

Abstract
We characterized the full-length genomes for nine novel variants of HCV genotype 4 (HCV-4), representing a new subtype 4s and eight unclassified lineages. They were obtained from patients who resided in Canada but all had origins in Africa. An extended maximum clade credibility (MCC) tree was reconstructed after the inclusion of 30 reference sequences. It differentiated 18 assigned subtypes and 10 unclassified lineages within HCV-4. Similar analysis of 102 partial NS5B sequences resulted in another MCC tree that revealed 22 assigned subtypes (4a–4t, 4w, and 4v) and 30 unclassified lineages at the subtype level. Our study shows that HCV-4 is taxonomically complex and it displays high genetic diversity to support an African origin.
Keywords: HCV, Full-length genome, Genotype 4, Sequence
Introduction
Hepatitis C virus (HCV) is a blood-borne pathogen that infects an estimated 115 million people worldwide or approximately 1.3–2.1% of the global population (Gower et al., 2014). HCV infection is characterized by the establishment of chronic hepatitis in about 70–85% of the infected individuals among whom many develop hepatocellular carcinoma, liver cirrhosis, and liver failure (Hoofnagle, 2002; Zoulim et al., 2003). Eventually, these end-stage liver diseases cause substantial morbidity and mortality (Razavi et al., 2014).
HCV possesses a single stranded, positive-sense RNA genome of about 9600 nucleotides in length. HCV is classified into the Hepacivirus genus of the Flaviviridae family and has a high degree of genetic diversity (Thiel et al., 2005). Accordingly, HCV can be divided into seven genotypes and each genotype, excluding genotypes 5 and 7, is further divided into a number of subtypes. Currently, 82 subtypes of HCV have been confirmed. Each have at least one full-length ORF sequence characterized and encompass a minimum of three epidemiologically unrelated isolates. Among them, genotype 4 (HCV-4) represents one of the most complex lineages for which 21 subtypes have been designated in addition to a number of variants that remain unclassified (Smith et al., 2014) (http://talk.ictvonline.org/ictv_wikis/w/sg_flavi/56.hcv-classification.aspx). The HCV genotypes have different geographic distribution patterns and respond in a different way to antiviral therapy. Generally, subtypes 1a, 1b, 2a, 2b, and 3a are globally distributed. In contrast, most other subtypes are restricted to certain regions (Simmonds et al., 2005). Clinically, genotypes 2 and 3 have shown better responses than genotypes 1 and 4 to interferon-α and ribavirin standard combination therapy (Feld and Hoofnagle, 2005; Manns et al., 2006). Although the advent of direct-acting antivirals (DAAs) may show improved genotype coverage and response, their approval remain restricted to specific genotypes (Delang et al., 2013).
Based on full-length genome sequence data from our previous study, a total of 17 HCV-4 subtypes (4a–4d, 4f, 4g, 4k–4r, 4v, 4w) are now confirmed (Li et al., 2009; Smith et al., 2014). Two distinct HCV-4 variants not assigned to a subtype also have had their genomes completely sequenced. However, there remain a number of HCV-4 variants that show a considerable degree of genetic diversity from the other known HCV-4 lineages based on partial genome sequences (Murphy et al., 2007). In the present study, we determined the full-length genome sequences for nine such variants further revealing the extensive diversity and complexity of HCV-4.
Results
Analysis of the full-length genomes
The full-length HCV-4 genome was characterized for nine isolates represented by QC108, QC127, QC132, QC147, QC215, QC253, QC352, QC361, and QC58, each from 16–20 overlapping fragments (data not shown). These genomes were 9426–9563 nucleotides (nt) in length, starting from the extreme 5′-UTR through to the variable region of the 3′-UTR (Table 1). For a more comprehensive analysis of the nine full-length genome sequences, a maximum clade credibility (MCC) tree was reconstructed with the inclusion of 30 reference sequences (Fig. 1). Genotype 4, the major cluster, is represented by a total of 33 sequences and the remaining 6 genotypes are each represented by a solitary branch (genotypes 1–3 and 5–7). In the MCC tree, the genotype 4 cluster was supported with a clade probability of 0.99 and proved more closely related to genotype 1 than to the other five genotypes. Within this cluster, a total of 28 taxonomic lineages could be differentiated including 18 confirmed subtypes (4a–4d, 4f, 4g, 4k–4t, 4v, and 4w), two unclassified lineages represented by P025 and BID-G1253, and eight other unclassified lineages represented by the genomes characterized in this study. Among the 18 confirmed subtypes, 4s represented by QC361, was newly assigned in this study. Analysis of Core-E1 and NS5B genome sequences revealed that QC361 showed a high degree of genetic similarity to three other QC-isolates (QC348, QC409, and QC547) thus meeting current criteria for subtype assignment (see Fig. S1 and Fig. 2). Fig. 1 shows that the HCV-4 cluster can be divided into three major subsets. The first subset is the most complex, containing 13 assigned subtypes (4a down to 4t) and five unclassified variants represented by QC352, QC215, QC127, QC132, and QC147. Among the latter five, QC352 and QC127 each showed a relatively shorter branch, with QC352 positioned between subtypes 4a and 4c and QC127 between subtypes 4v and 4q. In contrast, QC215, QC132, and QC147 each showed a relatively longer branch, with QC215 adjacently grouped with subtype 4s and QC132 and QC147 grouped around subtype 4l. The second subset contained two assigned subtypes, 4b and 4w, and four unclassified variants represented by QC253, QC108, QC58, and P026. Among the latter four, the three QC-genomes determined in this study grouped closest to subtype 4b. P026 was initially classified as subtype 4b (Koletzki et al., 2009), but has later been recategorized as an unclassified variant (Smith et al, 2014). In contrast to the above two subsets, the third subset contained no isolates from this study. Phylogenies based on the maximized parsimony method and the maximum likelihood method were also reconstructed, which showed topologies consistent to that in Fig. 1. For simplicity, these phylogenies are not shown in this paper.
Table 1.
Patient information for the 10 genotype 4 isolates and the number of nucleotides/amino acids in each genomic region.
| ID | Sex | Originb | Viral load (Log IU/ml) | Full | ORF | 5′UTR | E2 | NS5A | 3′UTR |
|---|---|---|---|---|---|---|---|---|---|
| H77a | M | US | 9646 | 9036/3011 | 341 | 1089/363 | 1344/448 | 269 | |
| 4_QC108 | M | CD | 5.98 | 9441 | 9036/3012 | 340 | 1089/363 | 1344/448 | 65 |
| 4_QC127 | M | CD | 5.82 | 9426 | 9021/3007 | 340 | 1086/362 | 1332/444 | 65 |
| 4_QC132 | M | RW | 6.04 | 9430 | 9024/3008 | 340 | 1086/362 | 1335/445 | 66 |
| 4_QC147 | M | BI | 6.36 | 9426 | 9021/3007 | 341 | 1086/362 | 1332/444 | 64 |
| 4_QC215 | F | EG | – | 9535 | 9129/3043 | 340 | 1089/363 | 1437/479 | 66 |
| 4_QC253 | F | CD | 6.70 | 9437 | 9036/3012 | 340 | 1089/363 | 1344/448 | 61 |
| 4_QC352 | M | CF | 6.67 | 9431 | 9024/3008 | 340 | 1089/363 | 1332/444 | 67 |
| 4_QC361 | F | AF | 7.04 | 9426 | 9021/3007 | 341 | 1089/363 | 1329/443 | 64 |
| 4_QC58 | M | CF | 6.53 | 9563 | 9039/3013 | 340 | 1092/364 | 1344/448 | 184 |
The H77 genome (Genebank accession no. AF009606) of subtype 1a was included as a standard for comparison. Comparing with the H77 genome, eight protein encoding regions of the 10 genotype 4 isolates showed no differences in their numbers of nucleotide: Core (573 nt), E1 (576 nt), NS2 (651 nt), NS3 (1893 nt), NS4A (162 nt), NS4B (783 nt), NS5B (1776 nt).
Country codes officially assigned in ISO 3166-1: AF: East Africa; BI: Burundi; CD: Democratic of the Congo; CF: Central African Republic; EG: Egypt; RW: Rwanda; US: United States.
Fig. 1.
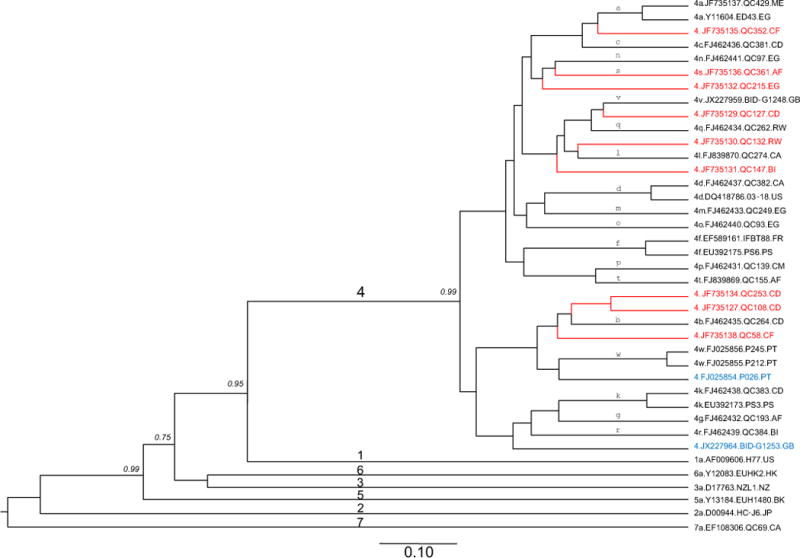
MCC tree estimated using full-length genome sequences of HCV. Reference sequences from the confirmed subtypes of genotypes 1, 2, 3, 4, 5, 6, and 7 are included (black tips), together with the nine new genomes determined in this study (red tips) and two additional unclassified variants (blue tips). Each genotype is denoted with a single digit number on the major branches of the tree while all subtypes of genotype 4 are labeled with single lower case letters above the related branches in proximity to the right tips. Isolates are named using the following format: genotype or subtype.accession number.isolate name.sampling country. To simplify the tree, only the clade posterior probability supports of less than 1 are shown in italics, otherwise they are equal to 1 but are not shown. A scale bar at the bottom represents 0.10 units of nucleotide substitution per site. Country codes: BI: Burundi; CA: Canada; CD: Congo; CF: Central Africa; CM: Cameroon; EG: Egypt; FR: France; GB: United Kingdom; HK: Hong Kong; JP: Japan, ME, Montenegro; NZ: New Zealand; PT: Portugal; RW: Rwanda; US: the United States.
Fig. 2.
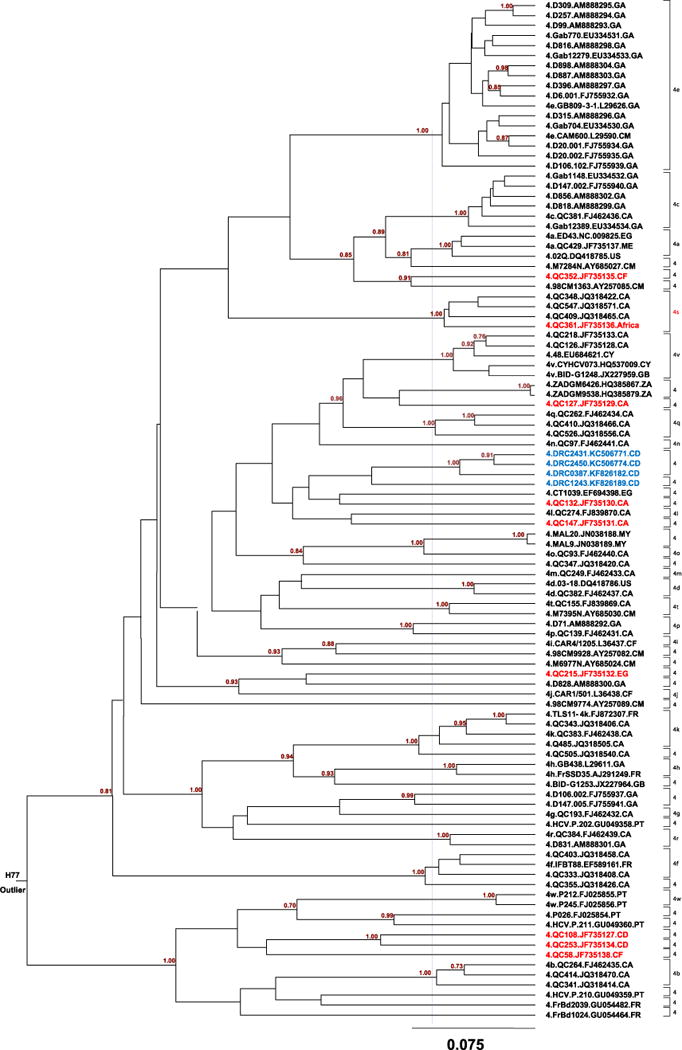
MCC tree of HCV-4 estimated using 102 partial NS5B sequences corresponding to nucleotide positions 8288–8610 in the referenced H77 genome. Each subtype or subtype analogy is denoted by a half bracket at the right hand side of the tree. These brackets are vertically aligned and each is labeled with single digit number 4 if it refers to an unclassified lineage or is followed by a lower case letter if it refers to an assigned subtype. A purple vertical dash line in the middle of the tree delimits the genetic distance at which the 22 assigned HCV-4 subtypes are separable from each other and for which an additional 30 unclassified lineages can be also differentiated. At the root of the tree, the outlier H77 is indicated. A scale bar at the bottom represents 0.075 units of nucleotide substitution per site. The nine genomes from this study are shown in red tips whereas those four sequences from the report by Iles et al. (2014) are indicated with blue tips. In addition to country codes indicated in Fig. 1, the other codes are: GA: Gabon; CY: Cyprus; ZA: South Africa; MA: Malaysia.
Pairwise nucleotide similarities were calculated for the full-length HCV-4 genome sequences. When the nine genomes determined in this study were compared to each other, the largest similarity of 86.2% was observed between QC108 and QC253, while the smallest similarity of 77.5% was seen between QC132 and QC58. When these nine genomes were compared with the 24 reference HCV-4 sequences, the highest similarity of 86.5% was shown between QC352 and QC381/4c, while the lowest similarity of 77.3% was seen between QC58 and ED43/4a. Smith et al. (2014) recently observed that members of the same subtype almost exclusively displayed nucleotide differences of <13% while members of different subtypes virtually displayed differences of >15%. In this study, however, we did identify nucleotide differences that fell into the 13–15% gap. When QC127 was compared either to QC262/4q or to BID-G1248, a nucleotide difference of 14.1% was observed. When QC352 was compared to QC429/4a and QC381/4c, the nucleotide differences were 14.1% and 13.5%, respectively. Similarly, comparing QC429/4a to QC381/4c, QC139/4p to QC155/4t, and QC262/4q to BID-G1248/4v, nucleotide differences of 14.9%, 14.7%, and 14.0%, respectively, were observed. Because these intermediate results do not arise from technical problems or a recent recombination event, they would reflect the actual variation as recently discussed (Li et al., 2015). Pairwise comparison of four subtype 4s isolates was also performed in their partial Core-E1 and NS5B regions that showed nucleotide similarities of 7.6–9.6% and 4.6–7.7%, respectively, confirming members of the same subtype.
Analysis of partial sequences
A segment of the NS5B region, corresponding to the nucleotides numbered 8276-8615 in H77 genome has been found to reliably differentiate HCV genotypes and subtypes (Murphy et al., 2007). For a better understanding of the genetic variation and epidemiological distribution patterns relating to the nine isolates characterized in this study, we also analyzed the above NS5B sequence from 102 isolates representing each of the 22 assigned subtypes (counting subtype 4s designated in this study) and all unassigned variants of HCV-4. This resulted in the second MCC tree shown in Fig. 2, which differentiated 52 lineages at the subtype level. Among these lineages, 22 assigned subtypes 4a–4t, 4v, and 4w are well separated (Koletzki et al., 2009; Simmonds et al., 2005; Smith et al., 2014). Of the nine genomes we determined in this study, excluding QC361 that we assigned as a new subtype 4s, the other eight each represent one unassigned lineage. Each of these unassigned lineages contained no more than two individual isolates, indicating their low frequencies in the populations that were sampled (Fig. 2). We are now unable to assign these eight isolates new subtypes, because they do not meet the presently recommended criteria. These criteria suggest that future subtype assignments will be only made for lineages containing sequence data from three or more isolates and for which the coding region sequence of at least one must be complete or nearly complete (Smith et al., 2014).
Except for those characterized in the NS5B region, a number of HCV-4 isolates characterized only in other genomic regions were also analyzed. However, none of them grouped with the eight unclassified variants we determined in this study (data not shown). The results suggest that all the unclassified HCV-4 lineages lack sufficient numbers of closely related isolates for allowing their assignment to new subtypes.
Similarity plotting
To exclude the possibility of viral recombination, pairwise nucleotide similarity curves were plotted along HCV genomes. Upon comparison of the nine genomes from this study with each other, and with the 49 reference sequences shown in Fig. 1, no such evidence was detected (data not shown).
Discussion
In this study, the full-length genomes were characterized for nine HCV-4 isolates. Excluding QC361 assigned to subtype 4s, the other eight genomes each represent an unclassified lineage differentiable at the subtype level. This was supported not only by the analysis of full-length genome sequences but also by the analysis of partial sequences in both Core-E1 and NS5B regions. In previous reports, two other unclassified variants of HCV-4 each representing a distinct lineage, also had their full-length genomes determined (Newman et al., 2013; Smith et al., 2014). With the inclusion of these two variants, there are now a total of 10 unclassified lineages for which a complete genome sequence is available further indicating the high genetic complexity of HCV-4. Analysis of partial NS5B sequences revealed many additional unclassified lineages of HCV-4 at the subtype level. Such a finding implies that HCV-4 is far more complex than that presently sampled. However, if these variants are not highly prevalent, their assignment to a new subtype has a little meaning.
Although sampled in Canada, the nine new genomes were all from patients that had their origins in Africa (Table 1). Likewise, the majority of sequences in both Figs. 1 and 2 also showed an African origin. The ancestral origin of HCV-4 in Africa is supported by studies from sub-Saharan African countries that showed high HCV seroprevalences and local epidemics in association with multiple HCV-4 lineages. For example, 11.2% of people screened in rural Gabon were positive for HCV, among whom 92% were infected with HCV-4 strains (Njouom et al., 2012). In Cameroon, HCV seroprevalence was 11% in a group of high-HIV risk individuals and 16% of the isolates were classified as HCV-4 (Ndjomou et al., 2003). In another study from Cameroon on individuals aged >60, about 56% of the infections were due to HCV-4 strains (Pépin et al., 2010). In a recent study based on 1999 members of the uniformed services in the Democratic Republic of Congo, the seroprevalence of HCV was 3% and all the classified HCV sequences belonged to genotype 4, comprising subtypes 4c, 4k, 4h and 4r, as well as a potential new subtype candidate (Iles et al., 2014). In each of these studies, however, the seroprevalence of HCV was associated with patient age and multiple HCV lineages were detected, indicating the indigenous and long-term endemic circulation of HCV-4. Diverse HCV-4 isolates are also common in Europe. However, this could have resulted from the historical roles played by the European explorers in Africa (Markov et al., 2012) and the fact that many immigrants from Africa are now residing in Europe. In addition, there has been rapid growth in the prevalence of HCV subtypes 4a and 4d in Europe in recent decades, particularly among IDUs (Ciccozzi et al., 2012; de Bruijne et al., 2009; van Asten et al., 2004). Subtypes 4a and 4d have also been found in North America among the local population likely acquired through injection drug use (Murphy et al., 2007).
Financial disclosure
The study described was supported by a Grant from The National Institute of Allergy and Infectious Diseases (5 R01 AI080734-03A). The funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Materials and methods
Subjects and specimens
In the Canadian province of Quebec, serum samples from patients, who were positive for HCV, are generally submitted from hospital laboratories or private clinics to the Laboratoire de Santé Publique du Québec for routine HCV genotyping. This resulted in a large collection of samples that had their partial HCV sequences determined and classified into various genotypes and subtypes (Murphy et al., 2007). Among them, nine samples, QC108, QC127, QC132, QC147, QC215, QC253, QC352, QC361, and QC58, collected from 2003–2009 were found to contain unique HCV-4 strains and were selected for further characterization in this study. Detailed information about the patients’ gender, geographic origin and HCV viral load are shown in Table 1.
PCR amplification and sequencing
Full-length HCV-4 genomes were each determined from a 100 μl serum sample. Briefly, the RNA extraction (Qiagen Viral RNA extraction kit, Qiagen, Valencia, CA) and the cDNA synthesis (RevertAid First Strand cDNA Synthesis Kit, Fermentas Life Science, EU) were performed according to the manufacturer’s protocols. Genomic fragments overlapping the full-length HCV-4 genomes were amplified in conventional PCR using degenerate primers as previously described (Li et al., 2009) or in combination with those specific primers designed based on the obtained sequences. Standard procedures were adopted to avoid potential carryover contamination (Kwok and Higuchi, 1989). Amplicons were sequenced as previously described (Li et al., 2009).
Sequence datasets and inspection
The obtained nine full-length genomes were annotated according to the standard nucleotide numbering in the H77 genome, from the extreme 5′-end through to the 3′-UTR (Kuiken et al., 2006). To determine the phylogenetic relationship, we retrieved 24 full-length HCV-4 sequences representing subtypes 4a–4d, 4f–4g, 4k–4r, 4t, 4v–4w, the two HCV-4 unassigned variants BID-G1253 and P026 (Smith et al, 2014) and an additional six sequences representing each of the other six genotypes for a total of 39 full-length genome sequences.
To better explore the epidemiology and genetic relationship of HCV-4 variants related to our new isolates, an additional NS5B sequence dataset was assembled representing each assigned subtypes and all unassigned subtype variants of genotype 4 (http://hcv.lanl.gov/content/index). As a result, 102 sequences of HCV-4 were included, each of which has approximately 323 nucleotides in length, corresponding to nucleotide positions 8288–8610 in the H77 genome.
The two sequence datasets were then aligned using the BioEdit software (Tippmann, 2004) followed by visual inspection and manual adjustments. To exclude possible recent viral recombination events, the RDP3 software (Martin et al., 2010) was run with settings as previously described (Lu et al., 2007) for the full length sequence dataset.
Phylogenetic analyses
The BEAST software was used to analyze the two datasets under the combination of the GTR+I+Γ6 substitution model, uncorrelated lognormal clock model, and the Bayesian skyline model to reconstruct the maximum clade credibility (MCC) trees (Drummond and Rambaut, 2007). For this purpose, we selected a rate of 1.0 in the panel of “Clock Models”, which would result in the nodes and branches of the tree being estimated in units of substitution/sites based on the Markov Chain Monte Carlo (MCMC) algorithm. Except for that above mentioned, all the other BEAST procedures were the same as that we have recently described (Li et al., 2014).
Genbank accession numbers
The nucleotide sequences reported in this study were deposited in Genbank with the following accession numbers: JF735127, JF735129-JF735132, JF735134-JF735138.
Supplementary Material
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2015.03.038.
References
- Bukh J, Purcell RH, Miller RH. Sequence analysis of the core gene of 14 hepatitis C virus genotypes. Proc Natl Acad Sci USA. 1994;91:8239–8243. doi: 10.1073/pnas.91.17.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccozzi M, Equestre M, Costantino A, Marascio N, Quirino A, Lo Presti A, Cella E, Bruni R, Liberto MC, Focà A, Pisani G, Zehender G, Ciccaglione AR. Hepatitis C virus genotype 4d in Southern Italy: reconstruction of its origin and spread by a phylodynamic analysis. J Med Virol. 2012;84:1613–1619. doi: 10.1002/jmv.23384. [DOI] [PubMed] [Google Scholar]
- de Bruijne J, Schinkel J, Prins M, Koekkoek SM, Aronson SJ, van Ballegooijen MW, Reesink HW, Molenkamp R, van de Laar TJ. Emergence of hepatitis C virus genotype 4: phylogenetic analysis reveals three distinct epidemiological profiles. J Clin Microbiol. 2009;47:3832–3838. doi: 10.1128/JCM.01146-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delang L, Neyts J, Vliegen I, Abrignani S, Neddermann P, De Francesco R. Hepatitis C virus-specific directly acting antiviral drugs. Curr Top Microbiol Immunol. 2013;369:289–320. doi: 10.1007/978-3-642-27340-7_12. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- Fu Y, Qin W, Cao H, Xu R, Tan Y, Lu T, Wang H, Tong W, Rong X, Li G, Li C, Abe K, Lu L. HCV 6a prevalence in Guangdong Province had the origin from Vietnam and recent dissemination to other regions of China: a phylogeography analysis. PLoS ONE. 2012;7:e28006. doi: 10.1371/journal.pone.0028006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61:S45–S57. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36:S21–S29. doi: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- Hraber PT, Leach RW, Reilly LP, Thurmond J, Yusim K, Kuiken C, Los Alamos HIV database team Los Alamos hepatitis C virus sequence and human immunology databases: an expanding resource for antiviral research. Antivir Chem Chemother. 2007;18:113–123. doi: 10.1177/095632020701800301. [DOI] [PubMed] [Google Scholar]
- Iles JC, Raghwani J, Harrison GL, Pepin J, Djoko CF, Tamoufe U, LeBreton M, Schneider BS, Fair JN, Tshala FM, Kayembe PK, Muyembe JJ, Edidi-Basepeo S, Wolfe ND, Simmonds P, Klenerman P, Pybus OG. Phylogeography and epidemic history of hepatitis C virus genotype 4 in Africa. Virology. 2014:464–465. doi: 10.1016/j.virol.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koletzki D, Dumont S, Vermeiren H, Peixe P, Nina J, Camacho RJ, Stuyver LJ. Full genome sequence of three isolates of hepatitis C virus subtype 4b from Portugal. Arch Virol. 2009;154:127–132. doi: 10.1007/s00705-008-0270-z. [DOI] [PubMed] [Google Scholar]
- Kuiken C, Combet C, Bukh J, Shin IT, Deleage G, Mizokami M, Richardson R, Sablon E, Yusim K, et al. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology. 2006;44:1355–1361. doi: 10.1002/hep.21377. [DOI] [PubMed] [Google Scholar]
- Kwok S, Higuchi R. Avoiding false positives with PCR. Nature. 1989;339:490. doi: 10.1038/339237a0. [DOI] [PubMed] [Google Scholar]
- Li C, Lu L, Wu X, Wang C, Bennett P, Lu T, Murphy D. Complete genomic sequences for HCV subtypes 4b, 4c, 4d, 4g, 4k, 4l,4m, 4n, 4o, 4p, 4q, 4r and 4t. J Gen Virol. 2009;90:1820–1826. doi: 10.1099/vir.0.010330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Cao H, Lu L, Murphy D. Full-length sequences of 11 hepatitis C virus genotype 2 isolates representing five subtypes and six unclassified lineages with unique geographical distributions and genetic variation patterns. J Gen Virol. 2012;93:1173–1184. doi: 10.1099/vir.0.038315-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Njouom R, Pépin J, Bennett P, Pybus OG, Lu L. Full-length genome sequences of seven genotype 1 isolates of HCV representing subtypes 1e, 1h, and 1l, and a novel variant. J Gen Virol. 2013;94:1780–1790. doi: 10.1099/vir.0.048835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Lu L, Murphy DG, Negro F, Okamoto H. The origin of HCV genotype 3 in Africa was estimated by evolutionary analysis of the full-length genome sequences for nine subtypes including 3d and 3e first determined. J Gen Virol. 2014;95:1677–1688. doi: 10.1099/vir.0.065128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Barnes E, Newton PN, Fu Y, Vongsouvath M, Klenerman P, Okamoto H, Abe K, Pybus OG, Lu L. An expanded taxonomy of hepatitis C virus geno type 6: characterization of 22 new full-length viral genomes. Virology. 2015 doi: 10.1016/j.virol.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Li C, Fu Y, Gao F, Pybus OG, Abe K, Okamoto H, Hagedorn CH, Murphy D. Complete genomes of hepatitis C virus (HCV) subtypes 6c, 6l,6o, 6p and 6q: completion of a full panel of genomes for HCV genotype 6. J Gen Virol. 2007;88:1519–1525. doi: 10.1099/vir.0.82820-0. [DOI] [PubMed] [Google Scholar]
- Lu L, Li C, Yuan J, Lu T, Okamoto H, Murphy DG. Full-length genome sequences of five hepatitis C virus isolates representing subtypes 3g, 3h, 3i and 3k, and a unique genotype 3 variant. J Gen Virol. 2013;94:543–548. doi: 10.1099/vir.0.049668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Li C, Xu Y, Murphy DG. Full-length genomes of 16 hepatitis C virus genotype 1 isolates representing subtypes 1c, 1d, 1e, 1g, 1h, 1i, 1j and 1k, and two new subtypes 1m and 1n, and four unclassified variants reveal ancestral relationships among subtypes. J Gen Virol. 2014;95:1479–1487. doi: 10.1099/vir.0.064980-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiorkinis G, Magiorkinis E, Paraskevis D, Ho SYW, Shapiro B, Pybus OG, Allain JP, Hatzakis A. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLoS Med. 2009;6:e1000198. doi: 10.1371/journal.pmed.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns MP, Wedemeyer H, Cornberg M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut. 2006;55:1350–1359. doi: 10.1136/gut.2005.076646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markov PV, van de Laar TJ, Thomas XV, Aronson SJ, Weegink CJ, van den Berk GE, Prins M, Pybus OG, Schinkel J. Colonial history and contemporary transmission shape the genetic diversity of hepatitis C virus genotype 2 in Amsterdam. J Virol. 2012;86:7677–7687. doi: 10.1128/JVI.06910-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–6243. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McOmish F, Yap PL, Dow BC, Follett EA, Seed C, Keller AJ, Cobain TJ, Krusius T, Kolho E, Naukkarinen R, Lin C, Lai C, Leong S, Medgyesi GA, Hejjas M, Kiyokawa H, Fukada K, Cuypers T, Saeed AA, Al-Rasheed AM, Lin M, Simmonds P. Geographical distribution of hepatitis C virus genotypes in blood donors: an international collaborative survey. J Clin Microbiol. 1994;32:884–892. doi: 10.1128/jcm.32.4.884-892.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DG, Willems B, Deschenes M, Hilzenrat N, Mousseau R, Sabbah S. Use of sequence analysis of the NS5B region for routine genotyping of hepatitis C virus with reference to C/E1 and 5′ untranslated region sequences. J Clin Microbiol. 2007;45:1102–1112. doi: 10.1128/JCM.02366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RM, Kuntzen T, Weiner B, Berical A, Charlebois P, Kuiken C, Murphy DG, Simmonds P, Bennett P, Lennon NJ, Birren BW, Zody MC, Allen TM, Henn MR. Whole genome pyrosequencing of rare hepatitis C virus genotypes enhances subtype classification and identification of naturally occurring drug resistance variants. J Infect Dis. 2013;208:17–31. doi: 10.1093/infdis/jis679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndjomou J, Pybus OG, Matz B. Phylogenetic analysis of hepatitis C virus isolates indicates a unique pattern of endemic infection in Cameroon. J Gen Virol. 2003;84:2333–2341. doi: 10.1099/vir.0.19240-0. [DOI] [PubMed] [Google Scholar]
- Njouom R, Caron M, Besson G, Ndong-Atome GR, Makuwa M, Pouillot R, Nkoghe D, Leroy E, Kazanji M. Phylogeography, risk factors and genetic history of hepatitis C virus in Gabon, central Africa. PloS ONE. 2012;7:e42002. doi: 10.1371/journal.pone.0042002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier C, Njouom R, Ayouba A, Dubois M, Sartre MT, Vessiere A, Timba I, Thonnon J, Izopet J, Nerrienet E. Distribution and heterogeneity of hepatitis C genotypes in hepatitis patients in Cameroon. J Med Virol. 2005;77:390–398. doi: 10.1002/jmv.20468. [DOI] [PubMed] [Google Scholar]
- Pépin J, Labbé AC, Mamadou-Yaya F, Mbélesso P, Mbadingaï S, Deslandes S, Locas MC, Frost E. Iatrogenic transmission of human T cell lymphotropic virus type 1 and hepatitis C virus through parenteral treatment and chemoprophylaxis of sleeping sickness in colonial equatorial Africa. Clin Infect Dis. 2010;51:777–784. doi: 10.1086/656232. [DOI] [PubMed] [Google Scholar]
- Prescott LE, Simmonds P, Lai CL, Chan NK, Pike I, Yap PL, Lin CK. Detection and clinical features of hepatitis C virus type 6 infections in blood donors from Hong Kong. J Med Virol. 1996;50:168–175. doi: 10.1002/(SICI)1096-9071(199610)50:2<168::AID-JMV10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Pybus OG, Cochrane A, Holmes EC, Simmonds P. The hepatitis C virus epidemic among injecting drug users. Infect Genet Evol. 2005;5:131–139. doi: 10.1016/j.meegid.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Pybus OG, Markov PV, Wu A, Tatem AJ. Investigating the endemic transmission of the hepatitis C virus. Int J Parasitol. 2007;37:839–849. doi: 10.1016/j.ijpara.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet. 2009;10:540–550. doi: 10.1038/nrg2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi H, Waked I, Sarrazin C, Myers RP, Idilman R, Calinas F, Vogel W, Mendes Correa MC, Hézode C, et al. The present and future disease burden of hepatitis C virus (HCV) infection with today’s treatment paradigm. J Viral Hepat. 2014;21(Suppl 1):S34–S59. doi: 10.1111/jvh.12248. [DOI] [PubMed] [Google Scholar]
- Simmonds P. Genetic diversity and evolution of hepatitis C virus - 15 years on. J Gen Virol. 2004;85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- Smith DB, Pathirana S, Davidson F, Lawlor E, Power J, Yap PL, Simmonds P. The origin of hepatitis C virus genotypes. J Gen Virol. 1997;78:321–328. doi: 10.1099/0022-1317-78-2-321. [DOI] [PubMed] [Google Scholar]
- Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. Expanded classification of hepatitis C Virus into 7 genotypes and 67 Subtypes: updated criteria and assignment web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel HJ, Collett MS, Gould EA, Heinz FX, Houghton M, Meyers G, Purcell RH, Rice CM. Virus Taxonomy. VIIIth Report of the ICTV. 2005:993–p998. [Google Scholar]
- Tippmann HF. Analysis for free: comparing programs for sequence analysis. Brief Bioinformat. 2004;5:82–87. doi: 10.1093/bib/5.1.82. [DOI] [PubMed] [Google Scholar]
- van Asten L, Verhaest I, Lamzira S, Hernandez-Aguado I, Zangerle R, Boufassa F, Rezza G, Broers B, Robertson JR, Brettle RP, McMenamin J, Prins M, Cochrane A, Simmonds P, Coutinho RA, Bruisten S. European and Italian seroconverter studies. Spread of hepatitis C virus among European injection drug users infected with HIV: a phylogenetic analysis. J Infect Dis. 2004;189:292–302. doi: 10.1086/380821. [DOI] [PubMed] [Google Scholar]
- Wang H, Barnes E, Yuan M, Li C, Fu Y, Xia X, Pybus O, Murphy D, Abe K, Lu L. Eight novel hepatitis C virus genomes reveal the changing taxonomic structure of genotype 6. J Gen Virol. 2013;94:76–80. doi: 10.1099/vir.0.047506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F, Chevallier M, Maynard M, Trepo C. Clinical consequences of hepatitis C virus infection. Rev Med Virol. 2003;13:57–68. doi: 10.1002/rmv.371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.