

Abstract
Talin plays important role in regulating integrin-mediated signaling. Talin function is autoinhibited by intramolecular interactions between the integrin-binding F3 domain and the autoinhibitory domain (R9). We determined the crystal structure of a triple domain fragment R7R8R9, which contains R9 and the RIAM (Rap1-Interacting Adaptor Molecule) binding domain (R8). The structure reveals a crystallographic contact between R9 and a symmetrically related R8 domain, representing a homodimeric interaction in talin. Strikingly, we demonstrated that the α5 helix of R9 also interacts with the F3 domain, despite no interdomain contact involving the α5 helix in the crystal structure of an F2F3:R9 autoinhibitory complex reported previously. Mutations on the α5 helix significantly diminish the F3:R9 association and lead to elevated talin activity. Our results offer the biochemical and functional evidence of the existence of a new talin autoinhibitory configuration, thus providing a more comprehensive understanding of talin autoinhibition, regulation, and quaternary structure assembly.
eTOC
Zhang et al. offer the biochemical and functional evidence of the existence of a new talin autoinhibitory configuration, thus providing a more comprehensive understanding of talin autoinhibition, regulation, and quaternary structure assembly.
INTRODUCTION
Integrins mediate the interaction between cells and their surrounding extracellular matrix (ECM), thus serving essential functions in cell adhesion, survival, and proliferation (Alavi et al., 2003; Hood et al., 2003; Menter and Dubois, 2012; Parsons, 2003). Integrin dysfunction has been linked to thrombotic disorders, impaired immune responses, cardiovascular diseases, and cancer (Desgrosellier and Cheresh, 2010; Felding-Habermann et al., 2001; Garmy-Susini et al., 2010; Huveneers et al., 2007; Nip et al., 1992; Pinon and Wehrle-Haller, 2011). Unlike many cellular receptors that transduce signals only from the extracellular side into the cytoplasm, integrins are capable of transducing signals bi-directionally across the plasma membrane (PM). The interaction of integrins with ECM activates an outside-in signaling pathway, in which a series of cytoplasmic protein kinases including FAK, Src, PKC, and MEK promote downstream cell responses (Hood and Cheresh, 2002). Conversely, activation of the cytoplasmic GTPase Rap1 triggers an inside-out signaling pathway that allows the cytoskeletal protein talin to interact with the cytoplasmic region of integrins, leading to conformational changes in the integrin ectodomains, thereby activating the integrins (Ginsberg et al., 2005). Talin also modulates the clustering of integrins that enhances integrin avidity and subsequent outside-in signaling (Ellis et al., 2014). Thus, talin plays important regulatory roles in both types of integrin-mediated signaling pathways.
Talin is a large homodimeric protein. Each monomer contains a “head” region (talin-H) with a FERM domain (F1, F2, and F3) and an F0 domain, a “rod” region (talin-R) with 13 helical bundle domains (R1-R13), and a short C-terminal helical region (DH) for homodimerization. When talin translocates to the PM, the F3 domain of talin-H interacts with the cytoplasmic region of the integrin β-subunit. The interaction causes a packing mismatch and results in a separation of the tails of the α- and β- subunits, thereby switching the integrin ectodomains from an inactive state to an active state (Banno and Ginsberg, 2008; Margadant et al., 2013; Watanabe et al., 2008; Wegener et al., 2007). The PM-translocation of talin is mediated by a Rap1 effector protein RIAM (Rap1-interacting adaptor molecule) through its N-terminal talin-binding site (TBS1) (Chang et al., 2014; Lee et al., 2009). The helical bundle domains in talin-R serve multiple adaptor functions by interacting with various cytoplasmic components. Specifically, talin-R possesses an autoinhibitory domain, at least 11 binding sites for the focal adhesion protein vinculin and also interacts with RIAM and F-actin (Gingras et al., 2010). All of the helical domains in talin-R adopt a beads-on-a-string morphology except for the R8 domain, which is inserted between the α3 and α4 helices of the R7 domain (Gingras et al., 2010). Together with the R9 domain, the R7R8R9 triple-domain fragment plays central roles in regulating talin activity (Fig. 1A). The R7 domain interacts with vinculin through its α5 helix; the R8 domain binds to RIAM TBS1 during the PM-translocation (Chang et al., 2014); and the R9 domain is the autoinhibitory domain that binds to the F3 domain and blocks the integrin-binding site (Goksoy et al., 2008; Song et al., 2012). Moreover, structural studies of full-length talin have revealed a compact autoinhibitory dimer that suppresses its signaling functions, suggesting that talin must undergo conformational changes before it can promote integrin activation (Goult et al., 2013). Because the R7R8R9 fragment is located in the central region of the flexible talin-R and serves essential functions in regulating talin activity and maintaining its quaternary structure, the structure of this triple-domain subunit is important for understanding talin autoinhibition and domain organization.
Figure 1. Structure of talin R7R8R9.
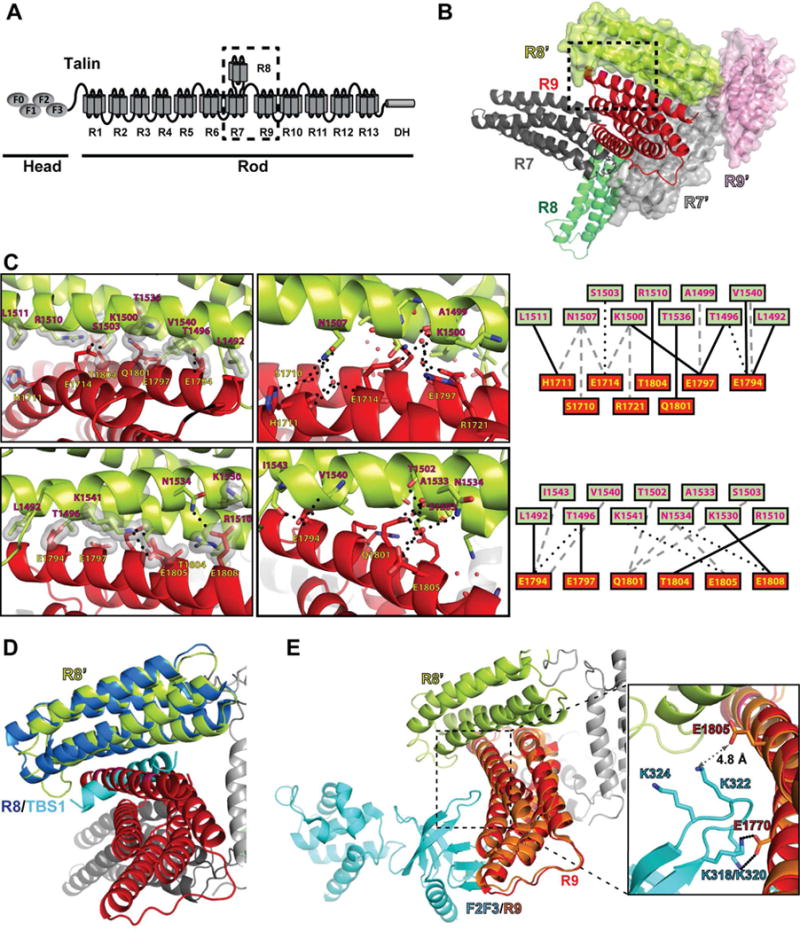
(A) Schematic diagram of talin. The head region possesses the F0, F1, F2, and F3 domains (ovals). The rod region contains 13 helical bundle domains (R1–R13) and a dimerization helix (DH). The R7R8R9 fragment is indicated by the dashed box. (B) Cartoon diagram of wild-type talin triple domains R7R8R9. The R7, R8, and R9 domains are colored in dark gray, green, and red, respectively. A symmetrically related R7R8R9 molecule is shown in surface representation (R7’ in light gray, R8’ in lime, and R9’ in pink). The interface formed by the R9 and R8’ domains is indicated by the dashed box. (C) Close-up views of the dashed box from (B) in both front view (upper panels) and back view (lower panels). Hydrogen bonds are indicated by dotted lines and van der Waals contacts are represented by surfaces. Interacting residues are labeled (R8’ in red-black text and R9 in yellow-black text). Schematic maps of the interactions are shown on the right with van der Waals contacts in solid line, direct hydrogen bonds in dotted lines, and water-mediated hydrogen bonds in gray dashed lines. (D) The crystal structure of R8:TBS1 (PDB ID: 4W8P. R8 in dark blue, TBS1 in cyan) is superimposed on the symmetrically related R8’ domain (lime) of the R7R8R9 structure. The TBS1 fragment sterically clashes with the R9 domain (red). (E) The crystal structure of F2F3:R9 (PDB ID: 4F7G. R9 in salmon, F2F3 in cyan) is superimposed on the R7R8R9 structure. The salt bridge interactions of K318/K320 in the lysine finger of the F3 domain and E1770 in the R9 domain are shown in the inset box. The amino group of Lys322 and the carboxyl group of Glu1805 is 4.8 Å apart, and thus does not form a salt bridge.
Here we report a 2.0-Å crystal structure of the R7R8R9 domains. The structure reveals a crystallographic intermolecular contact formed by the R9 domain and a symmetrically related R8 domain (R8’). This interface is compatible with the reported F3:R9 autoinhibitory interface but overlaps with the interface of the RIAM-TBS1:R8 complex that mediates the PM-translocation of talin by RIAM. Although R7R8R9 does not exhibit a substantial tendency to dimerize in solution, mutagenesis and functional studies of full-length talin confirm the physiological relevance of the R9:R8′ interface. We also identified several mutations in the R9 domain that enhance the dimerization of R7R8R9. Strikingly, although these residues are away from the F3:R9 interface revealed by a crystal structure of F2F3:R9 complex (Song et al., 2012), their mutations significantly diminish the autoinhibitory interaction between the R9 and F2F3 domains. Functional analysis also confirmed that full-length talin bearing these mutations exhibits elevated activity in integrin signaling. Together, these results demonstrate the existence of a new autoinhibitory configuration of talin and identify an additional F3-binding site in the R9 domain that mediates an alternative intramolecular binding mode.
RESULTS
Crystal structure of the R7R8R9 triple-domain module
The R7R8R9 triple-domain module mediates talin autoinhibition, PM translocation, and the talin-vinculin association. To elucidate the domain organization of this module, we determined the crystal structure of the talin R7R8R9 domains (residues 1357–1822) at 2.0 Å resolution (Table 1). The crystal belongs to the primitive monoclinic space group P21 with unit-cell parameters of a = 49.0 Å, b = 77.6 Å, c = 61.6 Å, and β = 109.8°. Each asymmetric unit contains one R7R8R9 molecule. The structure contains a five-helical-bundle R7 domain, a four-helical-bundle R8 domain, and a five-helical-bundle R9 domain (Fig. 1B). Linkers connecting the R7 domain and the inserted R8 domain (residues 1449–1461 and 1577–1590, respectively) are similar to that of the structure of R7R8 in complex with the RIAM TBS1 peptide (PDB ID: 4W8P), suggesting the structural integrity of each individual domain. The R7 domain is joined to the R9 domain by a short linker (residues 1655–1657) that allows minimal flexibility between the two domains.
Table 1.
X-ray data collection and refinement statistics
| R7R8R9-WT | R7R8R9-3Y | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Cell Dimensions | ||
| a, b, c (Å) | 49.0, 77.6, 61.6 | 60.6, 67.2, 66.0 |
| a, β, ɤ (°) | 90.0, 109.8, 90.0 | 90.0, 101.5, 90.0 |
| Resolution (Å) | 46.4–2.0 (2.03–2.00) | 48.9–2.2 (2.24–2.20) |
| Completeness (%) | 99.3 (98.3) | 99.8 (100.0) |
| Rsym (%) | 13.4 (52.8) | 10.1 (49.9) |
| I / σ (I) | 13.2 (2.4) | 17.9 (2.3) |
| Unique reflections | 29840 (1457) | 26339 (1325) |
| Redundancy | 3.0 (2.6) | 3.8 (3.8) |
| Refinement | ||
| Resolution (Å) | 46.4–2.0 | 48.9–2.2 |
| Rwork (%) | 20.0 (23.1) | 20.8 (26.1) |
| Rfree (%) | 22.9 (23.3) | 24.9 (29.9) |
| RMSD bonds (Å) | 0.006 | 0.007 |
| RMSD angle (°) | 0.932 | 1.026 |
| Protein atoms | 3524 | 3372 |
| Solvent atoms | 224 | 135 |
| Total residues | 465 | 464 |
| Average B-factors (Å2) | ||
| Protein | ||
| Main chain atoms | 14.8 | 29.6 |
| Side chain atoms | 16.0 | 30.8 |
| Solvent | 24.9 | 39.9 |
| Ramachandran | ||
| Favored regions (%) | 99.4 | 99.4 |
| Allowed regions (%) | 100.0 | 100.0 |
Rsym = Σ|Iobs − Iavg|/ΣIavg; Rwork = Σ||Fobs − Fcalc||/ΣFobs; Rfree was calculated using 5% of the data and the same sums. Values in parentheses are for highest-resolution shell.
Crystal contact analysis of the R7R8R9 structure reveals a large interface between the R9 domain and a symmetrically related R8 domain (R8′). The two domains share a surface area of 800 Å2 that involves the α5 helix of R9 and the α2 and α3 helices of R8’. This interface is primarily mediated by R9 domain residues Glu1794, Glu1797, Gln1801, Thr1804, Glu1805, and Glu1808 on the α5 helix with R8’ domain residues Thr1496, Lys1500, Ser1503, Arg1510, Lys1530, Asn1534, Thr1536, and Lys1541 (Fig. 1C). In addition, His1711 and Glu1714 in the α2 helix of R9 also contribute to this interaction. Interestingly, the same surface area in the R8 domain, including most of its interface residues, also mediates the interaction with the RIAM TBS1 region. Thus, the contacting R9 domain masks the binding site for the TBS1 fragment on the symmetrical R8’ domain (Chang et al., 2014) (Fig. 1D). In contrast, this interface is completely compatible with the reported autoinhibitory configuration of the F2F3:R9 complex (Song et al., 2012) (Fig. 1E). Although the interaction of the F3 domain lysine finger (K318/K320/K322/K324) with the R9 domain residue E1770 in the α4 helix positions the lysine finger close to the α5 helix of R9, none of the residues in the α5 helix makes direct contact with the F3 domain as seen in the reported F2F3:R9 complex structure (Fig. 1E).
Mutations in R9 enhance the R9:R8’ intermolecular contact in solution and in the context of full-length talin
To assess the physiological relevance of the intermolecular contacts, we mutated interface residues in the R9 α5 helix in the combinations of E1794Y/E1797Y (2Y) and E1794Y/E1797Y/Q1801Y (3Y) in an attempt to disrupt the R9:R8’ interface (Fig. 2A). Purified R7R8R9, R7R8R9-2Y, and R7R8R9-3Y proteins were subject to size exclusion chromatography with inline multi-angle light scattering (SEC-MALS) analysis. Surprisingly, both the R7R8R9-2Y and R7R8R9-3Y exhibited significantly enhanced dimerization (Fig. 2B). Both R7R8R9-2Y and R7R8R9-3Y proteins were eluted in two distinct peaks that correspond to their monomeric and dimeric forms, respectively, whereas the dimer peak for the wild-type R7R8R9 is barely detectable. Cross-linking experiments using these mutant proteins also exhibited stronger homodimer bands compared with the wild-type protein, confirming that these mutations enhance the dimerization of the R7R8R9 protein in solution (Fig. S1A).
Figure 2. Mutations enhance the R7R8R9 dimerization.
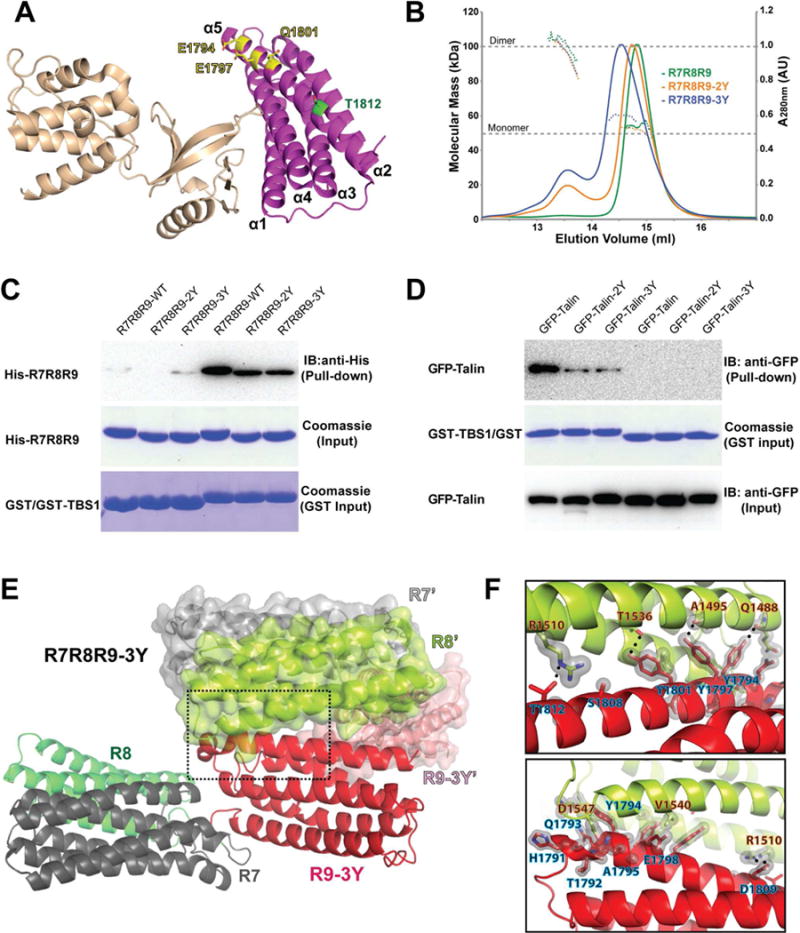
(A) Cartoon diagram of the F2F3:R9 complex structure. The side chain configurations of Glu1794, Glu1797, Gln1801 (yellow) and Thr1812 (green) are shown in stick. (B) UV traces from the HPLC elution for R7R8R9 (green), R7R8R9-2Y (orange), and R7R8R9-3Y (blue) are shown as colored solid lines with the scale shown on the right side. The MALS-derived molecular mass distribution are plotted as dotted lines in the colors corresponding to the UV traces, with the scale shown on the left side. The dashed lines at 50 kDa and 100 kDa indicate the molecular mass of the monomer and dimer forms of R7R8R9, respectively. (C) In vitro pull-down of His-tagged R7R8R9, -2Y and -3Y mutants by GST-TBS1. His-tagged proteins in the pull-down samples were detected by Western blot using anti-His antibody. Input samples of His-R7R8R9 and GST/GST-TBS1 were determined by Coomassie staining. (D) Pull-down of full-length GFP-talin and mutants expressed in HEK293T cells by purified GST-TBS1. Pull-down and input of GFP-talin were detected by Western blot using anti-GFP antibody. The input of GST-TBS1 was determined by Coomassie staining. (E) Cartoon representation of the R7R8R9-3Y crystal structure. Each domain is colored and labeled. A symmetrically related R7R8R9-3Y molecule is shown in surface/cartoon representation. The interface formed by the R9-3Y and R8’ domains is indicated by the dashed box. (F) Close-up views of the box in (E). Hydrogen bonds are denoted by a dotted line, and van der Waals contacts are represented by a light gray surface. Interacting residues are labeled (R8’ in orange-black text and R9 in blue-black text).
See also: Figure S1.
To examine whether the dimeric assembly masks the TBS1-binding site in R8 as seen in the R7R8R9 crystal structure, we performed a pull-down assay using a GST-tagged RIAM TBS1 fragment with purified R7R8R9-2Y and R7R8R9-3Y and with full-length talin bearing these mutations. The mutations diminish the association of R7R8R9 with GST-TBS1 (Fig. 2C), and more significant effect is observed on the association of full-length talin with the TBS1 fragment (Fig. 2D). Evidently, the changes in TBS1 association observed in R7R8R9 mutant proteins are less significant compared with those in mutants of the full-length talin because of the lack of the C-terminal DH region, which mediates the intrinsic dimerization in the full-length talin (Gingras et al., 2008; Goult et al., 2013), thus facilitating the intermolecular interaction of the R8 and R9 domains. This result suggests that the -2Y and -3Y mutations enhance intermolecular contacts between the R8 and R9 domains and mask the TBS1-binding site.
Crystal structure of R7R8R9-3Y
To verify the integrity of the mutant protein and the R9:R8’ intermolecular interface, we determined the crystal structure of R7R8R9-3Y at 2.2 Å resolution (Table 1). The structure reveals a virtually identical R9 domain between the wild-type and the mutant with a root mean square deviation (RMSD) of 0.34 Å. The interface between the R9 domain and the R8’ domain in the R7R8R9-3Y structure is similar to that of the wild-type R7R8R9 structure, in which the TBS1-binding site in the R8’ domain is masked by the R9 domain (Fig. 2E). The three tyrosine mutations (E1794Y/E1797Y/Q1801Y) in the R9 domain are situated in the groove between the α2 and α3 helices of R8’ and form extensive contacts with the side chains of the R8’ helices (Fig. 2F). The contact area of the R9-3Y and R8’ domains increased to 890 Å2 as a result, thus enhancing the intermolecular interaction (Fig. 2F). Notably, the symmetrical R8 domain in the R7R8R9-3Y structure undergoes a ~30° rotation in relation to that of the wild-type protein, whereas the F3-binding interface in the R9 domain remains intact despite the rearrangement of the interacting R8’ domain (Fig. S1B).
Mutations in the R9 domain diminish its association with the F2F3 domains
We then assessed the effect of the dimer-enhanced mutations on the autoinhibitory configuration of talin. We first examined the F2F3-binding property of the mutant R7R8R9 proteins. Because the mutated residues (E1794, E1797, and Q1801) do not make direct contact with the F3 domain in the crystal structure of F2F3:R9 complex and the dimeric configuration of R7R8R9-3Y is compatible with the F2F3:R9 interaction (Fig. 2A), the mutations were not expected to affect on the F3:R9 association. Surprisingly, the mutant R7R8R9 proteins exhibited reduced association with GST-tagged F2F3 domains (Fig. 3A). There are two possible explanations: 1) Glu1794, Glu1797, and Gln1801 directly interact with the F2F3 domains or 2) the dimerization of the mutant protein masks other unidentified residues that interact with the F2F3 domains. To test these possibilities, we examined the binding of the F2F3 domains using GST-tagged R9 and its 3Y and E1770A mutant counterparts to eliminate the dimerization effect caused by the R9:R8’ interaction. The well-characterized E1770A mutation completely abolished the F2F3:R9 interaction, whereas the R9-3Y also exhibited significantly reduced F2F3-binding capability (Fig. 3B). The result suggests that Glu1794, Glu1797, and Gln1801 in the α5 helix of the R9 domain indeed make direct contact with the F2F3 domains. Thus, this binding mode is distinct from the previously reported F2F3:R9 complex structure, in which the R9 domain only contacts the F3 domain through the α1 and α4 helices.
Figure 3. Additional F3:R9 interaction site.
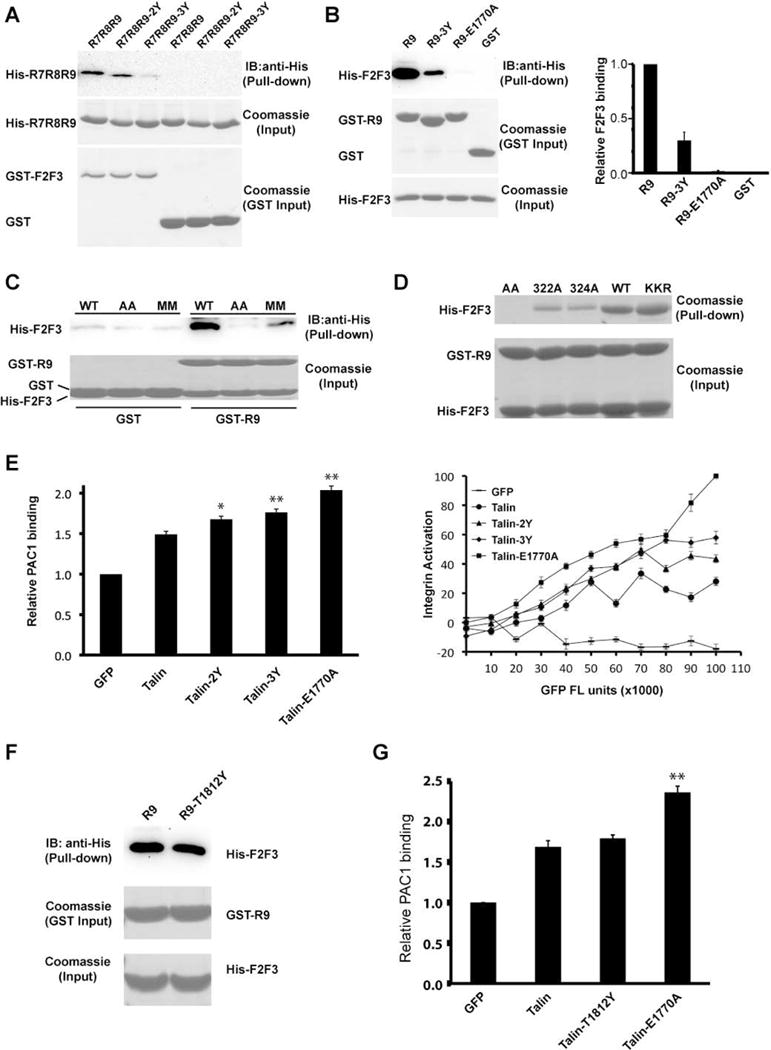
(A) In vitro pull-down of His-tagged R7R8R9 and mutants by GST-F2F3. Bound R7R8R9 was detected by Western blot using anti-His antibody. All input samples were examined by Coomassie staining for equal loading. (B) Left: in vitro pull-down of His-tagged F2F3 by GST-R9 and mutants. Bound F2F3 was detected by Western blot using an anti-His antibody. GST/GST-R9 pull-down and His-F2F3 input were shown by Coomassie staining. Right: the intensity of each band derived by Western blot was quantified by ImageJ software and shown in histogram. The F2F3 binding to wild-type R9 was defined as 1 and data are shown as the means ± SD from three experiments. (C) In-vitro pull-down of His-F2F3 and two double mutants (AA: K322A/K324A; MM: K322M/K324M) by GST-R9. Bound F2F3 was detected by Western blot using anti-His antibody. His-F2F3 input and GST/GST-R9 pull-down were shown by Coomassie staining. (D) In-vitro pull-down of His-F2F3 and its assorted mutants by GST-R9. His-F2F3 input and GST/GST-R9 pull-down were shown by Coomassie staining. (E) Integrin activity analyses of various talin mutants. CHO-A5 cells were transfected with GFP empty vector or GFP-talin and mutants. αIIbβ3 integrin activity was determined by PAC-1 antibody and FACS analysis. Left: histogram of relative PAC1 binding. PAC-1 binding of GFP transfected cells was defined as 1. Right: dose-dependent analysis of integrin activation. The relative PAC1 binding was plotted against GFP fluorescence intensity (FL unit) that indicates the expression level of each GFP-tagged talin construct. The PAC1 binding of talin-E1770A with maximal FL unit was defined as 100% and the fluorescence intensity of E1770A at minimal expression level was defined as 0%. *: P< 0.05, **: P< 0.01 compared with GFP-talin. All integrin activity data were represented by means ± SD, n=3. (F) Comparison of in vitro pull-down of His-F2F3 by GST-R9 and GST-R9-T1812Y. (G) The integrin activity of the cells transfected with various talin constructs was detected as described in (E). NS: not significant compared with GFP-talin.
See also: Figures S2 and S4.
In the F2F3:R9 complex structure, K318 and K320 in the F3 domain lysine finger form salt bridges with E1770 of the R9 domain (Fig. 1E). Consistently, K318A/K320A has also been shown to activate talin (Saltel et al., 2009). Interestingly, although the complex structure reveals no direct interaction between the neighboring K322 and K324 residues and the R9 domain, the K322A/K324A mutation diminishes the F3:R9 interaction and also activates talin (Goult et al., 2009; Saltel et al., 2009; Song et al., 2012). It was suggested that the removal of the hydrophobic portions of K322 and K324 side chains in the K322A/K324A mutation destabilizes the lysine finger (Song et al., 2012). Nevertheless, to examine whether K322 and K324 directly interact with the R9 domain through the alternative binding mode, we performed a pull-down assay using GST-R9 with F2F3-K322A/K324A and a K322M/K324M mutant that retains the hydrophobic portions. Both mutations significantly diminished the F2F3:R9 interaction (Fig. 3C), suggesting that K322 and K324 indeed interact with the R9 domain. We also examined K322A or K324A single mutations in F2F3 for their binding with R9. The single mutations also diminished the autoinhibitory interaction with R9, whereas a control mutant F2F3-KKR bearing K272A/K274A/R277A mutations in the F2 domain exhibited unchanged interaction with R9 compared to the wild-type F2F3 protein (Fig. 3D). This result confirms that both K322 and K324 residues directly interact with the R9 domain.
Mutations in the R9 domain enhance talin-induced integrin activation
The intramolecular interaction of the integrin-binding F3 domain and the R9 domain inhibits the integrin-activating function of talin (Goksoy et al., 2008; Goult et al., 2009; Song et al., 2012). We examined the effect of the mutations of Glu1794, Glu1797, and Gln1801 on talin-induced integrin activity using an antibody-based FACS assay (Chang et al., 2014; Lee et al., 2009; Yang et al., 2014). A CHO-A5 cell line stably expressing αIIbβ3 integrin was transfected with talin and various mutants. Cells expressing talin-2Y or talin-3Y exhibit elevated αIIbβ3 integrin activity compared with those expressing wild-type talin (Fig. 3E). To define the F3-interacting area in the R9 α5 helix, we mutated Thr1812 at the C-terminus of the α5 (Fig. 2A). The T1812Y mutation does not affect R9 association with F2F3 (Fig. 3F), nor does it affect talin-induced integrin activation (Fig. 3G). Together, these results demonstrate that besides the α1 and α4 helices, the N-terminal portion of the R9 α5 helix also engages with the F3 domain, representing an alternative autoinhibitory configuration that is distinct from the previously published crystal structure.
The intermolecular interaction in talin dimer affects its activity in integrin signaling
To further assess the biological relevance of the intermolecular interactions revealed by the R7R8R9 crystal structure, we generated a V1540Y mutation in the R8 domain to disrupt the R9:R8’ interface. SEC analysis reveals no significant change between R7R8R9-V1540Y and the wild-type protein, indicating that this intermolecular contact may not occur simultaneously in the isolated R7R8R9 fragment (Fig. 4A). This is also supported by the analysis of a control mutation of an exposed residue T1812Y in the R9 domain (Fig. 4A). Furthermore, no significant change in integrin activity is observed in cells expressing talin-V1540Y (Fig. S2A). Nevertheless, because full-length talin possesses a C-terminal DH region that promotes homodimerization and forms a compact dimer, it is possible that the mutation is tolerant due to the pre-existing dimeric assembly of talin and other interdomain interactions in the compact talin dimer. We then asked whether talin activity in integrin signaling is affected by its dimerization through the DH region. We first deleted the DH fragment and examined the activity of the resulting talin molecule (ΔDH) in activating αIIbβ3 integrins. Cells expressing high levels of talin-ΔDH exhibit reduced activity compared with the wild-type control, suggesting that disruption of talin dimerization promotes the alternative F3:R9 autoinhibitory interaction. Consistently, the 2Y and 3Y mutations exhibit substantially elevated activity of talin-ΔDH by disrupting the alternative intramolecular autoinhibition (Fig. 4B). To assess the impact of the intermolecular R9:R8’ interaction on talin activity in the absence of the DH fragment, we then introduced the V1540Y mutation into talin-ΔDH to disrupt the R9:R8’ interaction. Talin-ΔDH-V1540Y further reduced talin activity compared with talin-ΔDH (Fig. 4C). This result suggests that V1540Y inhibits talin activity likely by disrupting the residual intermolecular interaction in the absence of the DH fragment and promoting the alternative autoinhibitory configuration that is otherwise suppressed by the R9:R8’ interaction.
Figure 4. Impact of intermolecular interactions on talin activity and the alternative talin autoinhibitory configuration.
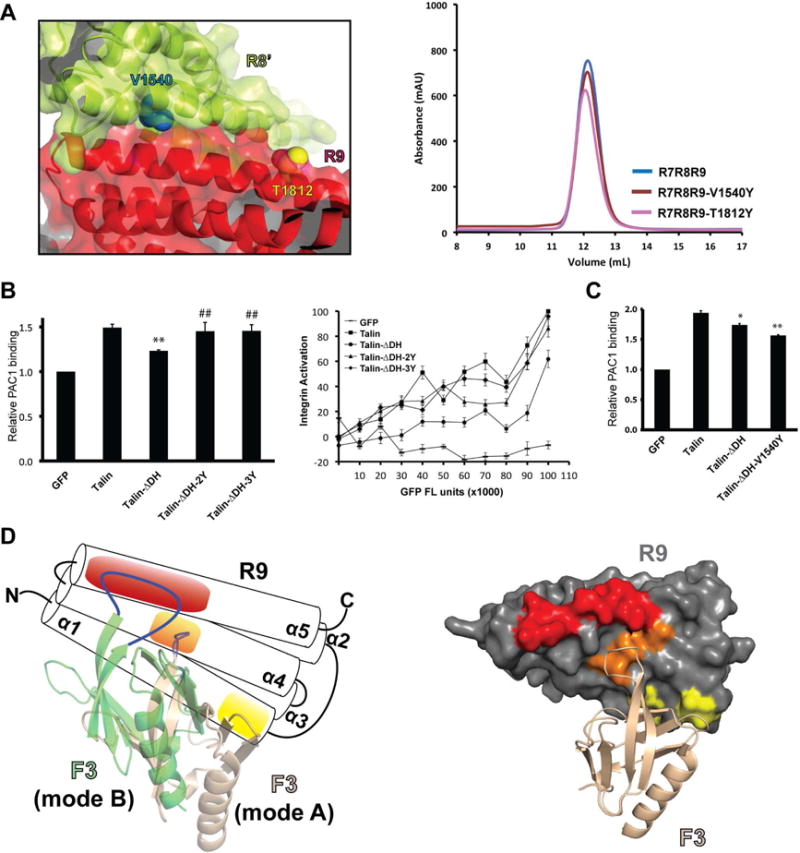
(A) Left: an interface residue V1540 (cyan) in the R8’ domain (lime) and an exposed residue T1812 (yellow) in the R9 domain (red) are shown in sphere representation. Right: Superose 12 elution profiles of R7R8R9, R7R8R9-V1540Y, and R7R8R9-T1812Y. (B) Integrin activity analyses of talin, talin-ΔDH, talin-ΔDH-2Y, and talin-ΔDH-3Y. Cells with GFP fluorescence intensity (FL unit) between 5×104 to 1×105 were gated to calculate the relative PAC1 binding for the histogram analysis. **: P< 0.01 compared with GFP-talin. ##: P< 0.01 compared with GFP-talin-ΔDH. (C) Integrin activity analyses of talin intermolecular interaction mutants. *: P< 0.05, **: P< 0.01 compared with GFP-talin. (D) Left: the five-helical-bundle R9 domain is shown in a schematic representation. The F3 domain (wheat) in the original autoinhibitory configuration (mode A) binds to the R9 domain via the binding sites on the α1 helix (yellow) and the α4 helix (orange). The F3 domain (green) in the alternative autoinhibitory configuration (mode B) binds to the R9 domain via the acidic patch on the α5 helix (red) and the α4 helix. Right: in the original autoinhibitory configuration, the F3 domain (wheat cartoon) does not interact with the acidic patch (red) of the R9 domain (gray surface).
See also Figure: S2 and S3.
DISCUSSION
Talin autoinhibition is mediated by the head-rod interaction and was first indicated by the observation that the cleavage of talin rod by calpain promotes talin binding to integrin (Yan et al., 2001). This autoinhibitory model was then confirmed by NMR spectroscopic study (Goksoy et al., 2008). It was later revealed that the R9 domain interacts with the talin F3 domain and masks the integrin-binding site (Goult et al., 2009). Song, et al. determined the crystal structure of an F2F3:R9 complex that reveals an autoinhibitory configuration (mode A) defined by the interaction between the α1 and α4 helices of the R9 domain and the F3 domain (Fig. 4D) (Song et al., 2012). Recently, atomic force microscopy and molecular dynamics (MD) simulations were used to analyze the talin autoinhibitory complex. The MD data suggested the possible existence of an alternative autoinhibitory configuration (mode B) that is not seen in the crystal structure (Zeng et al., 2015). We demonstrated that the R9 α5 helix also directly interacts with the F2F3 domains through an alternative autoinhibitory configuration. Mutation of the residues at the N-terminal portion of the R9 α5 helix leads to talin activation, confirming the existence of the alternative “mode B” autoinhibitory configuration of talin through these residues (Fig. 4D). The R9 α5 helix harbors a large acidic patch formed by Glu1794, Glu1797, Glu1798, Gln1801, Thr1804, Glu1805, Glu1808, and Asp1809. This acidic patch is electrostatically complementary to the lysine finger of the F3 domain, providing a structural foundation for the “mode B” configuration (Fig. 4D). Interestingly, while the 3Y mutant significantly diminishes the F2F3:R9 interaction, the E1770A mutation completely abolishes the F2F3:R9 interaction (Fig. 3B). This suggests that the “mode B” configuration contributes significantly to talin autoinhibition and the Glu1770 residue is essential for both autoinhibition modes. In the published crystal structure of F2F3:R9 complex, the F2 domain from a symmetrical molecule makes close contact with the N-terminus of the R9 α5 helix (Fig. S3). It is likely that this lattice contact prevents the formation of the “mode B” complex. Nevertheless, our data provide the first biochemical and functional evidence for an alternative talin autoinhibitory configuration.
Our result suggests that the K322/K324 lysine finger in the F3 domain directly interacts with the R9 domain. The lysine finger also interacts with the headgroup of PI(4,5)P2, leading to talin membrane association and integrin clustering (Saltel et al., 2009). However, when we examined the effect of PI(4,5)P2 on the F2F3:R9 interaction using inositol-1,4,5-trisphosphate (IP3), the headgroup of PI(4,5)P2, we only observed a slight weakening of the interaction at a high IP3 concentration (Fig. S4A). We also examined the effect of the PI(4,5)P2 headgroup on the interaction of R9 and the F2F3-KKR mutant that eliminates several PI(4,5)P2-binding sites in the F2 domain. Again we observed no significant effect on F2F3:R9 interaction in the presence of PI(4,5)P2 headgroup at up to 0.4 mM (Fig. S4B), suggesting the existence of additional PI(4,5)P2-binding sites in the F2 domain (Banno et al., 2012; Goksoy et al., 2008; Song et al., 2012). These sites dilute the effect of PI(4,5)P2 on the interaction of R9 and F3 at K322/K324 in solution. In addition, the effect of the PI(4,5)P2 headgroup on protein-protein interaction in solution is often significantly enhanced in the form of a lipid bilayer due to restrained PI(4,5)P2 diffusion and headgroup orientation that favors specific binding sites. Nevertheless, Goksoy, et al. had demonstrated that PI(4,5)P2 suppresses the interaction of talin F3 domain with the autoinhibitory domain (Goksoy et al., 2008).
Intramolecular interactions often regulate the biological functions of proteins. These interactions may be disrupted by the binding of other signaling components or by post-translational modifications such as phosphorylation, thus altering the activity states of the protein. To our knowledge, we demonstrate the first intramolecular interaction of the same domains at entirely different interfaces. For protein-protein interactions in general, there are examples, although very rare, that binding of two proteins differs substantially (Hamp and Rost, 2012). For instance, Ras binds to SOS (Son of Sevenless) at two distinct interfaces in a GDP-bound form and a GTP-bound form, respectively (Margarit et al., 2003). In addition, a group of proteins termed morpheein can adopt different quaternary structures of various multiplicities through distinct interfaces, representing a novel structural paradigm for allosteric regulation (Jaffe, 2005). Our study reveals an unusual scenario in which two talin domains can interact through two distinct binding interfaces. Nevertheless, although both two binding configurations result in talin autoinhibition, they may lead to different talin activity in binding to RIAM and changes in the dynamics of talin dimerization. Further study is required to elucidate these regulatory effects.
In summary, we determined the crystal structure of the talin R7R8R9 triple domain. The structure reveals an intermolecular contact between the R9 domain and the symmetrically related R8 domain. We further identified an alternative autoinhibitory configuration through an interaction between the F3 domain and the α5 helix of the R9 domain. This configuration is distinct from the reported crystal structure of F2F3:R9 complex. The new talin autoinhibitory configuration was then validated by biochemical and functional analyses. Our data provide a more comprehensive understanding of talin autoinhibition, regulation, and quaternary structure.
MATERIALS AND METHODS
Plasmid Construction and Protein Purification
Talin fragments containing the R7R8R9 (residues 1357–1822), R9 (residues 1655–1822), and F2F3 (residues 196–405) domains were subcloned into a modified pET28a expression vector with a His6-tag and a TEV protease cleavage site. The talin R9 and F2F3 domains were also inserted into pGEX-5X-1 vector to obtain GST-tagged proteins. All mutants were constructed by site-directed mutagenesis. Plasmids were transformed into the Escherichia coli BL21(DE3)-T1R strain for protein expression. Cells were grown to an A600 of 0.6–0.7 and then induced with 0.4 mM IPTG at 20°C overnight. Subsequent protein purif ication was carried out at 4°C. Cells were harvested and lysed in 20 mM Tris pH 7.5 and 500 mM NaCl (for His-tagged proteins) or 50 mM Tris pH 7.5, 150 mM NaCl, and 2 mM DTT (for GST-tagged proteins) using an EmulsiFlex-C3 homogenizer (AVESTIN, Inc.). Protein samples were extracted from the supernatant using HisTrap FF or GSTrap FF columns (GE Healthcare). For crystallization, proteins were treated by TEV protease to remove the His-tag and then further purified using a Resource Q column (GE Healthcare). GST-tagged proteins were dialyzed overnight in buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, and 2 mM DTT to remove glutathione.
Solution Characterization by Gel Filtration, SEC-MALS and Cross-linking
Purified R7R8R9 protein and its mutants were concentrated to 12 mg/mL, and subject to gel filtration analysis. 500 μL of the protein solutions were loaded onto a Superose 12 10/300 GL column (GE Healthcare) equilibrated in 20 mM Tris pH 7.5, 150 mM NaCl, and 2 mM DTT. For SEC-MALS analysis, 50 μL of R7R8R9 protein or its mutants at 12 mg/mL were loaded onto a Shodex KW-802.5 column (JM Science, Inc.) equilibrated in the aforementioned column buffer at a flow rate of 0.5 mL/min. The HPLC system (Waters) was connected in-line to a three-angle light-scattering detector (miniDAWN TREOS) and a refractive-index detector (Optilab rEX) (Wyatt Technology). MALS data were processed using the program ASTRA (Wyatt Technology). The cross-linking reaction was carried out at room temperature with protein concentration at 5 mg/mL (20 mM HEPES pH 7.0 and 150 mM NaCl). The reaction mixture contains 25 μg of His-tagged talin protein and 0.5 mM BS3 (Thermo Fisher Scientific). The reaction was terminated by addition of 2× Laemmli sample buffer at desired time points and subject to SDS-PAGE analysis.
GST Pull-Down and Western Blotting
Purified GST-tagged proteins were immobilized on glutathione agarose beads (Thermo Fisher Scientific) and incubated with purified His-tagged proteins in 250 μL of reaction buffer (50 mM Tris pH 7.5, 150 mM NaCl, and 2 mM DTT) on a rotator for 1 hr at 4°C. After washing three times with reaction buffer, bound protein was eluted by the addition of 10 mM glutathione. For cell lysate pull-down assays, HEK293T cells were transfected with GFP-talin constructs using calcium phosphate and lysed with buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100). Immobilized GST-tagged proteins were added to the clarified total lysates and incubated for 1 hr at 4°C. After washing, bound protein was eluted with 10 mM glutathione and detected by Western blot. For Western blot, protein samples were loaded onto an SDS-PAGE gel, transferred to an Immobilon-P membrane (EMD Millipore), blocked with TBST buffer containing 5% non-fat dry milk and incubated with anti-His (Sigma-Aldrich) or anti-GFP antibodies (Clontech Laboratories) for 1 hr at room temperature. SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) was applied to the incubated membrane and the protein bands were detected using a FluorChem E imager (ProteinSimple).
X-ray Crystallography
Purified talin proteins were concentrated to 5 mg/mL for wild-type R7R8R9 and 12 mg/mL for the R7R8R9-3Y mutant and crystallized using the hanging-drop vapor diffusion method at room temperature. Crystals of wild-type R7R8R9 were allowed to grow in 0.1 M MES pH 6.5, 21% (w/v) PEG 3350, 2% (w/v) benzamidine hydrochloride and were harvested after 3–5 days. Crystals of the R7R8R9-3Y protein grew in 0.2 M potassium phosphate and 20% (w/v) PEG 3350 at 4°C after 2 weeks. The crystals were transferred into cryo-solutions consisting of their respective well solutions supplemented with an additional 20% (v/v) ethylene glycol and flash-frozen in liquid nitrogen prior to the diffraction experiments.
X-ray diffraction data for the wild-type R7R8R9 crystal and the R7R8R9-3Y crystal were collected using beamline X25 of the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY) and beamline A1 of the Cornell High Energy Synchrotron Source (Ithaca, NY), respectively. Data were processed using the HKL-2000 package (Otwinowski and Minor, 1997). Structures of the talin R7R8 (PDB ID: 4W8P) and R9 (PDB ID: 4F7G) domains were used as the search models to determine the structures of wild-type R7R8R9 and R7R8R9-3Y by molecular replacement. Structural refinement was performed using REFMAC (Murshudov et al., 1997). Model building was performed using Coot (Emsley and Cowtan, 2004). The data collection and refinement statistics are listed in Table 1. The structural figures were generated using the PyMOL program suite (http://pymol.sourceforge.net). The final atomic models contain residues 1357–1819 for the wild-type R7R8R9 and residues 1357–1819 for the R7R8R9-3Y mutant. Examples of the electron densities at the R9:R8’ interface generated by PyMOL are shown (Fig. S1C). The atomic coordinates and structure factors have been deposited in the Protein Data Bank under the accession numbers 5IC0 (R7R8R9) and 5IC1 (R7R8R9-3Y).
Integrin Activation Assay
Activation of integrin αIIbβ3 was measured by PAC-1 mAb specifically recognizing activated αIIbβ3 integrin (Becton Dickinson Immunocytometry System) as described previously (Chang et al., 2014; Yang et al., 2014). Briefly, the CHO-A5 cells stably expressing integrin αIIbβ3 were transfected with GFP or GFP-tagged talin or mutant talin using Lipofectamine 2000 (Invitrogen). One day after transfection, the cells were resuspended in Tyrode’s buffer and incubated with PAC-1 mAb for 1 hr at 4°C. After three washes with Tyrode’s buffer, Alexa Fluor 647 labeled secondary antibody was added to the cells for 1 hr on ice. Stained cells were analyzed using an LSRII FACS instrument (BD Scientific). The data were processed by FlowJo software. Histogram analyses of the relative PAC1 binding were performed by gating cells with GFP fluorescence intensity (FL unit) between 5×104 to 1×105 unless otherwise specified. The mean fluorescence intensity (MFI) of the cells transfected with empty GFP vector was defined as 1 and all other constructs were compared with the GFP control. The data are shown as the means ± SD from three experiments repeated under the same conditions. Unpaired t test was determined by GraphPad software to calculate the P value. Talin were examined by Western blot to ensure equal expression levels in CHO-A5 cells (Fig. S2B).
Supplementary Material
Highlights.
Crystal structure of a talin triple domain module R7R8R9 was determined to 2.0 Å.
The structure reveals a dimeric interaction through the R8 and R9 domains.
Mutations on the α5 helix of the R9 domain lead to elevated talin activity.
Our results provide evidence for a new autoinhibitory configuration of talin.
Acknowledgments
We thank Drs. S.R. Hubbard and T. Ji (NYU School of Medicine) for the instrumental and technical support, and Drs. J.R. Peterson and M.D. Andrake for manuscript comments and discussions. We thank the beamline staff of X25 at the National Synchrotron Light Source, Brookhaven National Laboratory for technical support. Cornell High Energy Synchrotron Source (CHESS) is supported by the NSF & NIH/NIGMS via NSF award DMR-1332208, and the MacCHESS resource is supported by NIGMS award GM-103485. This work was supported by an NIH Core Grant CA006927, a Pennsylvania Department of Health Grant 4100068716, ACS RSG-15-167-01-DMC, and a CCSG Supported Pilot Projects award 5P30CA006927-51. H.Z. and Y.C.C were partially supported by the Elizabeth Knight Patterson Postdoctoral Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.Z., Y.C.C., and M.L.B performed the protein production and crystallization. Y.C.C., Q.H., and J.W. processed the crystallographic data and determined the structures. H.Z. and Y.C.C. performed biochemical experiments and contributed to the manuscript preparation. H.Z. performed the cell-based functional experiments. J.W. supervised the project and was the principal manuscript author.
References
- Alavi A, Hood JD, Frausto R, Stupack DG, Cheresh DA. Role of Raf in vascular protection from distinct apoptotic stimuli. Science. 2003;301:94–96. doi: 10.1126/science.1082015. [DOI] [PubMed] [Google Scholar]
- Banno A, Ginsberg MH. Integrin activation. Biochem Soc Trans. 2008;36:229–234. doi: 10.1042/BST0360229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banno A, Goult BT, Lee H, Bate N, Critchley DR, Ginsberg MH. Subcellular localization of talin is regulated by inter-domain interactions. J Biol Chem. 2012;287:13799–13812. doi: 10.1074/jbc.M112.341214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Zhang H, Franco-Barraza J, Brennan ML, Patel T, Cukierman E, Wu J. Structural and mechanistic insights into the recruitment of talin by RIAM in integrin signaling. Structure. 2014;22:1810–1820. doi: 10.1016/j.str.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis SJ, Lostchuck E, Goult BT, Bouaouina M, Fairchild MJ, Lopez-Ceballos P, Calderwood DA, Tanentzapf G. The talin head domain reinforces integrin-mediated adhesion by promoting adhesion complex stability and clustering. PLoS Genet. 2014;10:e1004756. doi: 10.1371/journal.pgen.1004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, Hughes PE, Pampori N, Shattil SJ, Saven A, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A. 2001;98:1853–1858. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmy-Susini B, Avraamides CJ, Schmid MC, Foubert P, Ellies LG, Barnes L, Feral C, Papayannopoulou T, Lowy A, Blair SL, et al. Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Res. 2010;70:3042–3051. doi: 10.1158/0008-5472.CAN-09-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AR, Bate N, Goult BT, Hazelwood L, Canestrelli I, Grossmann JG, Liu H, Putz NS, Roberts GC, Volkmann N, et al. The structure of the C-terminal actin-binding domain of talin. EMBO J. 2008;27:458–469. doi: 10.1038/sj.emboj.7601965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AR, Bate N, Goult BT, Patel B, Kopp PM, Emsley J, Barsukov IL, Roberts GC, Critchley DR. Central region of talin has a unique fold that binds vinculin and actin. J Biol Chem. 2010;285:29577–29587. doi: 10.1074/jbc.M109.095455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Goksoy E, Ma YQ, Wang X, Kong X, Perera D, Plow EF, Qin J. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol Cell. 2008;31:124–133. doi: 10.1016/j.molcel.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goult BT, Bate N, Anthis NJ, Wegener KL, Gingras AR, Patel B, Barsukov IL, Campbell ID, Roberts GC, Critchley DR. The structure of an interdomain complex that regulates talin activity. J Biol Chem. 2009;284:15097–15106. doi: 10.1074/jbc.M900078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goult BT, Xu XP, Gingras AR, Swift M, Patel B, Bate N, Kopp PM, Barsukov IL, Critchley DR, Volkmann N, et al. Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: implications for talin activation. J Struct Biol. 2013;184:21–32. doi: 10.1016/j.jsb.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamp T, Rost B. Alternative protein-protein interfaces are frequent exceptions. PLoS Comput Biol. 2012;8:e1002623. doi: 10.1371/journal.pcbi.1002623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- Hood JD, Frausto R, Kiosses WB, Schwartz MA, Cheresh DA. Differential alphav integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J Cell Biol. 2003;162:933–943. doi: 10.1083/jcb.200304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers S, Truong H, Danen HJ. Integrins: signaling, disease, and therapy. Int J Radiat Biol. 2007;83:743–751. doi: 10.1080/09553000701481808. [DOI] [PubMed] [Google Scholar]
- Jaffe EK. Morpheeins–a new structural paradigm for allosteric regulation. Trends Biochem Sci. 2005;30:490–497. doi: 10.1016/j.tibs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem. 2009;284:5119–5127. doi: 10.1074/jbc.M807117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margadant C, Kreft M, Zambruno G, Sonnenberg A. Kindlin-1 regulates integrin dynamics and adhesion turnover. PLoS One. 2013;8:e65341. doi: 10.1371/journal.pone.0065341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- Menter DG, Dubois RN. Prostaglandins in cancer cell adhesion, migration, and invasion. Int J Cell Biol. 2012;2012:723419. doi: 10.1155/2012/723419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nip J, Shibata H, Loskutoff DJ, Cheresh DA, Brodt P. Human melanoma cells derived from lymphatic metastases use integrin alpha v beta 3 to adhere to lymph node vitronectin. J Clin Invest. 1992;90:1406–1413. doi: 10.1172/JCI116007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Pinon P, Wehrle-Haller B. Integrins: versatile receptors controlling melanocyte adhesion, migration and proliferation. Pigment Cell Melanoma Res. 2011;24:282–294. doi: 10.1111/j.1755-148X.2010.00806.x. [DOI] [PubMed] [Google Scholar]
- Saltel F, Mortier E, Hytonen VP, Jacquier MC, Zimmermann P, Vogel V, Liu W, Wehrle-Haller B. New PI(4,5)P2- and membrane proximal integrin-binding motifs in the talin head control beta3-integrin clustering. J Cell Biol. 2009;187:715–731. doi: 10.1083/jcb.200908134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Yang J, Hirbawi J, Ye S, Perera HD, Goksoy E, Dwivedi P, Plow EF, Zhang R, Qin J. A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 2012;22:1533–1545. doi: 10.1038/cr.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Bodin L, Pandey M, Krause M, Coughlin S, Boussiotis VA, Ginsberg MH, Shattil SJ. Mechanisms and consequences of agonist-induced talin recruitment to platelet integrin alphaIIbbeta3. J Cell Biol. 2008;181:1211–1222. doi: 10.1083/jcb.200803094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- Yan B, Calderwood DA, Yaspan B, Ginsberg MH. Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J Biol Chem. 2001;276:28164–28170. doi: 10.1074/jbc.M104161200. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhu L, Zhang H, Hirbawi J, Fukuda K, Dwivedi P, Liu J, Byzova T, Plow EF, Wu J, et al. Conformational activation of talin by RIAM triggers integrin-mediated cell adhesion. Nat Commun. 2014;5:5880. doi: 10.1038/ncomms6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Zhang Y, Song X, Ji Q, Ye S, Zhang R, Lou J. The conformational states of talin autoinhibition complex and its activation under forces. Sci China Life Sci. 2015 doi: 10.1007/s11427-015-4873-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.