.2 | 0.035265 | 1.92 | 0.000306 |
| 20 | 212040_at | TGOLN2 | trans-golgi network protein 2 | 1.42 | 0.031182 | 1.15 | 0.032404 |
| 21 | 218113_at | TMEM2 | transmembrane protein 2 | 1.2 | 0.011507 | 1.25 | 0.048554 |
| 22 | 203050_at | TP53BP1 | tumor protein p53 binding protein 1 | 1.13 | 0.049225 | 1.11 | 0.044562 |
| 23 | 211950_at | UBR4 | ubiquitin protein ligase E3 component n-recognin 4 | 1.2 | 0.027953 | 1.47 | 0.006463 |
| 24 | 218022_at | VRK3 | vaccinia related kinase 3 | 1.2 | 0.002414 | 1.15 | 0.044567 |
| 25 | 207753_at | ZNF304 | zinc finger protein 304 | 1.18 | 0.045855 | 1.33 | 0.017485 |
| Down-regulated genes | |||||||
| 1 | 220208_at | ADAMTS13 | ADAM metallopeptidase with thrombospondin type 1 motif, 13 | −1.13 | 0.019926 | −1.05 | 0.04569 |
| 2 | 214993_at | ASPHD1 | aspartate beta-hydroxylase domain containing 1 | −1.09 | 0.014294 | −1.06 | 0.044282 |
| 3 | 207995_s_at | CLEC4M | C-type lectin domain family 4, member M | −1.06 | 0.025636 | −1.11 | 0.012449 |
| 4 | 215552_s_at | ESR1 | estrogen receptor 1 | −1.06 | 0.048591 | −1.1 | 0.04457 |
| 5 | 214890_s_at | FAM149A | family with sequence similarity 149, member A | −1.13 | 0.010875 | −1.15 | 0.00139 |
| 6 | 213056_at | FRMD4B | FERM domain containing 4B | −1.23 | 0.006893 | −1.79 | 0.022362 |
| 7 | 204753_s_at | HLF | hepatic leukemia factor | −1.11 | 0.023729 | −1.12 | 0.017527 |
| 8 | 217173_s_at | LDLR | low density lipoprotein receptor | −1.06 | 0.045027 | −1.14 | 0.021961 |
| 9 | 210437_at | MAGEA9 | melanoma antigen family A, 9 | −1.08 | 0.037905 | −1.15 | 0.013205 |
| 10 | 213863_s_at | OAZ3 | ornithine decarboxylase antizyme 3 | −1.12 | 0.04579 | −1.18 | 0.020273 |
| 11 | 214443_at | PVR | poliovirus receptor | −1.16 | 0.031949 | −1.24 | 0.033103 |
| 12 | 204558_at | RAD54L | RAD54-like (S. cerevisiae) | −1.15 | 0.028583 | −1.22 | 0.041756 |
| 13 | 211362_s_at | SERPINB13 | serpin peptidase inhibitor, clade B (ovalbumin), member 13 | −1.12 | 0.022658 | −1.18 | 0.049298 |
| 14 | 213200_at | SYP | synaptophysin | −1.18 | 0.015208 | −1.11 | 0.030883 |
| 15 | 208850_s_at | THY1 | Thy-1 cell surface antigen | −1.05 | 0.034518 | −1.34 | 0.014863 |
| 16 | 211834_s_at | TP63 | tumor protein p63 | −1.1 | 0.027354 | −1.1 | 0.039171 |
Confirmation of Microarray Data
We selected four genes (CD79A, IGHD, SPIB, and TCL1A) for validation by RT-qPCR, based on their overall significance according to fold change and P value. Confirmation was based on 20 independent samples from smoking project (samples not used for microarray analysis), including 7 heavy smokers and 13 light smokers (Figure 4).
Figure 4.
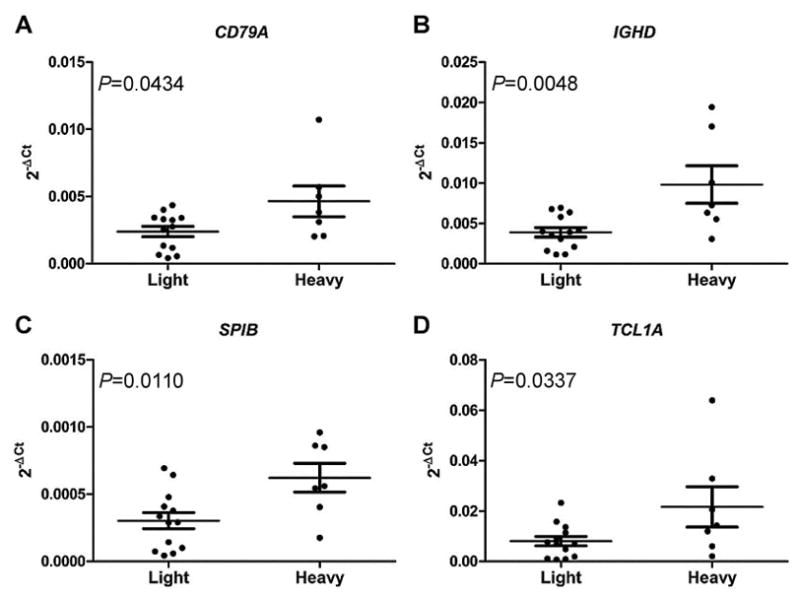
Expression levels of four significant genes between light smokers and heavy smokers. Gene expression was measured through RT-qPCR on 13 PBMCs from light smokers and seven PBMCs from heavy smokers. Expression levels are shown in 2−ΔCt on the Y-axis, smoking status is shown on the X-axis. The data are expressed as mean ± SEM. The P value of each gene is shown. (A) CD79A, (B) IGHD, (C) SPIB, and (D) TCL1A.
DISCUSSION
This study novelly assessed altered gene expression of human peripheral blood mononuclear cells (PBMCs) in vitro exposed to cigarette smoke condensate and seeks similar effects in humans. The goal is to identify new pathways affected by cigarette smoke in humans, and to develop new biomarkers of exposure and effect. The identification of those common genes affected in vitro and in vivo provides strong evidence that the altered gene expression in smokers is directly related to smoking. CSC, which contains thousands of chemicals, is known to perturb many cellular functions. The initial microarray analysis identified 1 178 genes associated with CSC treatment, and 614 genes associated with self-reported smoking status. Importantly, 41 genes were commonly affected in both in vitro and in vivo. A critical criterion for establishing a biomarker of smoke exposure is not only the demonstration that there are differences between smokers and non-smokers, but also that there is a difference among smokers of different smoking levels. This study identified those altered gene expressions with the level of smoking.
There are important reasons to identify new biomarkers of smoke exposure and risk. Better early detection methods for lung cancer are emerging, for example through CAT scan screening [36], but more rational methods are needed for identifying persons who would benefit most, or better selection methods for following—up abnormalities that may be false positives. Separately, in 2009, the United States Congress provided regulatory oversight to the Food and Drug Administration (FDA) for some tobacco products including cigarettes and smokeless tobacco [37]. The FDA is specifically directed to reduce the harm to the public by establishing performance standards to reduce tobacco-related toxicity and disease, and to approve new products to ensure that they are not more dangerous than existing products. Also, the tobacco industry continues to invent and market new tobacco products that they claim will reduce tobacco smoke exposure or toxicity, and they can ask the FDA to label such products to inform consumers that these are safer products. In 2014, the FDA is now proposing to extend their authority to additional tobacco products such as cigars and electronic cigarettes (https://www.federalregister.gov/articles/2014/04/25/2014-09491/deeming-tobacco-products-to-be-subject-to-the-federal-food-drug-and-cosmetic-act-as-amended-by-the). For the FDA to accomplish this mission, substantial research is needed to support and defend the best policy making and decisions by the FDA [38]. Critical to these efforts will be human studies using biomarkers [39,40]. Yet, there are only a few validated biomarkers of exposure and none are validated as biomarkers of lung cancer risk, although some exposure biomarkers may be risk markers too [3]. Thus, the need for new biomarkers to assess tobacco product toxicity is an important public health and FDA priority [41]. One approach to identifying new markers is to use a screening method that can assess broad changes in product design through the evaluation of gene expression [42].
Several genes identified herein to be affected by smoking have also been reported in other studies. van Leeuwen and colleagues demonstrated that three genes (ATF3, SERPINB2, and TGM2) that we report herein were among 14 genes identified by CSC treatment in PBMCs in vitro. Separately, Ryder and colleagues used an in vitro smoke system to treat PBMCs for 5 min and found 106 genes significantly elevated or depressed [43]. Thirteen genes from our study were also found in this gene list including ATF3, CTSL1, DNAJB1, EIF5, FUT1, JMJD6, MINK1, MLLT11, PHLDA1, PTEN, SERPINI11, SLC2A3, and ZNF410. Charlesworth and coworkers reported that 323 unique genes in lymphocytes were significantly correlated with smoking status including cigarettes per day and serum cotinine levels [44]. Eleven genes were also found in our in vivo study including EPB41L3, FAM53B, NCAM1, PALLD, PTPN6, RHOBTB3, SASH1, STK10, TBCD, TKTL1, and VPS37B. Buttner and colleagues identified 88 genes differentially expressed in T lymphocyte (CD3+/CD4+ and CD3+/CD8+) between three heavy smokers (> 20 cigarettes per day for at least 10 years) and three never smokers [19].
A premise for using blood biomarkers is that they reflect changes in target organs, such as the impact on lung, resulting from cigarette smoking, and are non-invasive. Beineke et al. constructed a five gene (CLDN1, LRRN3, MUC1, GOPC, LEF1) predictive model in whole blood to evaluate self-reported smoking status (current and recently quit smokers vs. former and never smokers) [45]. It should be noted that this study has focused on differences between current smokers, former smokers and never smokers, while our study examines smokers with different levels of exposure, and none sought cross-validation with an in vitro cell culture model. Among the genes found in our study to be associated with cigarette smoke status, seventy eight genes were positively/negatively correlated with number of cigarettes per day. Those 78 genes may serve as biomarkers of tobacco exposure. Further studies are needed to increase sample population, select candidate genes, and build a predictive model. There also are several studies of airway epithelium gene expression profiles on smokers, never-smokers and former smokers revealing a large number of altered gene expressions [46–51]. For example, in a series of studies by Spira and coworkers, gene transcription using bronchial cells obtained by bronchoscopy was able to distinguish never from former and current smokers, and several gene changes were found in the current smokers that no longer were present in the former smokers [46]. Forty-eight genes were tightly correlated with pack-years among current smokers (P < 0.0001). However, none of these 48 genes were found to be altered in our study. There are several possible reasons for this. First, airway epithelial cells are a direct target of smoking, whereas PBMCs can be considered a systemic or secondary target. Second, gene expression signatures have cell and/or tissue specific pattern.
In our study the most statistically significant pathway affected by CSC treatment is NRF2-mediated oxidative stress response (Supplementary Table S7). NRF2 pathway has emerged as a prime target of CSC-induced oxidative stress [52]. Smoking-induced NRF2 pathway activation is highly correlated to the lung diseases including lung cancer. Twenty five out of twenty six genes were significantly up-regulated after CSC exposure in PBMCs, only one gene (PIK3R5) was down-regulated (Supplementary Table S7). However, NRF2 (also known as NFE2L2) expression itself was not significantly affected by CSC, even though it was slightly increased after CSC exposure in the PBMCs. This may be because activation of NRF2 relies on the release from KEAP1, and then phosphorylation of the gene and translocation from cytoplasm to nucleus, rather than on gene expression [53]. Separately, we identified changes in several Phase I and II metabolizing genes that are known to be activated by CSC constituents through different mechanisms [54], namely up-regulated CYP1A1and CYP1B1. Both enzymes are controlled by the aryl hydrocarbon receptor (AhR). Recent investigations demonstrated that cigarette smoke contains high levels of agonists for AhR [55] and strongly activates the AhR signaling pathway [56]. However, the genes encoding other cytochrome P450 isoforms (e.g., CYP2B1) were not responsive or were much less responsive to CSC, which may reflect a tissue-specific context [31].
Unsupervised hierarchical clustering shows the wide interindividual variation among smokers and their gene expression patterns. Given that smokers have differing risks for different diseases, such variation in gene expression might be related to these phenotypes. The cluster analysis distinguished the heavy smokers from the light smokers, except for two light smokers who had patterns similar to the heavy smokers. It may be that these two light smokers did not report accurately and actually smoked more like heavy smokers, or that they are sensitive to cigarette smoke, for example, due to genetic susceptibilities and will have risk of a disease similar to heavier smokers. Several studies showed interindividual differences in DNA adduct levels [57], urine nicotine extraction, plasma cotinine [58], and gene expression [59] in adult cigarette smokers.
The comparison of gene expression in un-cultured PBMCs from smokers (in vivo) and cultured PBMCs from CSC exposure (in vitro) identified 41 common genes in both studies. The common genes between in vivo and in vitro suggest that smoking exposure and CSC treatment may trigger similar effects in PBMCs. Spira and his colleagues also showed that several genes are not reversible when smoking is discontinued, indicating persistent altered expression of genes among former smokers[60]. These 41 common genes demonstrated similar effects of smoking exposure not only in vitro but also in vivo. There are several reasons to explain why the gene list was different between in vitro and in vivo. First, CSC does not include all constituents of tobacco smoke; for example, the volatile organic compounds are not in the CSC. These compounds might play a role in modulation of gene expression in vivo. Second, PBMCs from smokers might also be influenced by other exposures that may interact with smoking (e.g., alcohol intake, food supplements), or smoking could be a surrogate for lifestyle, diet, and other exposures. Third, the kinetics of metabolism of smoking or CSC by humans may play a very important role in the gene expression profiling. Fourth, in vitro and in vivo provide totally different environments to study changes of gene expression. In the in vitro system, we can control variables to reduce the interaction between the CSC treatment and other factors. Culturing itself may also induce gene expression changes. However, those 41 (25, up-regulated; 16, down-regulated) concurrent differential genes may potential serve as biomarkers for smoke exposure. Further analysis of those 41 genes using Oncomine database (http://www.oncomin.org) demonstrated that seventeen out of twenty-five up-regulated genes and five out of sixteen down-regulated genes have the same expression pattern in non-small cell lung cancer (NSCLC) including (ABCG1, BSDC1, C12orf51, CHPF2, DYNC1H1, GALNT2, GM2A, GON4L, KLHDC10, MTF1, RABGAP1, SRD5A1, SSH1, TGOLN2, TP53BP1, UBR4, VRK3, CELC4M, ESR1, HLF, LDLR, and THY1) [61,62]. Among those genes, THY1 and ESR1 have been reported to serve as tumor suppressor genes in nasopharyngeal carcinoma and hepatocellular carcinoma, respectively [63,64]. Down-regulation of tumor suppressor genes may play an important role during tumorgenesis. Woenckhaus and colleagues also demonstrated that down-regulation of HLF was observed in the bronchial epithelium of smokers and squamous cell carcinoma [65]. Taken together, these results suggest that persistent alternations of 41 genes may not only serve as biomarkers for smoke exposure but also provide an evidence to demonstrate the relationship between smoke exposure and lung cancer.
The strength of this study was the use of isolated PBMCs. It is known that cigarette smoking significantly affects white blood cell (WBC) count, especially granulocytes [66]. Palmer and colleagues demonstrated that there is a different gene expression signatures between lymphocytes and granulocytes [67]. Our study used only mononuclear blood cells, which have a more uniform cell population, containing lymphocytes (~90%) and monocytes, and are the most transcriptionally active cells in blood. Another strength of this study was the comparison between heavy smokers and light smokers. Almost all previous studies employed a narrow spectrum of gene assessment and all compared current smoking and never smoking exposure; none have assessed gene expression as a function of dose. In addition, we also combined the analysis between in vitro and in vivo to select one set of genes that was persistently altered by smoking. This study also had a number of limitations. Self-reported smoking status is an imperfect measure because participants may not report their status correctly. The number of participants was small, leading to possible sampling errors that might have obscured important findings, which may have resulted in the inability to detect differentially expressed genes at a FDR <0.1 in the in vivo study. Still, the study identified 41 consistently changed genes by combining two different experiments. Finally, data relating to personal lifestyle history were limited; we lacked data for second-hand smoke exposure, and for food and supplement intake.
In conclusion, we have investigated the relationship between PBMCs from a cross-sectional epidemiology study of smokers and cultured blood cells exposed to CSC, in order to derive a concurrent differential gene set. With further, prospective investigations into the biological significance, these identified genes may provide insights into the underlying processes of smoking-related diseases and represent potential biomarkers for smoking exposure and disease response.
Supplementary Material
Acknowledgments
Grant sponsor: Transdisciplinary Tobacco Use Research Center; Grant number: P50 CA84718; Grant sponsor: National Cancer Institute and the National Institute on Drug Abuse; Grant number: P30 CA051008; Grant sponsor: National Cancer Institute, Laboratory Assessment of Tobacco Use Behavior and Exposure to Toxins; Grant number: N01 PC64402; Grant sponsor: National Cancer Institute; Grant number: K08 DA14276; Grant sponsor: National Institute on Drug Abuse
Footnotes
Conflict of interest: None.
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Glantz SA, Johnson KC. The surgeon general report on smoking and health 50 years later: Breast cancer and the cost of increasing caution. Cancer Epidemiol Biomarkers Prev. 2014;23:37–46. doi: 10.1158/1055-9965.EPI-13-1081. [DOI] [PubMed] [Google Scholar]
- 2.Hatsukami DK, Benowitz NL, Rennard SI, Oncken C, Hecht SS. Biomarkers to assess the utility of potential reduced exposure tobacco products. Nicotine Tob Res. 2006;8:169–191. doi: 10.1080/14622200600576628. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS, Murphy SE, Stepanov I, Nelson HH, Yuan JM. Tobacco smoke biomarkers and cancer risk among male smokers in the Shanghai Cohort Study. Cancer Lett. 2012;334:34–38. doi: 10.1016/j.canlet.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan JM, Butler LM, Stepanov I, Hecht SS. Urinary tobacco smoke-constituent biomarkers for assessing risk of lung cancer. Cancer Res. 2014;74:401–411. doi: 10.1158/0008-5472.CAN-13-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shenker NS, Ueland PM, Polidoro S, et al. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology. 2013;24:712–716. doi: 10.1097/EDE.0b013e31829d5cb3. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Schottker B, Florath I, et al. Smoking-associated DNA methylation biomarkers and their predictive value for all-cause and cardiovascular mortality. Environ Health Perspect. 2015 doi: 10.1289/ehp.1409020. http://dx.doi.org/10.1289/ehp.1409020. [DOI] [PMC free article] [PubMed]
- 7.Lowe FJ, Luettich K, Gregg EO. Lung cancer biomarkers for the assessment of modified risk tobacco products: An oxidative stress perspective. Biomarkers. 2013;18:183–195. doi: 10.3109/1354750X.2013.777116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiels MS, Katki HA, Freedman ND, et al. Cigarette smoking and variations in systemic immune and inflammation markers. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guzman L, Depix MS, Salinas AM, et al. Analysis of aberrant methylation on promoter sequences of tumor suppressor genes and total DNA in sputum samples: A promising tool for early detection of COPD and lung cancer in smokers. Diagn Pathol. 2012;7:87. doi: 10.1186/1746-1596-7-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gower AC, Steiling K, Brothers JF, Lenburg ME, Spira A. Transcriptomic studies of the airway field of injury associated with smoking-related lung disease. Proc Am Thorac Soc. 2011;8:173–179. doi: 10.1513/pats.201011-066MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu PC, Zhou B, Zhao Y, et al. Feasibility of identifying the tobacco-related global metabolome in blood by UPLC-QTOF-MS. J Proteome Res. 2013;12:679–691. doi: 10.1021/pr3007705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan D, Goerlitz DS, Dumitrescu RG, et al. Associations between cigarette smoking and mitochondrial DNA abnormalities in buccal cells. Carcinogenesis. 2008;29:1170–1177. doi: 10.1093/carcin/bgn034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Lenburg ME, Spira A. Comparison of nasal epithelial smoking-induced gene expression on Affymetrix Exon 1.0 and Gene 1.0. ST arrays. ScientificWorldJournal. 2013;2013:951416. doi: 10.1155/2013/951416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson MD, Schilz J, Djordjevic MV, Rice JR, Shields PG. Evaluation of in vitro assays for assessing the toxicity of cigarette smoke and smokeless tobacco. Cancer Epidemiol Biomarkers Prev. 2009;18:3263–3304. doi: 10.1158/1055-9965.EPI-09-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beane J, Vick J, Schembri F, et al. Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq. Cancer Prev Res (Phila) 2011;4:803–817. doi: 10.1158/1940-6207.CAPR-11-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Sebastiani P, Liu G, et al. Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol Genomics. 2009;41:1–8. doi: 10.1152/physiolgenomics.00167.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Leeuwen DM, Gottschalk RW, van Herwijnen MH, Moonen EJ, Kleinjans JC, van Delft JH. Differential gene expression in human peripheral blood mononuclear cells induced by cigarette smoke and its constituents. Toxicol Sci. 2005;86:200–210. doi: 10.1093/toxsci/kfi168. [DOI] [PubMed] [Google Scholar]
- 18.Lampe JW, Stepaniants SB, Mao M, et al. Signatures of environmental exposures using peripheral leukocyte gene expression: Tobacco smoke. Cancer Epidemiol Biomarkers Prev. 2004;13:445–453. [PubMed] [Google Scholar]
- 19.Buttner P, Mosig S, Funke H. Gene expression profiles of T lymphocytes are sensitive to the influence of heavy smoking: A pilot study. Immunogenetics. 2007;59:37–43. doi: 10.1007/s00251-006-0177-3. [DOI] [PubMed] [Google Scholar]
- 20.Wright WR, Parzych K, Crawford D, Mein C, Mitchell JA, Paul-Clark MJ. Inflammatory transcriptome profiling of human monocytes exposed acutely to cigarette smoke. PLoS ONE. 2012;7:e30120. doi: 10.1371/journal.pone.0030120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang JW, Sundar IK, Yao H, Sellix MT, Rahman I. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J. 2013;28:176–194. doi: 10.1096/fj.13-232629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niu J, Wang K, Kolattukudy PE. Cerium oxide nanoparticles inhibit oxidative stress and nuclear factor-kappaB activation in H9c2 cardiomyocytes exposed to cigarette smoke extract. J Pharmacol Exp Ther. 2011;338:53–61. doi: 10.1124/jpet.111.179978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moktar A, Ravoori S, Vadhanam MV, Gairola CG, Gupta RC. Cigarette smoke-induced DNA damage and repair detected by the comet assay in HPV-transformed cervical cells. Int J Oncol. 2009;35:1297–1304. doi: 10.3892/ijo_00000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Okuka M, Lu W, et al. Telomere shortening and DNA damage of embryonic stem cells induced by cigarette smoke. Reprod Toxicol. 2012;35:89–95. doi: 10.1016/j.reprotox.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Nordskog BK, Blixt AD, Morgan WT, Fields WR, Hellmann GM. Matrix-degrading and pro-inflammatory changes in human vascular endothelial cells exposed to cigarette smoke condensate. Cardiovasc Toxicol. 2003;3:101–117. doi: 10.1385/ct:3:2:101. [DOI] [PubMed] [Google Scholar]
- 26.Hellermann GR, Nagy SB, Kong X, Lockey RF, Mohapatra SS. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir Res. 2002;3:22. doi: 10.1186/rr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plottner S, Behm C, Bolt HM, Follmann W. Effects of cigarette smoke condensate on primary urothelial cells in vitro. J Toxicol Environ Health A. 2012;75:1194–1205. doi: 10.1080/15287394.2012.709166. [DOI] [PubMed] [Google Scholar]
- 28.Lin P, Hu SW, Chang TH. Correlation between gene expression of aryl hydrocarbon receptor (AhR), hydrocarbon receptor nuclear translocator (Arnt), cytochromes P4501A1 (CYP1A1) and 1B1 (CYP1B1), and inducibility of CYP1A1 and CYP1B1 in human lymphocytes. Toxicol Sci. 2003;71:20–26. doi: 10.1093/toxsci/71.1.20. [DOI] [PubMed] [Google Scholar]
- 29.Willey JC, Coy EL, Frampton MW, et al. Quantitative RT-PCR measurement of cytochromes p450 1A1, 1B1, and 2B7, microsomal epoxide hydrolase, and NADPH oxidoreductase expression in lung cells of smokers and nonsmokers. Am J Respir Cell Mol Biol. 1997;17:114–124. doi: 10.1165/ajrcmb.17.1.2783. [DOI] [PubMed] [Google Scholar]
- 30.Mace K, Bowman ED, Vautravers P, Shields PG, Harris CC, Pfeifer AM. Characterisation of xenobiotic-metabolising enzyme expression in human bronchial mucosa and peripheral lung tissues. Eur J Cancer. 1998;34:914–920. doi: 10.1016/s0959-8049(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 31.Benjamini Y, Hochberg Y. Controlling the false discovery rate -a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- 32.Saeed AI, Sharov V, White J, et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 33.Shannon P, Markiel A, Ozier O, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bindea G, Mlecnik B, Hackl H, et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felten MK, Knoll L, Schikowsky C, et al. Is it useful to combine sputum cytology and low-dose spiral computed tomography for early detection of lung cancer in formerly asbestos-exposed power industry workers? J Occup Med Toxicol. 2014;9:14. doi: 10.1186/1745-6673-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Husten CG, Deyton LR. Understanding the tobacco control act: Efforts by the US food and drug administration to make tobacco-related morbidity and mortality part of the USA’s past, not its future. Lancet. 2013;381:1570–1580. doi: 10.1016/S0140-6736(13)60735-7. [DOI] [PubMed] [Google Scholar]
- 38.Ashley DL, Backinger CL, van Bemmel DM, Neveleff DJ. Tobacco regulatory science: Research to inform regulatory action at the food and drug administration’s center for tobacco products. Nicotine Tob Res. 2014;16:1045–1049. doi: 10.1093/ntr/ntu038. [DOI] [PubMed] [Google Scholar]
- 39.Olson S, Robinson S, Rapporteurs RG. Accelerating the Development of Biomarkers for Drug Safety: Workshop Summary. The National Academies Press; 2009. [PubMed] [Google Scholar]
- 40.Micheel CM, Ball JR. Evaluation of Biomarkers and Surrogate Endpoints in Chronic Disease. The National Academies Press; 2010. [PubMed] [Google Scholar]
- 41.Ashley DL, Backinger CL. The food and drug administration’s regulation of tobacco: The center for tobacco products’ office of science. Am J Prev Med. 2012;43:S255–S263. doi: 10.1016/j.amepre.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Mattes WB, Richter P, Mendrick DL. An omics strategy for discovering pulmonary biomarkers potentially relevant to the evaluation of tobacco products. Biomark Med. 2012;6:849–860. doi: 10.2217/bmm.12.78. [DOI] [PubMed] [Google Scholar]
- 43.Ryder MI, Hyun W, Loomer P, Haqq C. Alteration of gene expression profiles of peripheral mononuclear blood cells by tobacco smoke: Implications for periodontal diseases. Oral Microbiol Immunol. 2004;19:39–49. doi: 10.1046/j.0902-0055.2003.00110.x. [DOI] [PubMed] [Google Scholar]
- 44.Charlesworth JC, Curran JE, Johnson MP, et al. Transcriptomic epidemiology of smoking: The effect of smoking on gene expression in lymphocytes. BMC Med Genomics. 2010;3:29. doi: 10.1186/1755-8794-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beineke P, Fitch K, Tao H, et al. A whole blood gene expression-based signature for smoking status. BMC Med Genomics. 2012;5:58. doi: 10.1186/1755-8794-5-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spira A, Beane J, Shah V, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA. 2004;101:10143–10148. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spira A, Beane JE, Shah V, et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med. 2007;13:361–366. doi: 10.1038/nm1556. [DOI] [PubMed] [Google Scholar]
- 48.Boelens MC, van den Berg A, Fehrmann RS, et al. Current smoking-specific gene expression signature in normal bronchial epithelium is enhanced in squamous cell lung cancer. J Pathol. 2009;218:182–191. doi: 10.1002/path.2520. [DOI] [PubMed] [Google Scholar]
- 49.Sridhar S, Schembri F, Zeskind J, et al. Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genomics. 2008;9:259. doi: 10.1186/1471-2164-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Lee JJ, Tang H, et al. Impact of smoking cessation on global gene expression in the bronchial epithelium of chronic smokers. Cancer Prev Res (Phila) 2008;1:112–118. doi: 10.1158/1940-6207.CAPR-07-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Dyck E, Nazarov PV, Muller A, et al. Bronchial airway gene expression in smokers with lung or head and neck cancer. Cancer Med. 2014;3:322–336. doi: 10.1002/cam4.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muller T, Hengstermann A. Nrf2: Friend and foe in preventing cigarette smoking-dependent lung disease. Chem Res Toxicol. 2012;25:1805–1824. doi: 10.1021/tx300145n. [DOI] [PubMed] [Google Scholar]
- 53.Kang KW, Lee SJ, Kim SG. Molecular mechanism of nrf2 activation by oxidative stress. Antioxid Redox Signal. 2005;7:1664–1673. doi: 10.1089/ars.2005.7.1664. [DOI] [PubMed] [Google Scholar]
- 54.Gebel S, Gerstmayer B, Bosio A, Haussmann HJ, Van Miert E, Muller T. Gene expression profiling in respiratory tissues from rats exposed to mainstream cigarette smoke. Carcinogenesis. 2004;25:169–178. doi: 10.1093/carcin/bgg193. [DOI] [PubMed] [Google Scholar]
- 55.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 56.Kitamura M, Kasai A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett. 2007;252:184–194. doi: 10.1016/j.canlet.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 57.Ketelslegers HB, Gottschalk RW, Godschalk RW, et al. Interindividual variations in DNA adduct levels assessed by analysis of multiple genetic polymorphisms in smokers. Cancer Epidemiol Biomarkers Prev. 2006;15:624–629. doi: 10.1158/1055-9965.EPI-05-0431. [DOI] [PubMed] [Google Scholar]
- 58.Liang Q, Sarkar M. Intra- and inter-individual variability in urinary nicotine excretion and plasma cotinine in adult cigarette smokers. Regul Toxicol Pharmacol. 2012;64:388–393. doi: 10.1016/j.yrtph.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 59.Dumeaux V, Olsen KS, Nuel G, Paulssen RH, Borresen-Dale AL, Lund E. Deciphering normal blood gene expression variation-The NOWAC postgenome study. PLoS Genet. 2010;6:e1000873. doi: 10.1371/journal.pgen.1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beane J, Sebastiani P, Liu G, Brody JS, Lenburg ME, Spira A. Reversible and permanent effects of tobacco smoke exposure on airway epithelial gene expression. Genome Biol. 2007;8:R201. doi: 10.1186/gb-2007-8-9-r201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hou J, Aerts J, den Hamer B, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Selamat SA, Chung BS, Girard L, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22:1197–1211. doi: 10.1101/gr.132662.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lung HL, Bangarusamy DK, Xie D, et al. THY1 is a candidate tumour suppressor gene with decreased expression in metastatic nasopharyngeal carcinoma. Oncogene. 2005;24:6525–6532. doi: 10.1038/sj.onc.1208812. [DOI] [PubMed] [Google Scholar]
- 64.Hishida M, Nomoto S, Inokawa Y, et al. Estrogen receptor 1 gene as a tumor suppressor gene in hepatocellular carcinoma detected by triple-combination array analysis. Int J Oncol. 2013;43:88–94. doi: 10.3892/ijo.2013.1951. [DOI] [PubMed] [Google Scholar]
- 65.Woenckhaus M, Klein-Hitpass L, Grepmeier U, et al. Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J Pathol. 2006;210:192–204. doi: 10.1002/path.2039. [DOI] [PubMed] [Google Scholar]
- 66.Smith MR, Kinmonth AL, Luben RN, et al. Smoking status and differential white cell count in men and women in the EPIC-Norfolk population. Atherosclerosis. 2003;169:331–337. doi: 10.1016/s0021-9150(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 67.Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. 2006;7:115. doi: 10.1186/1471-2164-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.