

Abstract
Age is the greatest risk factor for breast cancer, but the reasons underlying this association are unclear. While there is undeniably a genetic component to all cancers, the accumulation of mutations with age alone is insufficient to explain the age-dependent increase in breast cancer incidence. In this viewpoint we propose a multilevel framework to better understand the respective roles played by somatic mutation, microenvironment, and epigenetics making women more susceptible to breast cancer with age. The process of aging is associated with gradual breast tissue changes that not only corrupt the tumor suppressive activity of normal tissue, but also impose age-specific epigenetic changes that alter gene expression, thus reinforcing a continuum of age related tissue microenvironments. The evidence discussed here suggests that while the riddle of whether epigenetics drives microenvironmental changes, or whether changes in the microenvironment alter heritable cellular memory has not been solved, a path has been cleared enabling functional analysis leading to the prediction of key nodes in the network that link the microenvironment with the epigenome. The hypothesis that accumulation of somatic mutations with age drives the age-related increase in breast cancer incidence, if correct, has a somewhat nihilistic conclusion; that cancers will be impossible to avoid. Alternatively if microenvironment-driven epigenetic changes are key to explaining susceptibility to age-related breast cancers then there is hope that primary prevention is possible because epigenomes are relatively malleable.
Introduction
Phenotypes of aging tend to be tissue specific. For example, with age the skeletal muscle does not regenerate well, cognitive impairments in the brain are not uncommon, and in many epithelial tissues, including breast, there is an increased incidence of carcinomas. Indeed, more than 80% of breast cancers in the U.S. are diagnosed in women aged over 50 [1,2]. Although aging is generally associated with loss of function in tissues, age-related cancers may be paradoxical examples of gains of function in that there is uncontrolled growth and the appearance of novel functions, such as invasion and metastasis [3]. A long held and dominant view has been that progressive accrual of mutations in oncogenes and tumor suppressors accounts for increased cancer incidence [4]. While some cancers indeed show an exponential increase in incidence with age, consistent with the accumulated mutation hypothesis, the vast majority of breast cancers are age-related, whose incidence rates slow after age 50 [5]. Breast cancer has a bimodal distribution with respect to age that has modes at 50 and 70 years. There is undeniably a genetic component to all cancers, but mutation alone is insufficient to explain the age-dependent increases of breast cancer incidence. What is known of aging in human breast has been mainly the domain of pathologists who utilized normal tissues as controls for breast cancer studies. In order to develop a functional understanding of the effects of aging we have successfully used a combination of primary cell culture, bioengineering, and histology [6-8]. Based on an emerging understanding of the impact of tissue microenvironment on tumor genesis, and our approach to understanding consequences of aging in human mammary epithelia we propose an alternate hypothesis. Increased incidence of age-related breast cancers results from gradual loss of function changes at the level of tissue structure and organization that corrupt tumor suppressive activity of normal tissue architecture; and cause epigenetic changes that alter gene expression, thereby altering normal stem and somatic cell functions. These alterations lead to tissue-level phenotypes that make breast epithelia susceptible to transformation.
In this viewpoint, we aim to summarize the theoretical background of prevailing constructs, and expand the discussion of accumulation of somatic mutation and age dependent breast cancer incidence based on evidence that tissue microenvironments and epigenetic states strongly influence tumor genesis.
Aging and Breast Tissue Fitness
The term ‘breast cancer’ represents a diverse group of diseases, which are commonly classified as either luminal A and B, triple-negative/basal-like, or HER2- positive subtypes based on their expression of hormone receptors, HER2 amplification, and other biochemical and molecular markers. A full 80% of breast cancers in women over 50 are the luminal subtypes [9]. There are no particular patterns of gene mutations in these age-related cancers, but rather, they have the greatest transcriptional diversity and their transcriptomes exhibit age-specific expression patterns [10,11]. Increasing age correlates with shifting gene expression patterns in a number of healthy human tissues including mammary epithelia [6,12-14], but the sources and functional consequences of those changes are largely unknown. Age-dependent transcriptomes could be explained by mutational, epigenetic, and microenvironmental changes.
Tissue microenvironments, defined as the combinations of cell-cell, -ECM, and -soluble factor interactions surrounding each cell, exchange information with cells via a combination of physical, chemical, and electrical signals, frequently activating or deactivating the same pathways triggered by oncogenes [15-17]. Deleterious mutations can be amplified throughout a phenotypically normal epithelia, and only participate in the tumorigenic process in discrete locations where, presumably, additional deleterious events took place [18]. The influence of microenvironment can be so profound as to make frankly malignant cells behave in a phenotypically normal manner, so long as the normal tissue structure remains intact. Thus situations that challenge normal tissue architecture could unleash predisposed cells. Steady age-related decline in breast tissue fitness may explain why luminal subtype breast cancers carry the burden of risk of recurrence as far out as 10 to 20 years following their initial diagnosis [19].
Aging is a gradual process, but the change to our tissues is not subtle, even from a superficial perspective. The epidemiologist Malcolm Pike suggested that ‘breast tissue age' was a predictor of breast cancer risk that is distinct from chronological age [20]. This conceptually reasonable model mainly considered low-resolution changes such as hormones and childbirths, however, this model could not account for the cell- and molecular-level changes that arise with age in breast tissue. The breast consists of the gland that is a branching pseudostratified epithelium, which is surrounded by the stroma, composed of ECM, adipose cells, endothelial cells, fibroblasts, and blood cells. The vast majority of breast cancers originate in the epithelium. There are a number of systemic changes that occur in the transition into and during menopause, such as a loss of estrogen production. Hormone changes are coincident with other changes in breast tissue, such as decreased connective tissue, increased adipose, and discontinuities in the basement membrane that maintains normal polarity of the epithelium [21-23] (Fig 1). When we examined human mammary epithelia at high definition, aging was found to be associated with the accumulation of mammary epithelial progenitor cells, which are putative cells-of-origin for breast cancers, and with the presence of fewer myoepithelial cells, which can suppress malignant tumor-forming cells [6]. Thus during the aging process the population of cells potentially targeted for transformation is increased and there is a simultaneous decrease in tumor suppressive cells, which suggests a cell- and tissue-level mechanism that leads to increased susceptibility to malignant progression. The shape of the curve that best describes the rate of these changes will require more sample accumulation, and whether there is a biological age of breast tissue that is distinct from a chronological age is an open question.
Figure 1. A cartoon showing archetypical age-related states of breast tissue.
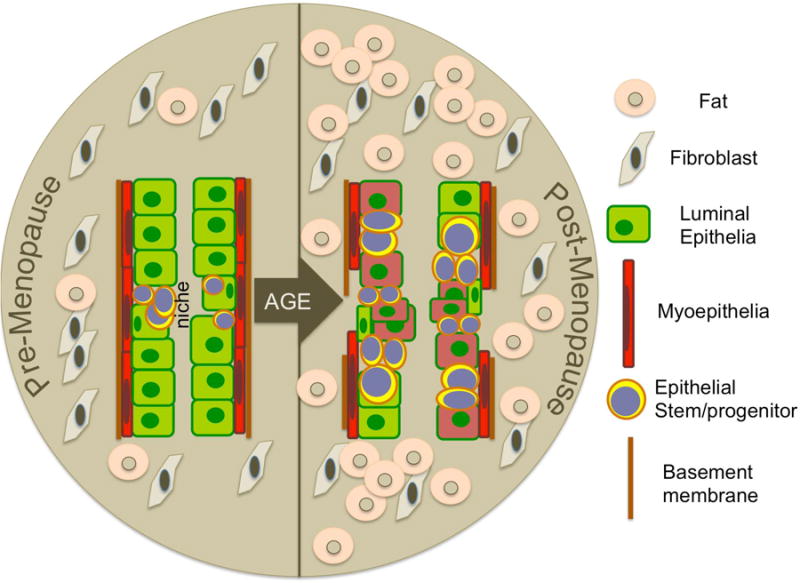
With increased age there is an increase in fat and a decrease in connective tissue in the stromal that surrounds the pseudostratified mammary epithelia. The mammary epithelium is a branching bilayered tissue, but the diagram shown is meant to convey the age-related changes that can arise at cellular resolution. With age, multipotent progenitors accumulate, luminal cells lose lineage fidelity and take on characteristics of the more basal myoepithelial cells. The proportions of tumor suppressive and contractile myoepithelial cells themselves also are diminished with age. Reduction of myoepithelial cells might help explain putative discontinuities in the basement membrane that are thought to arise with age because those cells produce many key basement membrane components.
The concept of dynamic-reciprocity posits that a cell's gene expression is modified by the microenvironment, and the cells in turn modify the microenvironment still further; creating cycles of information feedback [24]. Such information cycles would explain the age-dependence of tissue transcriptomes. The transcriptome changes that accompanies aging essentially altars the internal wiring diagram of cells, thus we should expect functional consequences. For most human tissues establishing links between age-related gene expression and specific functional behaviors has not been possible, but using primary culture systems we have had success with normal human mammary epithelial cells (HMEC). Mammary progenitor cells are tasked with continually renewing the various epithelial lineages throughout a woman's adult life. By exposing them to a range of engineered microenvironments we learned that after menopause they become less sensitive to tissue biomechanical cues that otherwise induce young progenitors to differentiate into tumor suppressive myoepithelial cells [7]. Reduced sensitivity to differentiation cues provides one explanation for the accumulation of progenitors with age and the loss of myoepithelial cells, but more broadly the evidence that the parameters that govern conversations between cells and microenvironments change with age sheds light on how successive homeostatic states might be established in mammary epithelia. A second interesting finding was that the same mechanically-activated transcription factors could be triggered in both younger and older progenitors, although at different mechanical thresholds, and they would initiate distinct differentiation responses suggesting that the genetic information read by the transcription factors was somehow different [7]. Maintaining healthy breast tissue requires the well-choreographed production of the correct proportions of differentiated cells, the organization of the cells into functional higher-order structures, and maintenance of communication between the epithelia and stroma. Such choreography relies on the individual cells accessing the correct information in their genomes based on cues from their microenvironment, and then responding as self-organizing collectives [25]. Based on transcriptome analyses of multiple human tissues, aging changes the internal wiring of cells, which must be traceable to corruption of the basic genetic information, resulting in tissues with suboptimal function. The prevailing dogma would suggest that genomes are corrupted by mutational changes. However age-related epigenetic states may provide a better explanation for the changes that arise in breast.
Testing the convention: accumulation of somatic mutations with age and cancer development
A decline in DNA repair efficiency, accumulation of somatic mutation, cellular mosaicism, and increasing senescent cells and epigenetic alterations are thought to be hallmarks of aging [26]. The foundation of the concept that age-related cancers are caused by accumulated mutations is that aging is not an acute, but rather a time-dependent process [27,28]. The gradual accumulation of somatic mutations is due to failures in removal of mutations over time, which leads to loss of the integrity and stability of the genome, so that malignant transformation occurs. This is why a large number of somatic mutations in tumors are detected [28-30], although it is not yet known whether the genomic instability observed in tumors is a cause or a consequence of tumor development.
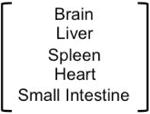
The available data do not show a clear picture that mutations accumulate with age (Table 1). Accumulations were observed in a number of tissues using lacZ transgenic mouse models [31-33], and the HPRT (hypoxanthine phosphoribosyltransferase 1) gene on the X-chromosome in clones of human T-lymphocytes [34]. While these studies indicated that accumulation of mutations with age occurs at those loci, they did not consider the phenomenon on a genome-wide scale. In addition, the accumulation of mutations in both cases was not reported to coincide with cancer incidence. Because human tissues present a number of challenges to studying the normal aging process, a common approach also has been to make a comparison of the number of somatic mutations among tumors from various age groups [35,36]. If the majority of somatic mutations in cancers are accumulated during the normal aging process, then cancers from older people are likely to have more somatic mutations than those from younger people. However, it must be noted that accumulation of mutations with age seems to occur in a tissue-specific manner. Thus chronic lymphocytic leukemia, uterine corpus endometrioid carcinoma, and colorectal cancer all have shown an association between the number of somatic mutations with age, but pancreatic cancer did not, it is thought, because it is not a self-renewing tissue[35]. It is suggested therefore that accumulation of mutations is correlated with the rate of cell proliferation rather than strictly with age. There is no correlation between age and the number of mutations in luminal subtype breast cancers even though the breast epithelium is a self-renewing tissue [35,36]. However, breast epithelia does undergo monthly cycles of proliferation, and can expand as much as ten-fold in preparation for lactation, thus there are ample opportunities for cell proliferation, and there is a distinct window of increased breast cancer risk for several years after childbirth [37]. These data suggest that the majority of the somatic mutations in the breast and ovarian genomes are introduced after the cancer initiation step. In addition, a general shortcoming of experiments to detect accumulation of somatic mutations in tissues were performed with different individuals, rather than longitudinally from the same person over time, and thus the possibility of individual genome variation cannot be ruled out.
Table 1. Summary of studies that addressed, directly or indirectly, the accumulation of somatic mutation with age in different tissues.
Single locus analysis suggested that mutations do accumulate with age. The only genome wide studies were performed in tumors, where accumulation seems to be tissue specific. Leukemia and colorectal cancer show correlation with the accumulation and exponential increase of cancer incidence with age. Uterine corpus endometrial, pancreatic, ovarian and breast cancer do not show correlation of mutations with increased age.
| Healthy Tissues | |||||
|---|---|---|---|---|---|
| Species | Tissue | Gene analyzed | Whole Genome Analysis | Accumulation of somatic mutation with age | reference |
| mouse |
|
LacZ | No | Yes | [31-33] |
| human | Blood | HPRT | No | Yes | [34] |
|
| |||||
| Cancer Tissues | |||||
|
| |||||
| Species | Tissue | Database | Whole Genome Analysis | Accumulation of somatic mutation with age | reference |
|
| |||||
| human |
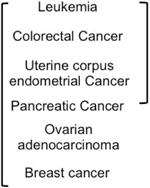
|
TCGA ICGC CLL |
Yes | Yes | [35] |
| Yes | Yes | [35] | |||
| Yes | Yes | [35] | |||
| Yes | No | [35] | |||
| TCGA | Yes | No | [35] | ||
| oiriginal sample set COSMIC | Yes | No | [36] | ||
There are a great number of mutations detected in tumors, but the majority of them, known as passenger mutations, have no deleterious effect on the cells. Multiple mutations are thought to be required for initiation and development of solid tumors [38-40]. However, not all tumors have a driver gene mutation [41]. Three errors – epigenetic and/or genetic errors - are minimally required to gain a malignant phenotype in the absence of passenger errors in otherwise normal HMEC [42]. Indeed, cancers appear to require much more than mutations in order to develop. Bissell and colleagues provided a powerful demonstration of this principle when they showed that the vast majority of cells in chickens that were infected with and expressing the v-src oncogene did not form tumors, but that a wound healing environment was required for the tumors to form from infected cells [43]. Loss of function mutations in the gene encoding BRCA1 (Breast cancer 1, early onset) carries an 80% lifelong risk for breast and ovarian cancers. Although BRCA1 is expressed in every cell, there is only a slightly increased cancer incidence in other tissues, and the effect is mainly seen in breast and ovary, which exhibits the tissue-specific nature of cancer development and the ability of most tissue microenvironments to suppress cancers even in the presence of a mutated tumor suppressor [44]. Thus genetic mutation alone is insufficient to understand tumor development in terms of aging, and at least in the breast, genetic mutations are unlikely to be the major factor that increases susceptibility to cancer with age.
Linking microenvironments to age-related epigenetic states
That normal primary HMEC isolated directly from breast tissues can be grown for multiple passages on plastic dishes in low stress media and still retain transcriptome, biochemical, and functional phenotypes characteristic of specific lineages and chronological age that are largely consistent with in vivo[6], suggests that aging phenotypes are metastable. Metastability denotes an extended equilibrium that can be changed in an energy-dependent manner. Epigenetics provides a reasonable mechanism for biological metastability. Broadly defined, epigenetics is heritable changes in gene expression or cellular memory not encoded by the underlying DNA sequence. The major epigenetic phenomena identified are DNA methylation, chromatin remodeling, histone modification, long non-coding RNAs and microRNAs. Determination of whether epigenetics drive microenvironmental change or, rather, changes in the microenvironment alter heritable cellular memory is a classic chicken and egg situation. It is clear that perturbation of one can lead to alteration of the other. Importantly, deregulation of either the microenvironment or epigenetic states can lead to oncogenesis. There are few reports showing that microenvironments can impact epigenetic states. For example, mesenchymal cells placed in embryonic versus adult tissue microenvironments [45] or in tumor core versus periphery regions [46] show specific patterns of DNA and histone modifications. Moreover, patterns of histone modification and DNA methylation in carcinoma cell lines are different in 2-D versus 3-D cultures versus xenografts [47-49] (Table 2). There is a general paucity in this area of study, perhaps due to the relatively few systems available where cells are analyzed in situ with their relevant stroma, especially in the context of chronological aging.
Table 2. Summary of studies that have addressed effects of the microenvironment on epigenetic states, or of epigenetic perturbations on microenvironments.
| Microenvironmental Perturbation | Epigenetic effect | Reference |
|---|---|---|
| Exposure of melanoma cells to embryonic microenvironment | Methylation of lefty B, inhibitor of Nodal | [44] |
| Laser-dissected tumor captured both center and peripheral cells | Hypermethylation of p16/lnk4a in center cells only | [45] |
| Growth of E-cadherin deficient carcinoma cells in 2D vs. 3D | Loss of E-cadherin hypermethylation in 3D | [46] |
| Growth of cp70 ovarian cancer cell line in 2D vs. 3D | Decrease in H3 acetylation, increase in H3K27 methylation in 3D | [47] |
| Growth of patient glioma stem cells in 2D vs. Xenograft | Epigenetic pattern of GSCs grown in xenograft more closely mimic parental tumor | [48] |
|
| ||
| Epigenetic Perturbation | Microenvironmental Effect | Reference |
|
| ||
| |DNA methylation of Cav-1 | Increased number of stromal cells | [56] |
| |DNA methylation of TIMP3 | Breast cancer/ increased progenitor pool | [59] |
Microenvironment-imposed epigenetic changes that drive susceptibility to age-related breast cancer have been largely unexplored because the studies addressing the role of aging and cancer interrogate the differences between young and old patients who already have cancer, rather than focusing on changes that occur normally during aging and subsequent consequences for breast tissue function. There appears to be a role for age-related epigenetic states in breast cancer etiology, as we have shown that immortal transformation of HMEC from younger women more often results in basal subtypes seen in younger cancer patients, whereas immortalized post-menopausal HMEC exhibited the luminal subtypes that are most often seen in postmenopausal patients [50]. It will be crucial to understand if there is a connection between aging microenvironments and epigenetic states because some age-related epigenetic changes are thought to promote cancers [51].
Perhaps the longest studied epigenetic phenomenon is DNA methylation, which involves the addition of a methyl group to the cytosine pyrimidine ring of DNA. DNA methylation functions to repress transcription by inhibition of transcription factor binding and through recruitment of co-repressor complexes. Whereas global DNA methylation decreases with age resulting in overall genomic hypomethylation, there are CpG-island containing regions that are hypermethylated with age [52]. It is well known that altering cellular microenvironments will alter gene expression patterns, causing some genes to be expressed and others to be turned off [53]. Because persistent transcriptional repression of a given CpG-containing promoter can favor increased methylation, whereas persistently active promoters favor unmethylated states [54], there is good reason to suspect that distinct microenvironments beget distinctive patterns of methylation. For example the Caveolin-1 deficient mouse model has been used as a model of accelerated aging as animals lacking expression of the Cav-1 gene have a significantly diminished lifespan and exhibit signs of premature aging, such as increased beta-amyloid production and neurodegeneration [55]. In the mammary gland, Cav-1 deficient mice exhibit increased stromal cells that can function similar to breast cancer associated fibroblasts [56]. Indeed, loss of expression of Cav-1, although context dependent has been observed in luminal subtype breast cancers and several cases were attributed to DNA methylation-related silencing, suggesting that loss of Cav-1 in the stroma promotes both aging and a tumor permissive microenvironment [57,58].
Extracellular metalloproteases are critical components of the mammary microenvironment that serve to facilitate the remodeling of the tissue at the interface of the stroma and the epithelium. These metalloproteases are regulated in part by tissue inhibitors of metalloproteases (TIMPs) that are expressed in both the breast stroma and epithelium [59]. It has been demonstrated that TIMP3 is often hypermethylated in breast cancer, although the effects of this silencing appear to be context-dependent as it is thought to both promote and prevent cancer genesis. Interestingly when a TIMP3 null mutation is coupled with loss of TIMP1 in mice, they exhibit an increased pool of stem/progenitor cells in their mammary glands, even in older mice where these cells normally are in decline [60]. These results suggest that epigenetic alteration of TIMP3 in the stroma can alter the microenvironment to provide an increased progenitor pool that, perhaps, increases the probability that molecular insults may lead to oncogenesis. It should be noted, however, that much of our understanding of the interplay between microenvironment and epigenetics are derived from mouse models, which do not fully capture the complexity of mammary gland biology observed in humans.
Taken together, these examples illustrate how epigenetic states might arise from age-related changes in the breast microenvironment. The importance of identifying the specific environmental and microenvironmental drivers of epigenetic states cannot be understated because that information will have broad impact well beyond understanding age-related breast cancers.
Conclusions
There is undeniably a genetic component to all cancers, but the accumulation of mutations with age alone is insufficient to explain the age-dependent increase in breast cancer incidence. From our viewpoint, aging causes a gradual loss of function at the levels of the breast microenvironmental structure and tissue organizational features that normally suppress tumor formation. The tissue changes not only corrupt the tumor suppressive activity of normal tissue, but the effects of the changes are reinforced by consequent epigenetic changes that alter gene expression. Thus the spectrum of normal cellular functions is gradually altered in the cells that maintain tissue integrity. While the evidence discussed here suggests that the riddle of whether epigenetics drives microenvironmental changes, or whether changes in the microenvironment alter heritable cellular memory has not been solved, a path has been cleared enabling functional analysis leading to the prediction of key nodes in the network that link the microenvironment with the epigenome. The hypothesis that accumulation of somatic mutations with age drives the age-related increase in breast cancer incidence, if correct, has a somewhat nihilistic conclusion; that cancers will be impossible to avoid. Alternatively if microenvironment-driven epigenetic changes are key to explaining the tissue-level changes that make older women more susceptible to breast cancer there is hope that primary prevention is possible. Whereas genomes are intractable to change, there is translational promise for preventing (or altering) epigenomes with chemoprevention, nutrition, stress and exercise [61,62].
Finally, a new tool set for dissecting the cellular and molecular mechanisms of age-related breast cancers needs to be developed, for this is a situation in which the most common models for aging research are unlikely to be useful. Yeast, flies, and worms do not have mammary glands. And three relevant limitations of rodent models are: (i) major tumor-suppressive barriers are absent in mice, (ii) the tumor incidence curves differ by inbred strain and do not even mimic human populations, and (iii) there is a striking paucity of luminal-type mammary tumor mouse models, which is the subtype principally concerned with aging. There is some hope that genetically diverse murine cohorts will better model population level disease patterns [63], and there may be promise for the STAT1 knockout mouse model, which has a characteristic slow growing, estrogen receptor-expressing, luminal subtype [64]. Although humans are notoriously challenging experimental systems, it does not represent an intractable problem. We need to consider how to develop human model systems that can better address this issue, such as establishment of cell culture systems, using normal healthy tissues serially collected from the same individual over a span of time. To understand age-related cancers, which represent the majority of human cancers, we need to determine conclusively the relative roles played by accumulated mutations, and altered microenvironments and epigenetic states in different tissues. Achieving this goal will require the selfless establishment of biobanking programs that span multiple investigator-generations, whose aim will be to collect tissue samples longitudinally from healthy volunteers. We believe that through literature review and a critical appraisal of the respective roles of somatic mutation, environmental and epigenetic influences, we lay out new conceptual issues that future research on breast cancer, beyond the age of mutation, needs to address.
Acknowledgments
We are grateful for the generous support received from: the National Institutes of Aging (NIA R01AG040081), the California Breast Cancer Research Program and Anita Tarr Turk Fund for Breast Cancer Research (20IB-0109), and the Era of Hope Scholar Award from the Congressionally Directed Medical Research Program's Breast Cancer Research Program.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Smigal C, Jemal A, Ward E, Cokkinides V, Smith R, Howe HL, Thun M. Trends in breast cancer by race and ethnicity: update 2006. CA Cancer J Clin. 2006;56:168–183. doi: 10.3322/canjclin.56.3.168. [DOI] [PubMed] [Google Scholar]
- 3.Campisi J, Yaswen P. Aging and cancer cell biology, 2009. Aging Cell. 2009;8:221–225. doi: 10.1111/j.1474-9726.2009.00475.x. [DOI] [PubMed] [Google Scholar]
- 4.DePinho RA. The age of cancer. Nature. 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 5.Anderson WF, Rosenberg PS, Prat A, Perou CM, Sherman ME. How many etiological subtypes of breast cancer: two, three, four, or more? J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garbe JC, Pepin F, Pelissier FA, Sputova K, Fridriksdottir AJ, Guo DE, Villadsen R, Park M, Petersen OW, Borowsky AD, et al. Accumulation of multipotent progenitors with a basal differentiation bias during aging of human mammary epithelia. Cancer Res. 2012;72:3687–3701. doi: 10.1158/0008-5472.CAN-12-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pelissier FA, Garbe JC, Ananthanarayanan B, Miyano M, Lin C, Jokela T, Kumar S, Stampfer MR, Lorens JB, LaBarge MA. Age-related dysfunction in mechano-transduction impairs differentiation of human mammary epithelial progenitors. Cell Reports. 2014 doi: 10.1016/j.celrep.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sputova K, Garbe JC, Pelissier FA, Chang E, Stampfer MR, Labarge MA. Aging phenotypes in cultured normal human mammary epithelial cells are correlated with decreased telomerase activity independent of telomere length. Genome Integr. 2013;4:4. doi: 10.1186/2041-9414-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenkins EO, Deal AM, Anders CK, Prat A, Perou CM, Carey LA, Muss HB. Age-specific changes in intrinsic breast cancer subtypes: a focus on older women. Oncologist. 2014;19:1076–1083. doi: 10.1634/theoncologist.2014-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yau C, Fedele V, Roydasgupta R, Fridlyand J, Hubbard A, Gray JW, Chew K, Dairkee SH, Moore DH, Schittulli F, et al. Aging impacts transcriptomes but not genomes of hormone-dependent breast cancers. Breast Cancer Res. 2007;9:R59. doi: 10.1186/bcr1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, et al. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004;2:e427. doi: 10.1371/journal.pbio.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006;2:e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haakensen VD, Lingjaerde OC, Luders T, Riis M, Prat A, Troester MA, Holmen MM, Frantzen JO, Romundstad L, Navjord D, et al. Gene expression profiles of breast biopsies from healthy women identify a group with claudin-low features. BMC Med Genomics. 2011;4:77. doi: 10.1186/1755-8794-4-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- 16.DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nat Rev Mol Cell Biol. 2011;12:308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science. 1996;274:2057–2059. doi: 10.1126/science.274.5295.2057. [DOI] [PubMed] [Google Scholar]
- 19.Esserman LJ, Moore DH, Tsing PJ, Chu PW, Yau C, Ozanne E, Chung RE, Tandon VJ, Park JW, Baehner FL, et al. Biologic markers determine both the risk and the timing of recurrence in breast cancer. Breast Cancer Res Treat. 2011;129:607–616. doi: 10.1007/s10549-011-1564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pike MC, Krailo MD, Henderson BE, Casagrande JT, Hoel DG. ‘Hormonal’ risk factors, ‘breast tissue age' and the age-incidence of breast cancer. Nature. 1983;303:767–770. doi: 10.1038/303767a0. [DOI] [PubMed] [Google Scholar]
- 21.Howeedy AA, Virtanen I, Laitinen L, Gould NS, Koukoulis GK, Gould VE. Differential distribution of tenascin in the normal, hyperplastic, and neoplastic breast. Lab Invest. 1990;63:798–806. [PubMed] [Google Scholar]
- 22.Milanese TR, Hartmann LC, Sellers TA, Frost MH, Vierkant RA, Maloney SD, Pankratz VS, Degnim AC, Vachon CM, Reynolds CA, et al. Age-related lobular involution and risk of breast cancer. J Natl Cancer Inst. 2006;98:1600–1607. doi: 10.1093/jnci/djj439. [DOI] [PubMed] [Google Scholar]
- 23.Well D, Yang H, Houseni M, Iruvuri S, Alzeair S, Sansovini M, Wintering N, Alavi A, Torigian DA. Age-related structural and metabolic changes in the pelvic reproductive end organs. Semin Nucl Med. 2007;37:173–184. doi: 10.1053/j.semnuclmed.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Bissell MJ. The differentiated state of normal and malignant cells or how to define a “normal’ cell in culture. Int Rev Cytol. 1981;70:27–100. doi: 10.1016/s0074-7696(08)61130-4. [DOI] [PubMed] [Google Scholar]
- 25.Chanson L, Brownfield D, Garbe JC, Kuhn I, Stampfer MR, Bissell MJ, Labarge MA. Self-organization is a dynamic and lineage-intrinsic property of mammary epithelial cells. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1019556108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vijg J. Somatic mutations, genome mosaicism, cancer and aging. Curr Opin Genet Dev. 2014;26:141–149. doi: 10.1016/j.gde.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Cancer Genome Atlas network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buettner VLHK, Halangoda A, Sommer SS. Tandem-base mutations occur in mouse liver and adipose tissue preferentially as G:C to T:A transversions and accumulate with age. Environ Mol Mutagen. 1999;33:320–324. doi: 10.1002/(sici)1098-2280(1999)33:4<320::aid-em9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 32.Dollé MESW, Gossen JA, Lohman PH, Vijg J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci U S A. 2000;97:8403–8408. doi: 10.1073/pnas.97.15.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dollé MESW, Dunson DB, Vijg J. Mutational fingerprints of aging. Nucleic Acids Res. 2002;30:545–549. doi: 10.1093/nar/30.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Branda RFSL, O'Neill JP, Falta MT, Nicklas JA, Hirsch B, Vacek PM, Albertini RJ. Measurement of HPRT mutant frequencies in T-lymphocytes from healthy human populations. Mutat Res. 1993;285:267–279. doi: 10.1016/0027-5107(93)90115-v. [DOI] [PubMed] [Google Scholar]
- 35.Tomasetti CVB, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A. 2013;110:1999–2004. doi: 10.1073/pnas.1221068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinson HA, Lyons TR, Giles ED, Borges VF, Schedin P. Developmental windows of breast cancer risk provide opportunities for targeted chemoprevention. Exp Cell Res. 2013;319:1671–1678. doi: 10.1016/j.yexcr.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moolgavkar SHDA, Venzon DJ. A stochastic two-stage model for cancer risk assessment. I. The hazard function and the probability of tumor. Risk Anal. 1988;8:383–392. doi: 10.1111/j.1539-6924.1988.tb00502.x. [DOI] [PubMed] [Google Scholar]
- 39.Armitage PaDR. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci U S A. 2015;112:118–123. doi: 10.1073/pnas.1421839112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogelstein BPN, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garbe JC, Vrba L, Sputova K, Fuchs L, Novak P, Brothman AR, Jackson M, Chin K, LaBarge MA, Watts G, et al. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle. 2014;13:3423–3435. doi: 10.4161/15384101.2014.954456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stoker AWHC, Bissell MJ. The embryonic environment strongly attenuates v-src oncogenesis in mesenchymal and epithelial tissues, but not in endothelia. J Cell Biol. 1990 doi: 10.1083/jcb.111.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, Ghadirian P, Foulkes WD, Armel S, Eisen A, et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: results from a follow-up study. Br J Cancer. 2014;110:530–534. doi: 10.1038/bjc.2013.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costa FF, Seftor EA, Bischof JM, Kirschmann DA, Strizzi L, Arndt K, de Fatima Bonaldo M, Soares MB, Hendrix MJ. Epigenetically reprogramming metastatic tumor cells with an embryonic microenvironment. Epigenomics. 2009;1:387–398. doi: 10.2217/epi.09.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jie G, Zhixiang S, Lei S, Hesheng L, Xiaojun T. Relationship between expression and methylation status of p16INK4a and the proliferative activity of different areas' tumour cells in human colorectal cancer. Int J Clin Pract. 2007;61:1523–1529. doi: 10.1111/j.1742-1241.2006.01033.x. [DOI] [PubMed] [Google Scholar]
- 47.DesRochers TM, Shamis Y, Alt-Holland A, Kudo Y, Takata T, Wang G, Jackson-Grusby L, Garlick JA. The 3D tissue microenvironment modulates DNA methylation and E-cadherin expression in squamous cell carcinoma. Epigenetics. 2012;7:34–46. doi: 10.4161/epi.7.1.18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bapat SA, Jin V, Berry N, Balch C, Sharma N, Kurrey N, Zhang S, Fang F, Lan X, Li M, et al. Multivalent epigenetic marks confer microenvironment-responsive epigenetic plasticity to ovarian cancer cells. Epigenetics. 2010;5:716–729. doi: 10.4161/epi.5.8.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baysan M, Woolard K, Bozdag S, Riddick G, Kotliarova S, Cam MC, Belova GI, Ahn S, Zhang W, Song H, et al. Micro-environment causes reversible changes in DNA methylation and mRNA expression profiles in patient-derived glioma stem cells. PLoS One. 2014;9:e94045. doi: 10.1371/journal.pone.0094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JK, Garbe JC, Vrba L, Miyano M, Futscher BW, Stampfer MR, LaBarge MA. Age and the means of bypassing stasis influence the intrinsic subtype of immortalized human mammary epithelial cells. Front Cell Dev Biol. 2015;3:13. doi: 10.3389/fcell.2015.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Issa JP, Garber JE. Time to think outside the (genetic) box. Cancer Prev Res (Phila) 2011;4:6–8. doi: 10.1158/1940-6207.CAPR-10-0348. [DOI] [PubMed] [Google Scholar]
- 52.Fraga MF, Agrelo R, Esteller M. Cross-talk between aging and cancer: the epigenetic language. Ann N Y Acad Sci. 2007;1100:60–74. doi: 10.1196/annals.1395.005. [DOI] [PubMed] [Google Scholar]
- 53.LaBarge MA, Nelson CM, Villadsen R, Fridriksdottir A, Ruth JR, Stampfer M, Petersen OW, Bissell MJ. Human mammary progenitor cell fate decsions are products of interactions with combinatorial microenvironments. Integrative Biology. 2009;1:70–79. doi: 10.1039/b816472j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oyer JA, Chu A, Brar S, Turker MS. Aberrant epigenetic silencing is triggered by a transient reduction in gene expression. PLoS One. 2009;4:e4832. doi: 10.1371/journal.pone.0004832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Head BP, Peart JN, Panneerselvam M, Yokoyama T, Pearn ML, Niesman IR, Bonds JA, Schilling JM, Miyanohara A, Headrick J, et al. Loss of caveolin-1 accelerates neurodegeneration and aging. PLoS One. 2010;5:e15697. doi: 10.1371/journal.pone.0015697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sotgia F, Del Galdo F, Casimiro MC, Bonuccelli G, Mercier I, Whitaker-Menezes D, Daumer KM, Zhou J, Wang C, Katiyar S, et al. Caveolin-1-/- null mammary stromal fibroblasts share characteristics with human breast cancer-associated fibroblasts. Am J Pathol. 2009;174:746–761. doi: 10.2353/ajpath.2009.080658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rao X, Evans J, Chae H, Pilrose J, Kim S, Yan P, Huang RL, Lai HC, Lin H, Liu Y, et al. CpG island shore methylation regulates caveolin-1 expression in breast cancer. Oncogene. 2013;32:4519–4528. doi: 10.1038/onc.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, Fanelli MA, Cuello-Carrion FD, Gago FE, Anderson RL. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–2043. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khokha R, Werb Z. Mammary gland reprogramming: metalloproteinases couple form with function. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jackson HW, Waterhouse P, Sinha A, Kislinger T, Berman HK, Khokha R. Expansion of stem cells counteracts age-related mammary regression in compound Timp1/Timp3 null mice. Nat Cell Biol. 2015;17:217–227. doi: 10.1038/ncb3118. [DOI] [PubMed] [Google Scholar]
- 61.McGowan PO, Meaney MJ, Szyf M. Diet and the epigenetic (re)programming of phenotypic differences in behavior. Brain Res. 2008;1237:12–24. doi: 10.1016/j.brainres.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGowan PO, Szyf M. The epigenetics of social adversity in early life: implications for mental health outcomes. Neurobiol Dis. 2010;39:66–72. doi: 10.1016/j.nbd.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 63.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 64.Chen JQ, Mori H, Cardiff RD, Trott JF, Hovey RC, Hubbard NE, Engelberg JA, Tepper CG, Willis BJ, Khan IH, et al. Abnormal Mammary Development in 129:STAT1-Null Mice is Stroma-Dependent. PLoS One. 2015;10:e0129895. doi: 10.1371/journal.pone.0129895. [DOI] [PMC free article] [PubMed] [Google Scholar]