

Abstract Abstract
Pulmonary arterial hypertension (PAH) is a devastating vasculopathy that predominates in women and has been associated with dysregulated estrogen and serotonin signaling. Overexpression of the serotonin transporter (SERT+) in mice results in an estrogen-dependent development of pulmonary hypertension (PH). Estrogen metabolism by cytochrome P450 1B1 (CYP1B1) contributes to the pathogenesis of PAH, and serotonin can increase CYP1B1 expression in human pulmonary arterial smooth muscle cells (hPASMCs). We hypothesized that an increase in intracellular serotonin via increased SERT expression may dysregulate estrogen metabolism via CYP1B1 to facilitate PAH. Consistent with this hypothesis, we found elevated lung CYP1B1 protein expression in female SERT+ mice accompanied by PH, which was attenuated by the CYP1B1 inhibitor 2,3′,4,5′-tetramethoxystilbene (TMS). Lungs from female SERT+ mice demonstrated an increase in oxidative stress that was marked by the expression of 8-hydroxyguanosine; however, this was unaffected by CYP1B1 inhibition. SERT expression was increased in monocrotaline-induced PH in female rats; however, TMS did not reverse PH in monocrotaline-treated rats but prolonged survival. Stimulation of hPASMCs with the CYP1B1 metabolite 16α-hydroxyestrone increased cellular proliferation, which was attenuated by an inhibitor (MPP) of estrogen receptor alpha (ERα) and a specific ERα antibody. Thus, increased intracellular serotonin caused by increased SERT expression may contribute to PAH pathobiology by dysregulation of estrogen metabolic pathways via increased CYP1B1 activity. This promotes PASMC proliferation by the formation of pathogenic metabolites of estrogen that mediate their effects via ERα. Our studies indicate that targeting this pathway in PAH may provide a promising antiproliferative therapeutic strategy.
Keywords: pulmonary, hypertension, estrogen, cytochrome P450 1B1, serotonin
Pulmonary arterial hypertension (PAH) is a fatal cardiovascular disorder with a poorly understood etiology. It is characterized by obliteration of the small and medium pulmonary arteries, resulting in increased pulmonary pressures and subsequent enlargement and failure of the right ventricle. Female sex is possibly the strongest and most established risk factor for PAH, with more than 70% of patients diagnosed being female.1-3 Estrogen (17β-estradiol) is recognized as a pathogenic mediator in numerous diseases, through aberrant estrogen receptor (ER)-mediated signaling and/or metabolism.4 More recently, increasing evidence suggests that PAH may also be affected by abnormal activity of the estrogen pathway to adversely affect pulmonary vascular cell homeostasis.5-12 It was initially hypothesized that estrogens and/or their metabolites may promote disease development and progression. However, in the “classical” animal models of PAH (monocrotaline [MCT] and hypoxia), exogenous administration of estrogen was shown to have a beneficial effect by, in part, promoting cardiac function.13-16 This is in line with the extensive literature that reports cardioprotective effects of estrogens.17 More recently, we have provided compelling evidence that the endogenous synthesis of estrogen within the pulmonary arteries by the cytochrome P450 (CYP) enzyme aromatase promotes the development of pulmonary hypertension (PH) in female rodents.6,12
One criticism of utilizing the “classical” animal models of PAH to study the effects of sex is that they do not recreate the sex disparity that is observed in clinic. We have recently described several novel murine models of PH that are specific to female mice. These include SERT+ mice (mice that overexpress the serotonin transporter),13 mice that overexpress the S100A4 calcium-binding protein18 (which requires cooperation between the 5-hydroxytryptamine receptor and SERT19), and mice administered dexfenfluramine (an indirect serotinergic agonist).20 Ovariectomy was shown to reverse PH in these mice, confirming a role for sex hormones in these female-specific models.13,18,20 Of interest, these models all depend on upregulation of the serotinergic pathway, suggesting interplay of the serotonin and estrogen pathways in disease development.
The metabolic fate of estrogen is regulated by the activity of CYP enzymes to yield biologically active metabolites.21 We have previously shown that at least one CYP enzyme, CYP1B1, is highly expressed within the pulmonary arterial lesions of patients with PAH, while minimal expression is observed in patients without any reported evidence of PAH.5 CYP1B1 metabolizes estrogen primarily to the 4-hydroxylated estrogens and to a lesser extent to the 2- and 16α-hydroxylated estrogens (among others).22-24 CYP1B1 polymorphisms that result in a reduced 2-OHE1 (2-hydroxyestrone)/16α-OHE1 estrogen metabolite ratio have been reported in patients with PAH associated with mutations in the bone morphogenic protein receptor type II (BMPR2)7—the predominant genetic basis for the development of heritable PAH.25 Importantly, inhibition of estrogen metabolism by CYP1B1 can attenuate the development of experimental PH induced by hypoxia, SU5416 plus hypoxia,5 or dexfenfluramine.20
Pulmonary arterial smooth muscle cell (PASMC) proliferation is an established and dominant feature associated with PAH pathobiology.26,27 Estrogen induces proliferation of PASMCs, and this is attenuated by CYP1B1 inhibition.5 CYP1B1 expression is increased in PASMCs isolated from patients with idiopathic PAH and in female SERT+ mice,28 and serotonin can increase the expression of CYP1B1 in healthy PASMCs.28 We therefore hypothesized that CYP1B1 may play a causative role in the development of PH in female SERT+ mice by promoting pathologic estrogen metabolism. To test this hypothesis, we assessed the therapeutic potential of CYP1B1 inhibition in female SERT+ mice. We also tested this in the MCT model of PAH, which has elevated SERT expression,29 and examined the molecular mechanisms that may mediate the pathogenic signaling associated with elevated CYP1B1.
Methods
Ethics
All experimental procedures were carried out in accordance with the United Kingdom Animal Procedures Act (1986) and with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication 85-23, revised 1996). Rodents were housed in a 12-hour light-dark cycle with access to food and water ad libitum. Experimental procedures using human PASMCs (hPASMCs) conform to the principles outlined in the Declaration of Helsinki. All animals were randomly allocated to groups, and all measurements and analysis were done in a blinded fashion, where possible.
Animal models and in vivo CYP1B1 inhibition with 2,3′,4,5′-tetramethoxystilbene
Female mice 5–6 months old ubiquitously overexpressing the human SERT gene (SERT+ mice) were generated as previously described on a C57Bl/6J CBA background.30 These were a kind gift from the late Professor Tony Harmar (University of Edinburgh). Age-matched littermates (hereafter referred to as wild-type) were studied as controls. Mice were subdivided into groups to receive either the highly potent and selective CYP1B1 inhibitor 2,3′,4,5′-tetramethoxystilbene (TMS; 1.5 mg/kg/day by intraperitoneal [i.p.] injection; Tocris Biosciences, Bristol, UK) or vehicle (∼5% [v/v] ethanol in 0.9% saline) for 14 days before assessment of hemodynamic parameters.
Male and female Wistar rats (Harlan Laboratories, Bicester, UK), 10–12 weeks old at euthanasia received either the pneumotoxin MCT pyrrole (Sigma-Aldrich, Gillingham, UK) or its vehicle at a dose of 60 mg/kg by subcutaneous injection in the flank at day 0. MCT was dissolved in 1 mol/L hydrogen chloride (HCl) at a concentration of 100 mg/mL, neutralized with 1 mol/L sodium hydroxide (NaOH), and diluted with sterile distilled water to 6 mg/mL. All animals were weighed every day and monitored for any signs of discomfort. A single injection of MCT is sufficient to induce a PH phenotype in rats (MCT does not induce PH in mice). MCT mediates damage to the pulmonary endothelium and promotes the extension of smooth muscle cells into normally nonmuscular pulmonary arteries, promoting PH and right ventricular (RV) heart failure.31 Fourteen days after MCT injection, animals were administered the CYP1B1 inhibitor TMS, at 3 mg/kg/day, or its vehicle (∼5% [v/v] ethanol in 0.9% saline) by i.p. injection for an additional 14 days before hemodynamic analysis.
In vivo hemodynamic measurements
Rats and mice were anesthetized with isoflurane supplemented with a constant flow of oxygen. RV systolic pressures (RVSPs) were obtained by transdiaphragmatic puncture of the right ventricle with a saline-filled 25-gauge needle (in mice) and by jugular vein cannulation of the right ventricle with a fluid-filled catheter (in rats). To monitor off-target effects on the systemic vasculature, mean systemic arterial pressures (SAPs) were monitored by microcannulation of the left common carotid artery.
RV hypertrophy
RV hypertrophy was assessed by the Fulton index32 by calculating the ratio of the dry weight of the right ventricle to the dry weight of the left ventricle plus septum.
Lung histopathology
The inferior lobe of the lung was fixed in 10% neutral buffered formalin for a minimum of 4 hours with gentle agitation before embedding in paraffin. Sections (3-μm) were cut and stained with elastin and counterstained with Picro Sirius Red for identification of vascular thickening in the small arteries with an internal diameter up to 80 μm. This was characterized by an increase in the vessel wall diameter in more than 50% of the arterial wall. The total number of remodeled vessels was expressed as a ratio of the total number of vessels present within a lung section.
CYP1B1 immunolocalization
CYP1B1 immunolocalization was determined in mouse, rat, and human pulmonary arteries by immunohistochemistry, as described previously.5 Briefly, 3-μm sections were deparaffinized and rehydrated through a xylene-ethanol-to-water gradient. After antigen retrieval with citric acid buffer, endogenous peroxidase and biotin activity was blocked, followed by a 1-hour incubation with 2.5% normal horse serum to block nonspecific binding. Sections were incubated overnight at 4°C with an antibody against CYP1B1 (0.6 μg/mL, ab32649, Abcam, Cambridge, UK), proliferating cell nuclear antigen (PCNA; 0.2 μg/mL, ab2426, Abcam), SERT (1∶200, whole antiserum, ab44520, Abcam) or α-smooth muscle cell actin (α-SMA; 0.4 μg/mL, ab5694, Abcam). Excess primary antibody was removed by 3 consecutive washes in Tris-buffered saline with Tween 20 (TBS-T) and then incubated with a secondary anti-rabbit antibody for 1 hour at room temperature. Cells positive for CYP1B1 were visualized by VIP peroxidase (which stains pink; Vector Laboratories, Peterborough, UK) in mice and diaminobenzidine (DAB, which stains dark brown; Vector Laboratories) in rats and humans. Cells positive for PCNA, SERT, and α-SMA were visualized by DAB. Rat sections were counterstained with hematoxylin. Images were captured with a Zeiss Axio Imager M1. For all antibodies, an immunoglobulin G control was performed at the same concentration to control for nonspecific binding.
PCNA quantification
The percentage of PCNA-expressing nuclei was calculated with a free web application (ImmunoRatio) used for automated analysis of immunostained tissue sections. The application segments DAB-stained and hematoxylin-stained nuclei regions from the user-submitted image and calculates the percentage of DAB-stained nuclear area in the total nuclear area. A total of 3 or 4 pulmonary arteries were analyzed per lung section.
Determination of reactive oxygen species in lung sections by immunofluorescence
Immunohistochemistry of the reactive oxygen species (ROS) marker 8-hydroxyguanosine (8-OHG) was determined in the lungs of wild-type, SERT+, and TMS-treated SERT+ mice. Sections were deparaffinized in a xylene-ethanol-to-water gradient. Antigen retrieval was performed by boiling slides in ethylenediaminetetraacetic acid (pH 8.0) for 15 minutes before a 1-hour incubation in a humidified chamber at room temperature with 10% donkey serum/1% bovine serum albumin in 1× TBS-T to block nonspecific binding. Sections were incubated with an antibody against 8-OHG overnight at 4°C (5 μg/mL, ab10802, Abcam). For identification of 8-OHG-positive cells, sections were incubated with Alexa-fluor-488-conjugated donkey anti-goat secondary antibody (A-11055, Molecular Probes, Life Technologies, Warrington, UK) for 45 minutes at room temperature in the dark. Lipofuscin-mediated autofluorescence was removed with 0.1% Sudan Black B (Sigma-Aldrich) in methanol w/v for 10 minutes. Slides were mounted with ProLong Gold antifade mounting media containing DAPI (4′,6-diamidino-2-phenylindole; P-36931, Molecular Probes, Life Technologies). Fluorescence imaging was measured in an Axiovert 200M microscope with a laser-scanning module LSM 510 (Carl Zeiss, Jena, Germany). DAPI was excited at 405 nm and Alexa-fluor 488 at 488 nm. Images were captured with the LSM 510 evaluation software physiology (Zeiss, Cambridge, UK). Mean fluorescence intensity of 8-OHG was measured on at least 5 different calibrated images of lung sections of the different groups at ×40 magnification with ImageJ software. An auto threshold for each image was applied by using the Huang method in Image J.
Protein analysis
Whole-lung samples were homogenized and lysed in ice-cold 0.1% lauryl maltoside solution in phosphate-buffered saline (PBS; v/v), and protein concentrations were measured by a bicinchoninic acid (BCA) assay (Thermo Scientific, Cramlington, UK). Protein (30 μg) was loaded for whole-lung homogenates, and samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Protein expression of CYP1B1 (2 μg/mL; sc-32882, Santa Cruz Biotechnology, Dallas) and SERT expression (2 μg/mL; AB1594P, Millipore, Livingston, UK) was determined by overnight incubation at 4°C and normalized to GAPDH (0.2 μg/mL; ab8264, Abcam).
Estrogen immunoassay
The levels of estrogen were determined by enzyme-linked immunosorbent assay (ELISA) in whole-lung homogenate samples from female wild-type and SERT+ mice and SERT+ mice treated with TMS. Lung samples were homogenized in ice-cold 1% lauryl maltoside solution in PBS (v/v) and solubilized on ice; cellular fractionation followed. Protein concentrations were determined by a BCA assay. Protein (400 μg) was loaded for mouse lung samples and assayed in duplicate as per manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI). The plate was read at a wavelength of 405 nm for end point measurements (SpectraMax M2 plate reader, Molecular Devices, Sunnyvale, CA). Data were normalized to per microgram of protein.
Human PASMCs
Human PASMCs (hPASMCs) were provided by Professor Nicholas W. Morrell (University of Cambridge). Female hPASMCs were explanted from the distal pulmonary microvasculature from subjects with no reported presence of PAH. Three independent female cell lines were used in this study and characterized by morphological assessment and staining for the smooth muscle cell marker α-SMA. Assays were performed between passages 2 and 7. Cells were seeded in 24-well plates (cell counts) and 96-well plates (BrdU [bromodeoxyuridine] incorporation) at a density of 7,500 cells/cm2. Cells were grown to ∼60% confluency and then cell-cycle synchronized by serum deprivation (0.2% charcoal-stripped fetal bovine serum [FBS]) in phenol red–free Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Loughborough, UK) for 24 hours.
Charcoal-stripped FBS
FBS (Sera Labs, Haywards Heath, UK) was charcoal stripped twice to remove estrogens. Dextran-coated charcoal (Sigma-Aldrich) was added to FBS at a concentration of 1 g/100 mL and left overnight at 4ºC under gentle agitation. Samples were centrifuged at 1,811g at 4ºC for 30 minutes. The stripped serum was decanted and filtered through a 0.22-μm filter. ELISA analysis of 50 μL of FBS confirmed successful removal of estrogen (as described above).
Cell proliferation assays
Proliferation of hPASMCs was assessed by two approaches: live cell counts (cell viability) with a hemocytometer and a BrdU incorporation assay, as per manufacturers protocol (Millipore). Cells were treated for 72 hours, in phenol red–free 2% charcoal-stripped FBS DMEM with 1 nM 16α-OHE1 (Steraloids, Newport, RI) and 0.1 μM MPP dihydrochloride (estrogen receptor alpha [ERα] antagonist; Tocris) or 0.002 pg/mL ERα antibody (sc-7207, Santa Cruz Biotechnology). Antagonists/antibodies were added 30 minutes before the addition of 16α-OHE1. Cell culture media and drugs were replaced after 48 hours, and PASMC proliferation was assessed at 72 hours.
Statistics
Data are represented as the group mean ± SEM. Data were analyzed by either a Student 2-tailed t test or a 1-way analysis of variance followed by a Tukey post hoc test. The statistical test used is indicated in the figure legends.
Results
Female SERT+ mice develop PH via CYP1B1
Female mice overexpressing the human SERT gene develop a spontaneous PH phenotype that is dependent on estrogen.13 As serotonin can modulate CYP1B1 expression28 and CYP1B1 contributes to the development of PH,5 we investigated whether the development of PH in these mice was related to increased CYP1B1 activity. Overexpression of SERT resulted in elevated RVSPs (reflective of PH) compared to those in wild-type mice, and CYP1B1 inhibition with the highly potent and selective inhibitor TMS attenuated this effect (Fig. 1A). As previously reported,13 female SERT+ mice did not develop RV hypertrophy, despite elevated RVSPs. This is consistent with observations in numerous transgenic models of PH in normoxic conditions,12,18,33-35 and no further effects were observed in mice treated with TMS (Fig. 1B). Overexpression of SERT and CYP1B1 inhibition had no effect on mean SAPs (Fig. S1). SERT+ mice had lower heart rates than wild-type mice, and this was normalized by CYP1B1 inhibition (Fig. 1C). SERT+ mice had a significant degree of pulmonary vascular remodeling within the distal vasculature (where arteries are normally composed of a thin vascular layer), and this was attenuated by CYP1B1 inhibition (Fig. 1D).
Figure 1.
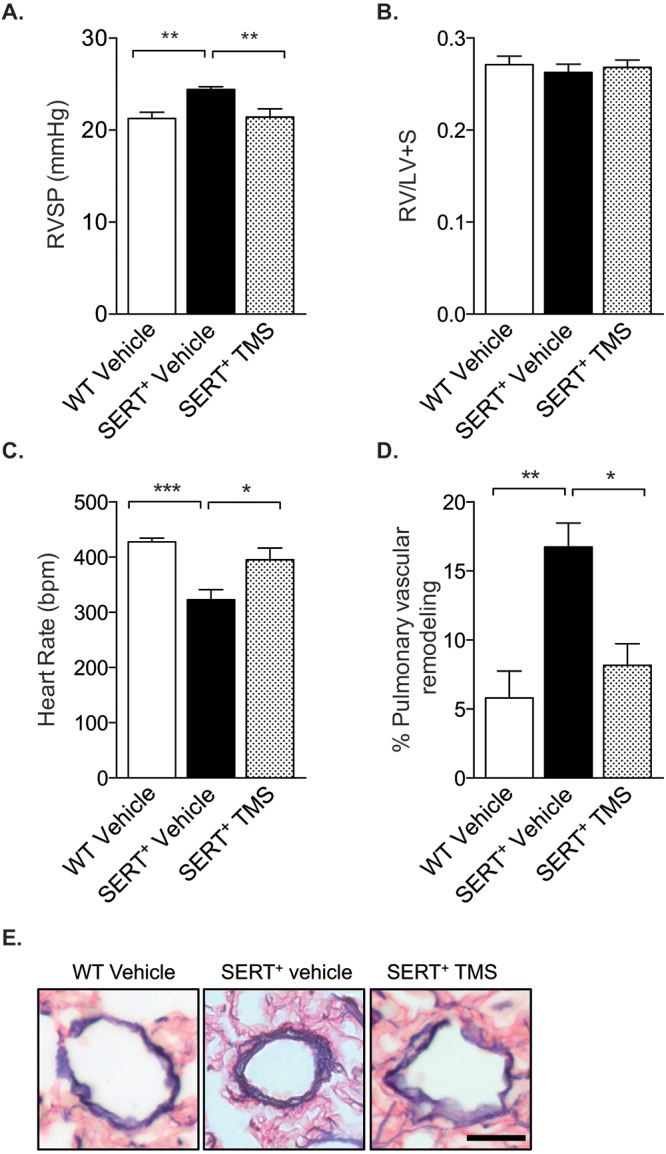
Inhibition of cytochrome P450 1B1 (CYP1B1) with 2,3′,4,5′-tetramethoxystilbene (TMS) attenuates PH in female SERT+ mice. Female SERT+ mice were injected with 1.5 mg/kg TMS by intraperitoneal injection for 2 weeks and compared with wild-type (WT; C57Bl/6J CBA) and SERT+ vehicle-treated mice for its effects on PH indices. A, Right ventricular systolic pressure (RVSP) measurement per group, as measured by transdiaphragmatic puncture; n = 9–10. B, Right ventricular hypertrophy expressed as the ratio of the weight of the right ventricle (RV) to the combined weight of the left ventricle and septum (LV+S); n = 10–11. C, Heart rate; n = 7–9. D, Pulmonary vascular remodeling; n = 5–6. E, Representative images of pulmonary vascular remodeling in arteries for each group. Scale bar: 20 μm. Data were analyzed by a 1-way analysis of variance followed by a Tukey post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001. SERT+: serotonin transporter is overexpressed.
CYP1B1 expression was increased in SERT+ mice, and CYP1B1 inhibition increased estrogen levels in the lung
Overexpression of the human SERT gene in mice increased CYP1B1 protein expression in whole-lung homogenates compared with that in wild-type mice. CYP1B1 expression in SERT+ TMS-treated mice was not significantly different from that in wild-type mice, suggesting that TMS reduced CYP1B1 expression. These findings were confirmed by immunohistochemistry staining for CYP1B1 (Fig. 2A, 2B).
Figure 2.
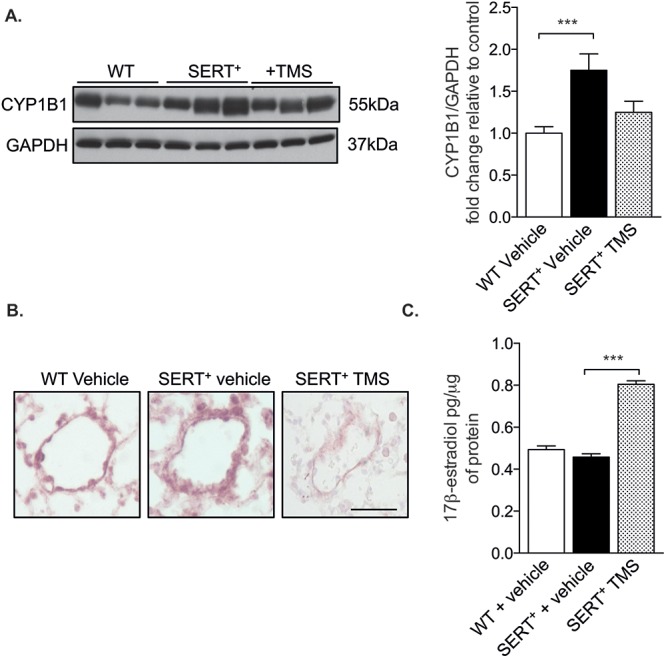
Cytochrome P450 1B1 (CYP1B1) expression is increased in female SERT+ mice, and CYP1B1 inhibition increases estrogen concentrations in lung homogenates. Female SERT+ mice were injected with 1.5 mg/kg 2,3′,4,5′-tetramethoxystilbene (TMS) by intraperitoneal injection for 2 weeks and compared with wild-type (WT; C57Bl/6J CBA) and SERT+ vehicle-treated mice. A, Whole-lung homogenates (30 μg) were analyzed for CYP1B1 expression by Western blotting and normalized to GAPDH; n = 5–8. B, Representative CYP1B1 immunolocalization in pulmonary arteries, as determined by VIP peroxidase–positive cells (pink). Scale bar: 20 μm. C, Estrogen (17β-estradiol) concentrations as determined by enzyme-linked immunosorbent assay in 400 μg of whole-lung homogenates; n = 4–5. Data were analyzed by a 1-way analysis of variance followed by a Tukey post hoc test. ***P < 0.001. SERT+: serotonin transporter is overexpressed.
A recent estrogen metabolic analysis of Cyp1b1-knockout mice reported increased lung concentrations of estrogen in Cyp1b1-knockout mice compared with wild-type mice.36 CYP enzymes regulate estrogen metabolism and it is therefore plausible that the increase in estrogen concentrations in these mice is attributed to reduced metabolism by CYP1B1. It was therefore of interest to assess estrogen concentrations in the lungs of SERT+ mice to determine the effect of SERT overexpression and CYP1B1 inhibition. Whole-lung estrogen concentrations were determined by ELISA and found to be similar in SERT+ mice and wild-type mice. However, CYP1B1 inhibition in SERT+ mice increased estrogen concentrations (Fig. 2C).
Oxidative stress is increased in SERT+ lungs
The formation of ROS by the metabolism of estrogen by CYP1B1 has been previously described,37 and increased oxidative stress may be associated with the pathobiology of PAH.38 We therefore investigated whether oxidative stress was regulated by CYP1B1 activity in our model by immunohistochemical analysis of 8-OHG (a sensitive measure of ROS). Female SERT+ mice had increased levels of 8-OHG compared to wild-type mice, and CYP1B1 inhibition with TMS had no effect on 8-OHG levels (Fig. 3). There was expression of 8-OHG within numerous cellular compartments in SERT+ mice treated with vehicle and TMS, including alveoli, bronchi, bronchioles, and vessels, whereas expression was visually less in control mice (Fig. 3). And 8-OHG was observed in both the cytoplasm and the nucleus, indicating increased RNA and DNA oxidation.
Figure 3.
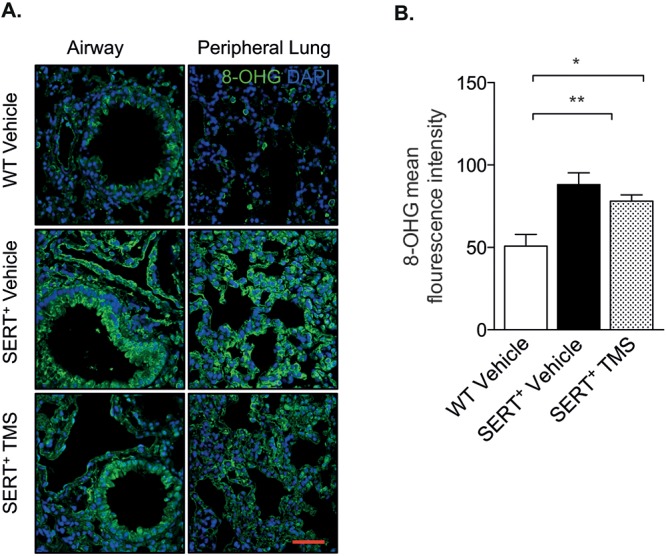
SERT+ mice have increased RNA and DNA oxidative damage in the lungs. A, Lung sections obtained from wild-type (WT) and SERT+ mice and SERT+ mice treated with the cytochrome P450 1B1 (CYP1B1) inhibitor 2,3′,4,5′-tetramethoxystilbene (TMS) were immunostained for the oxidative-stress marker 8-hydroxyguanosine (8-OHG). Sections were counterstained with the nuclear stain DAPI (4′,6-diamidino-2-phenylindole). Scale bar: 50 μm. B, 8-OHG mean fluorescence intensity was measured in whole-lung sections with ImageJ software; n = 4–5. Data were analyzed by a 1-way analysis of variance followed by a Tukey post hoc test. *P < 0.05; **P < 0.01. SERT+: serotonin transporter is overexpressed.
CYP1B1 inhibition did not improve RV function in MCT-induced PH but prolonged survival
Previous studies have reported increased SERT expression in the MCT model of PH.29 Because the overexpression of human SERT increased CYP1B1 expression in our mouse model, we rationalized that CYP1B1 expression would also be increased by MCT. Whole-lung expression of SERT protein was increased in female but not in male rats (Fig. 4A). However, CYP1B1 protein expression (Fig. 4B) was unchanged in the lungs from both male and female rats that had been treated with MCT for 28 days (all tissue examined was taken from animals that survived until day 28). MCT induced significant increases in RVSP (Fig. 5A) and RV hypertrophy (Fig. 5B). TMS had no effects on RVSP (Fig. 5A) and only moderate, yet significant, effects in reducing RV hypertrophy in male rats, while no effects were observed in female rats (Fig. 5B). Male rats developed more severe RV hypertrophy than female rats in response to MCT, despite developing a similar degree of increased RVSP (Fig. 5A, 5B). MCT and TMS treatment had no effects on mean SAPs in female and male rats (Fig. S2). MCT lowered the heart rate in male rats, and this was normalized by TMS to pressures similar to those in vehicle-treated rats. No changes in heart rate were observed in female rats (Fig. 5C). Mortalities were reported 23 and 26 days after MCT administration in male and female rats, respectively. TMS prolonged survival in male rats, and no mortalities were reported in female rats treated with TMS 2 weeks after administration of MCT (Fig. 5D). MCT induced a significant degree of pulmonary vascular remodeling in the distal vasculature, and this was not affected by CYP1B1 inhibition (Fig. 6A, 6B). CYP1B1 immunopositive cells were predominantly observed in pulmonary arterial endothelial cells (Fig. 6C).
Figure 4.
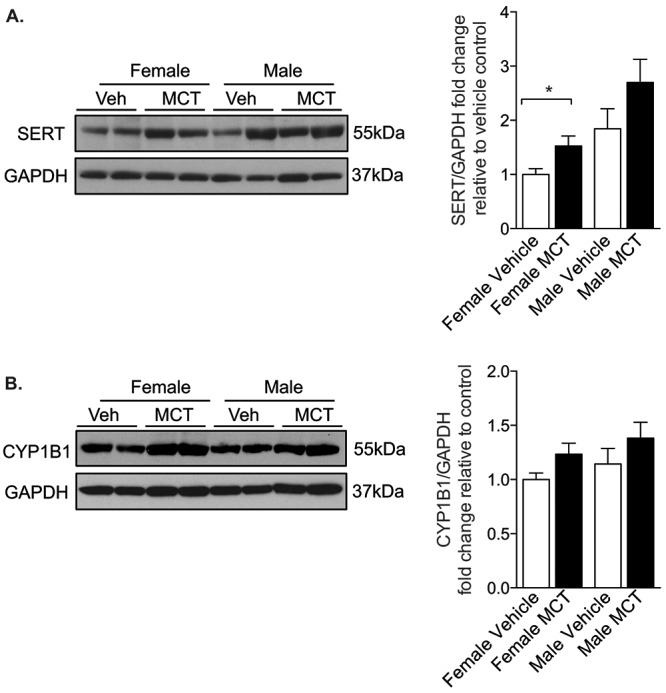
Serotonin transporter (SERT) and cytochrome P450 1B1 (CYP1B1) expression in monocrotaline (MCT)-treated rats. Female and male rats were given a single dose of monocrotaline (MCT; 60 mg/kg subcutaneously), and 28 days later whole-lung homogenates (30 μg) were analyzed for protein expression by Western blotting for SERT, n = 6 (A), and CYP1B1, n = 8 (B). Data were normalized to GAPDH. Data were analyzed by an unpaired t test. *P < 0.05. Veh: vehicle.
Figure 5.
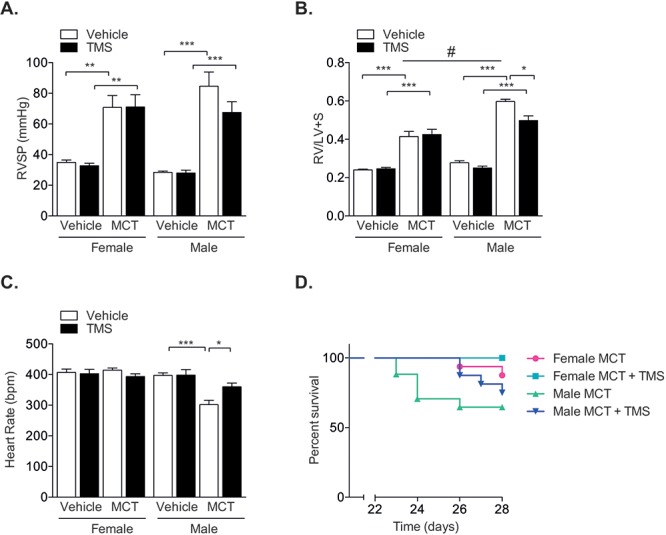
Inhibition of cytochrome P450 1B1 (CYP1B1) with 2,3′,4,5′-tetramethoxystilbene (TMS) does not reverse monocrotaline (MCT)-induced PH yet improves survival. Female and male rats were given a single dose of MCT (60 mg/kg subcutaneously) and 14 days later were given TMS (3 mg/kg/day intraperitoneally) or vehicle (∼5% ethanol in saline) for an additional 14 days and then assessed for indices of PH. A, Right ventricular systolic pressures (RVSP) as assessed by jugular vein cannulation; n = 5–11. B, Right ventricular hypertrophy as assessed by ratio of the weight of the right ventricle (RV) to the combined weight of the left ventricle and septum (LV+S); n = 9–14. C, Heart rate; n = 6–12. D, Kaplan-Meier survival analysis in rats treated with MCT (total of 16 animals per group at the start of the experiment). Data were analyzed by a 1-way analysis of variance followed by a Tukey post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001, vehicle versus MCT; #P < 0.05, male versus female.
Figure 6.
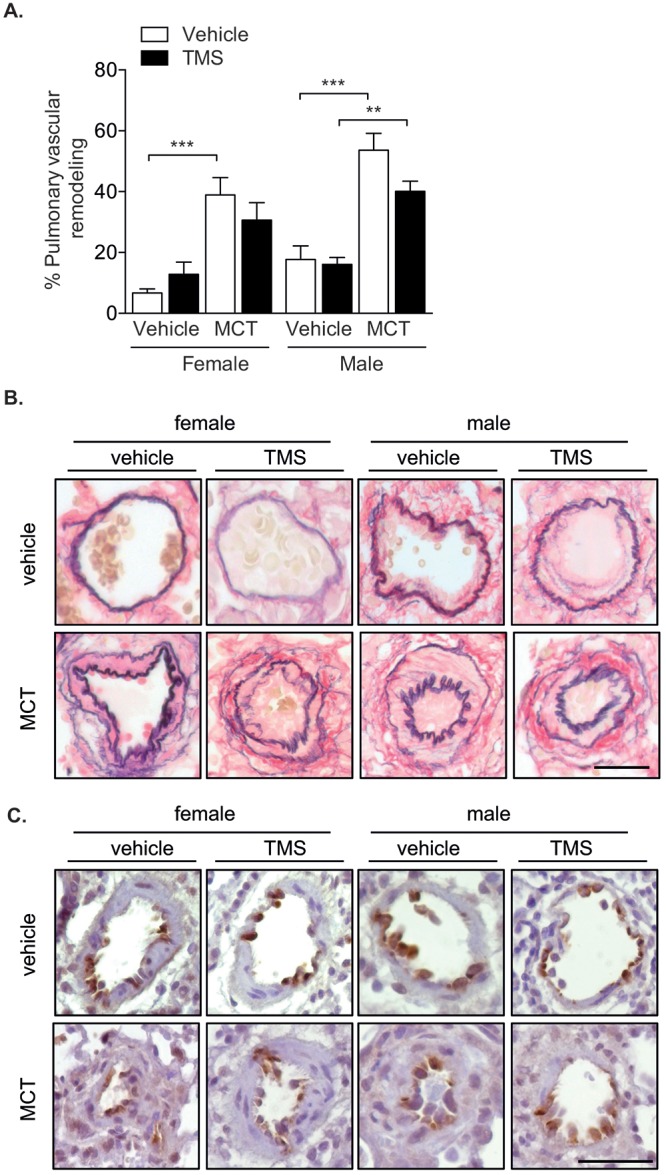
Inhibition of cytochrome P450 1B1 (CYP1B1) with 2,3′,4,5′-tetramethoxystilbene (TMS) does not reverse monocrotaline (MCT)-induced pulmonary arterial remodeling. Female and male rats were given a single dose of MCT (60 mg/kg subcutaneously) and 14 days later were given TMS (3 mg/kg/day intraperitoneally) or vehicle (∼5% ethanol in saline) for an additional 14 days and then assessed for indices of PH. A, Pulmonary vascular remodeling; n = 6–7. Data were analyzed by a 1-way analysis of variance followed by a Tukey post hoc test. **P < 0.01; ***P < 0.001. B, Representative pulmonary arteries for each group. Scale bar: 20 μm. C, CYP1B1 immunolocalization in representative pulmonary arteries indicates positive cells in the endothelial cell layer and periadventitial layer, as visualized by diaminobenzidine (dark brown). Sections were counterstained with hematoxylin. Scale bar: 50 μm.
The CYP1B1 metabolite 16α-OHE1 stimulates PASMC proliferation via ERα
PASMC proliferation is an established and dominant feature associated with PAH pathobiology,26,27 and we have previously shown that estrogen induces hPASMC proliferation via CYP1B1.5 Using immunohistochemistry, we determined that both SERT and CYP1B1 are highly expressed in α-SMA-positive cells (PASMCs) in pulmonary arteries from patients with PAH, whereas minimal expression is observed in non-PAH arteries (Fig. 7). SERT overexpression in mice increased the expression of CYP1B1, suggesting that the PH phenotype observed in these mice may be mediated by increased cellular proliferation. To investigate this, we performed immunohistochemistry analysis of the DNA replication marker PCNA. PCNA expression was increased in SERT+ mice, and this was attenuated by CYP1B1 inhibition (Fig. 8A, 8B). We next investigated whether the proliferative CYP1B1 estrogen metabolite 16α-OHE1 promoted proliferation via the estrogen receptor ERα by selectively blocking ERα with a specific antagonist and antibody. The 16α-OHE1 induced significant increases in PASMC proliferation, and this was completely attenuated by blocking of ERα signaling by both a specific antagonist and an antibody, as assessed by cell counts (Fig. 8C, 8E) and BrdU incorporation (Fig. 8D, 8F). To prove that these effects were directly related to inhibition of 16α-OHE1 signaling through ERα and not by indirect effects mediated by the antagonist or antibody, we stimulated hPASMCs with each treatment alone and observed no effects (data not shown).
Figure 7.
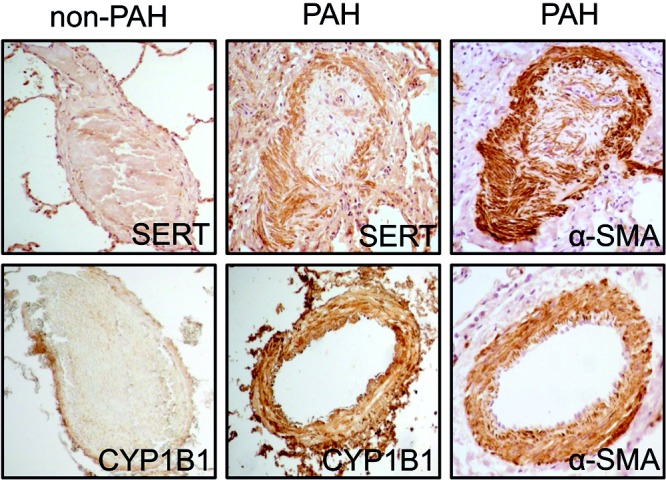
Serotonin transporter (SERT) and cytochrome P450 1B1 (CYP1B1) are expressed in α-smooth muscle actin (α-SMA)-positive cells in human pulmonary arteries. Immunolocalization of SERT, CYP1B1 and α-SMA in human pulmonary arteries of non-PAH and PAH lung sections as visualized by diaminobenzidine (dark brown). PAH lung samples were from a female aged 26 years with heritable PAH (top) and a female aged 51 years with idiopathic PAH (bottom). Scale bar: 200 μm. PAH: pulmonary arterial hypertension.
Figure 8.
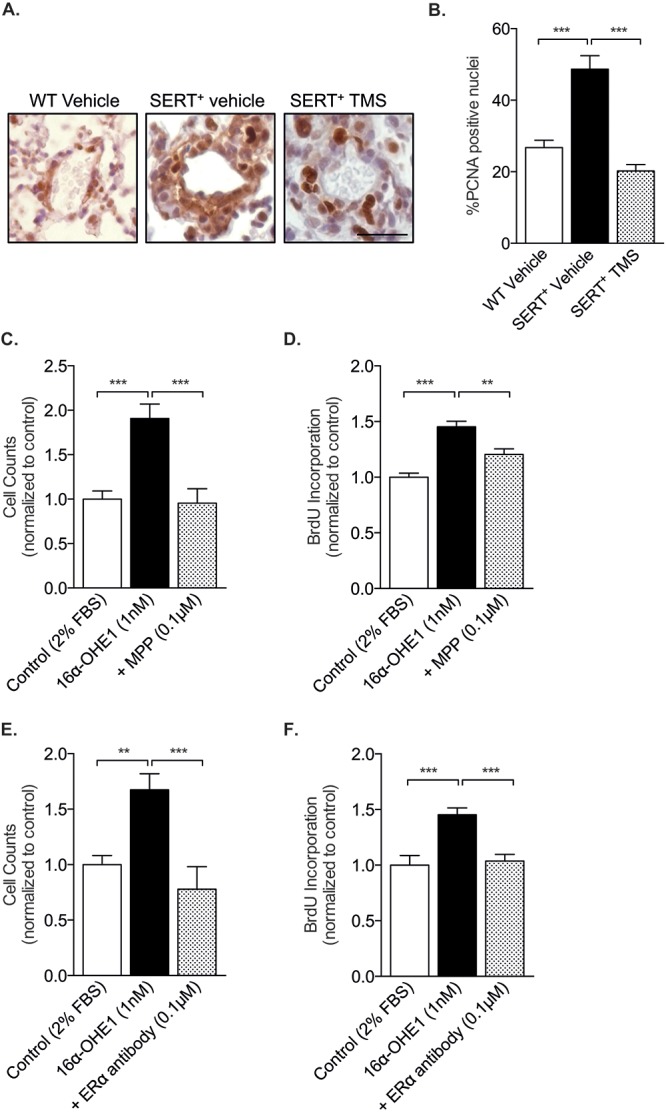
The cytochrome P450 1B1 (CYP1B1) metabolite 16α-hydroxyestrone (16α-OHE1) induces cell proliferation via estrogen receptor α (ERα). A, Representative images of immunohistochemistry analysis of the DNA replication marker PCNA in wild-type (WT) mice and in SERT+ mice treated with vehicle or the CYP1B1 inhibitor 2,3′,4,5′-tetramethoxystilbene (TMS). Scale bar: 20 μm. B, Percentage PCNA-positive nuclei; n = 3–6, 3 or 4 pulmonary arteries analyzed per lung. Data were analyzed by a 1-way analysis of variance (ANOVA) followed by a Tukey post hoc test. C, D, Female human pulmonary arterial smooth muscle cells (PASMCs) were incubated with 16α-OHE1 for 72 hours in 2% charcoal-stripped serum in the presence or absence of ERα inhibition and assessed for proliferation by cell counts and BrdU incorporation. 16α-OHE1 induced PASMC proliferation in the presence of a selective ERα antagonist MPP dihydrochloride (0.1 μM), as assessed by cell counts, n = 15 replicates per group (C), and BrdU incorporation, n = 11 replicates per group (D). E, F, 16α-OHE1 induced PASMC proliferation in the presence of a specific ERα antibody (0.002 pg/mL) as assessed by cell counts, n = 6–9 replicates per group (E), and BrdU incorporation, n = 16 replicates per group (F). Data were analyzed by a 1-way ANOVA followed by a Tukey post hoc test. **P < 0.01; ***P < 0.001. BrdU: bromodeoxyuridine; FBS: fetal bovine serum; PCNA: proliferating cell nuclear antigen; SERT+: serotonin transporter is overexpressed.
Discussion
The incidence of PAH is more common in women than in men.1-3 For example, in the largest PAH registry in the world, the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL), more than 80% of patients diagnosed were females.3 This suggests that estrogen and/or its metabolites may be associated with the pathobiology of PAH. Understanding the biological basis of this sex disparity may offer a new treatment paradigm in this devastating cardiovascular disorder that currently has a high unmet clinical need.
We have previously shown that serotonin can upregulate the expression of CYP1B1 in hPASMCs.13 Accordingly, female mice that overexpress the human SERT gene have increased CYP1B1 expression, and inhibition of CYP1B1 reduces CYP1B1 expression and can attenuate PH in these mice. This is consistent with the literature, where TMS has been shown to inhibit CYP1B1 messenger RNA and protein expression in a concentration-dependent manner.39 Of note, female SERT+ mice do not develop RV hypertrophy in the face of increased RVSP and muscularization of the pulmonary arteries. This is consistent with numerous transgenic models where mice develop a spontaneous PH phenotype in normoxic conditions.12,18,33-35 Although the reasons for this observation in the PH field remains undetermined, we speculate that this may be related to an inability of mice to develop compensatory RV hypertrophy in the face of moderate PH.
In the 1980s there was a PAH epidemic in women who had taken the anorexigens dexfenfluramine and aminorex.40,41 Both of these drugs are substrates for SERT and act as indirect serotinergic agonists.42 This outbreak formulated the “serotonin hypothesis of PAH” and suggested a direct interaction between SERT and the development of PAH. More recently, we have shown that administration of dexfenfluramine in mice results in the development of PH in female mice only and that this is dependent on intact ovaries and CYP1B1.20 These observations suggest that SERT facilitates the development of estrogen-related PH via CYP1B1. This may be relevant to human PAH, as CYP1B1 is highly upregulated in disease5 and polymorphisms of CYP1B1 confer increased susceptibility among patients with a BMPR2 mutation.7 In addition, we recently demonstrated that proliferation of female hPASMCs is increased via a reduction in BMPR2 signaling and that the CYP1B1 metabolite 4-hydroxyestradiol can modulate BMPR2 signaling in a sex-specific fashion.12
Overexpression of SERT increased CYP1B1 expression in vivo, and this was accompanied by increased expression of the proliferative marker PCNA. Inhibition of CYP1B1 in SERT+ mice reduced CYP1B1 and PCNA expression in the pulmonary arteries and the degree of pulmonary vascular remodeling. Estrogen promotes proliferation of hPASMCs,5,13 and CYP1B1 inhibition can attenuate this effect in a dose-dependent fashion.5 Proliferative screens of the most prominent CYP1B1 metabolites of estrogen revealed that the 16α-hydroxylated estrogens promote PASMC proliferation.5 Inhibition of estrogen synthesis and of its receptor ERα can attenuate experimental PH in female rodents.6,43 ERα is predominantly regarded as a nuclear receptor, but its expression may also be observed at the cell membrane. Upon ligand binding, the nuclear receptor dimerizes and translocates to the nucleus, where it acts as a regulator of gene transcription. Previous studies have shown that BMPR2-mutant pulmonary microvascular endothelial cells signal through noncanonical pathways by reduced trafficking of ERα to the cell nucleus.8 Furthermore, ERα can suppress the transcriptional activation of the BMPR2 gene by binding to a conserved estrogen response element within its promoter.8 Here, we show that ERα mediates the proliferative effects of the CYP1B1 metabolite 16α-OHE1. This is of interest because the concentrations of 16α-OHE1 are elevated in the urine of mice exposed to hypoxia,5 BMPR2-mutant mice,9 and importantly also in patients with PAH.7 We rationalized to focus our studies on ERα, given its more prominent role in the pathobiology of PAH.6,8,43,44 However, it should be noted that 16α-OHE1 has a high binding affinity for ERβ45 and may therefore also be signaling through this receptor.
The role for estrogen in the pathobiology of PAH has been met with much speculation and controversial data. Indeed, numerous preclinical studies have reported compelling evidence that estrogen mediates protective effects in the cardiopulmonary unit.13,14,46,47 Estrogen is a potent vasodilator in the pulmonary vascular bed,48 and physiological increases in circulating estrogens (the estrous cycle) can attenuate pulmonary arterial vasoconstriction under both normoxic and hypoxic conditions.49 On the contrary, endogenous synthesis of estrogen contributes to the development of PH and associated RV hypertrophy in intact female rodents.6 Moreover, there is a positive correlation between circulating estrogen levels and the degree of RV hypertrophy, suggesting an association between estrogens and disease severity.6 The estrogen-synthesizing enzyme aromatase is expressed within the smooth muscle cell layer of the pulmonary arterial wall,6 suggesting that estrogens can be produced locally. Local estrogens would be predicted to have a much more profound effect than the relatively low concentrations circulating in the bloodstream.17 Here, we show that CYP1B1 inhibition increases estrogen concentrations in the lung, which is consistent with a previous study in Cyp1b1-knockout mice.36 This suggests that the pathobiology behind estrogen-associated PAH may be, at least in part, mediated by its downstream metabolism.
Increasing evidence suggests that altered redox signaling may be associated with the pathobiology of PAH, and elevated ROS have been observed in numerous animal models and in patients with PAH.50 Estrogen metabolism by CYP1B1 to the 4-hydroxylated estrogens is associated with the formation of ROS, which can generate quinones and semiquinones that can mediate DNA damage by the formation of DNA adducts.51,52 We had therefore anticipated that CYP1B1 inhibition might alleviate oxidative stress. Although we observed increased ROS expression in SERT+ mouse lungs, this was unaffected by CYP1B1 inhibition, suggesting that other pathways unrelated to CYP1B1 activity contribute to the elevated oxidative stress in SERT+ mice.
The MCT rat model has been and continues to be used as a model of PH despite having been criticized as a preclinical model of PAH.53 However, previous studies have shown that MCT increases lung expression of SERT and that inhibition of SERT can prevent the development of MCT-induced PH.29 We therefore questioned whether this was related to elevated CYP1B1 activity and hypothesized that CYP1B1 inhibition may be effective in reversing MCT-induced PH. Although we observed an increase in SERT expression in female rats, this was not accompanied by an increase in CYP1B1 expression. This is likely due to the relatively moderate increase of SERT expression in comparison to the mouse model overexpressing SERT. The lack of increased CYP1B1 expression and the difference in cellular localization compared with mice and humans (endothelial vs. smooth muscle cell) may underlie the relatively mild therapeutic effects observed. In male rats, CYP1B1 inhibition reduced RV hypertrophy and improved survival. In female rats, no effect was reported on PH indices, yet a small improvement in survival was observed. Clinically, survival in males with PH is worse than that in females; this is thought to be related to a worse RV function,54 and indeed, RV function is a strong predictor of survival.55 In this study, the degree of RV hypertrophy was more profound in male rats treated with MCT than in females, and this was associated with poorer survival. MCT is bioactivated by CYPs to its active compound MCT pyrrole in the liver. Sex-specific CYPs may regulate MCT metabolism in the liver and underlie the sex difference in response to MCT. Of interest, the exogenous administration of estrogen has been shown to be therapeutic in MCT-treated rats, by activation of ERβ.16 This may explain the reduced degree of RV hypertrophy and the increased survival in female rats compared with male rats. However, we have recently reported pathogenic effects of endogenous estrogens in females in other models of PH.6 The influence of estrogens on RV function is still controversial in the literature. In healthy females receiving hormone replacement therapy, increased estrogen levels correlated with an improved RV function.56 However, recently it has been shown that men with idiopathic PAH have elevated estrogen levels and that this is correlated with poorer RV function and 6-minute walking test results.57
Serotonin and SERT are mediators in the pathogenesis of PAH. Overactivity of the estrogen and serotonergic pathways may be associated with the development of PAH, particularly in women, by increasing CYP1B1 expression promoting proproliferative estrogen metabolism. Targeting CYP1B1 is currently being explored therapeutically in clinical trials in cancer patients, using a vaccination with a DNA plasmid encoding an inactivated form of CYP1B1 (ZYC300). Upon delivery, the immune system elicits a cytotoxic T lymphocyte–mediated response against CYP1B1, resulting in the lysis of CYP1B1-expressing cells.58,59 This may be a strong therapeutic candidate in female patients who have elevated CYP1B1 expression. The subset of patients who would respond to this therapy could be identified by measuring the urinary 2-OHE1/16α-OHE1 ratio, as previously reported.7 Current PAH therapies do not encompass this well-defined sex disparity that occurs in PAH or the potential divergent response to current therapies that may be affected by sex. Future therapies that target dysregulated estrogen metabolism may have a favorable outcome in this highly neglected, devastating cardiovascular disease.
Acknowledgements
We thank Professor Nicholas W. Morrell for providing hPASMCs for these studies. The SERT+ mice were provided by the late Professor Tony Harmar.
Appendix. Supplementary figures
Figure S1.
Inhibition of cytochrome P450 1B1 (CYP1B1) with 2,3′,4,5′-tetramethoxystilbene (TMS) has no effects on mean systemic arterial pressures (SAPs). Female SERT+ mice were injected with 1.5 mg/kg TMS intraperitoneally for 2 weeks and compared with wild-type (WT; C57Bl/6J CBA) and SERT+ vehicle-treated mice. Mean SAPs were measured by cannulation of the carotid artery; n = 7–10. Data were analyzed by a 1-way analysis of variance followed by a Tukey post hoc test. SERT+: serotonin transporter is overexpressed.
Figure S2.
Inhibition of cytochrome P450 1B1 (CYP1B1) with 2,3′,4,5′-tetramethoxystilbene (TMS) and monocrotaline (MCT) has no effects on mean systemic arterial pressures (SAPs). Female and male rats were given a single dose of MCT (60 mg/kg subcutaneously) and 14 days later were given TMS (3 mg/kg/day intraperitoneally) or vehicle (∼5% ethanol in saline) for an additional 14 days. Mean SAPs were measured by cannulation of the carotid artery; n = 5–11. Data were analyzed by a 1-way analysis of variance followed by a Tukey post hoc test.
Source of Support: The British Heart Foundation.
Conflict of Interest: None declared.
Supplements
Appendix: Supplementary figuresPulmCirc-006-082.s001.pdf (620.5KB, pdf)
References
- 1.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186(8):790–796. [DOI] [PubMed]
- 2.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaïci A, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006;173(9):1023–1030. [DOI] [PubMed]
- 3.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL registry. Chest 2010;137(2):376–387. [DOI] [PubMed]
- 4.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest 2006;116(3):561–570. [DOI] [PMC free article] [PubMed]
- 5.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension/clinical perspective. Circulation 2012;126(9):1087–1098. [DOI] [PubMed]
- 6.Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, Fullerton J, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med 2014;190(4):456–467. [DOI] [PMC free article] [PubMed]
- 7.Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, Wheeler LA, Parl FF, Loyd JE, Phillips JA. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J 2009;34(5):1093–1099. [DOI] [PMC free article] [PubMed]
- 8.Austin E, Hamid R, Hemnes A, Loyd J, Blackwell T, Yu C, Phillips JA III, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ 2012;3:6. doi:10.1186/2042-6410-3-6. [DOI] [PMC free article] [PubMed]
- 9.Fessel JP, Chen X, Frump A, Gladson S, Blackwell T, Kang C, Johnson J, et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ 2013;3(3):564–577. [DOI] [PMC free article] [PubMed]
- 10.Austin ED, Lahm T, West J, Tofovic SP, Johansen AK, MacLean MR, Alzoubi A, Oka M. Gender, sex hormones and pulmonary hypertension. Pulm Circ 2013;3(2):294–314. [DOI] [PMC free article] [PubMed]
- 11.Mair KM, Johansen AK, Wright AF, Wallace E, MacLean MR. Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. Br J Pharmacol 2013;171(3):567–579. [DOI] [PMC free article] [PubMed]
- 12.Mair KM, Yang XD, Long L, White K, Wallace E, Ewart M-A, Docherty CK, Morrell NW, MacLean MR. Sex affects bone morphogenetic protein type II receptor signaling in pulmonary artery smooth muscle cells. Am J Respir Crit Care Med 2015;191(6):693–703. [DOI] [PMC free article] [PubMed]
- 13.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17β oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 2011;90(2):373–382. [DOI] [PubMed]
- 14.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, Justice MJ, et al. 17β-estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med 2012;185(9):965–980. [DOI] [PMC free article] [PubMed]
- 15.Xu DQ, Luo Y, Liu Y, Wang J, Zhang B, Xu M, Wang YX, et al. Beta-estradiol attenuates hypoxic pulmonary hypertension by stabilizing the expression of p27kip1 in rats. Respir Res 2010;11(1):182. doi:10.1186/1465-9921-11-182. [DOI] [PMC free article] [PubMed]
- 16.Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, van der Laarse A, Eghbali M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med 2011;184(6):715–723. [DOI] [PMC free article] [PubMed]
- 17.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med 1999;340(23):1801–1811. [DOI] [PubMed]
- 18.Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, Rabinovitch M, MacLean M. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir Res 2011;12:159. doi:10.1186/1465-9921-12-159. [DOI] [PMC free article] [PubMed]
- 19.Lawrie A, Spiekerkoetter E, Martinez EC, Ambartsumian N, Sheward WJ, MacLean MR, Harmar AJ, Schmidt A-M, Lukanidin E, Rabinovitch M. Interdependent serotonin transporter and receptor pathways regulate S100A4/Mts1, a gene associated with pulmonary vascular disease. Circ Res 2005;97(3):227–235. [DOI] [PubMed]
- 20.Dempsie Y, MacRitchie NA, White K, Morecroft I, Wright AF, Nilsen M, Loughlin L, Mair KM, MacLean MR. Dexfenfluramine and the oestrogen-metabolizing enzyme CYP1B1 in the development of pulmonary arterial hypertension. Cardiovasc Res 2013;99(1):24–34. [DOI] [PMC free article] [PubMed]
- 21.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett 2005;227(2):115–124. [DOI] [PubMed]
- 22.Badawi AF, Cavalieri EL, Rogan EG. Role of human cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16α-hydroxylation of 17β-estradiol. Metabolism 2001;50(9):1001–1003. [DOI] [PubMed]
- 23.Hanna IH, Dawling S, Roodi N, Guengerich FP, Parl FF. Cytochrome P450 1B1 (CYP1B1) pharmacogenetics: association of polymorphisms with functional differences in estrogen hydroxylation activity. Cancer Res 2000;60(13):3440–3444. [PubMed]
- 24.Lee AJ, Cai MX, Thomas PE, Conney AH, Zhu BT. Characterization of the oxidative metabolites of 17β-estradiol and estrone formed by 15 selectively expressed human cytochrome P450 isoforms. Endocrinology 2003;144(8):3382–3398. [DOI] [PubMed]
- 25.Ma L, Chung WK. The genetic basis of pulmonary arterial hypertension. Hum Genet 2014;133(5):471–479. [DOI] [PubMed]
- 26.Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 2010;121(18):2045–2066. [DOI] [PMC free article] [PubMed]
- 27.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 2011;8(8):443–455. [DOI] [PMC free article] [PubMed]
- 28.White K, Loughlin L, Maqbool Z, Nilsen M, McClure J, Dempsie Y, Baker AH, MacLean MR. Serotonin transporter, sex, and hypoxia: microarray analysis in the pulmonary arteries of mice identifies genes with relevance to human PAH. Physiol Genomics 2011;43(8):417–437. [DOI] [PMC free article] [PubMed]
- 29.Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, Rideau D, Hamon M, Adnot S, Eddahibi S. Serotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in rats. Circulation 2005;111(21):2812–2819. [DOI] [PubMed]
- 30.MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen S, Sheward J, Colston J, et al. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation 2004;109(17):2150–2155. [DOI] [PubMed]
- 31.Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol Heart Circ Physiol 1988;255(6):H1484–H1491. [DOI] [PubMed]
- 32.Fulton RM, Hutchinson EC, Jones AM. Ventricular weight in cardiac hypertrophy. Br Heart J 1952;14(3):413–420. [DOI] [PMC free article] [PubMed]
- 33.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 2013;123(8):3600–3613. [DOI] [PMC free article] [PubMed]
- 34.Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X, Rudarakanchana N, Southwood M, James V, Trembath RC, Morrell NW. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res 2006;98(6):818–827. [DOI] [PubMed]
- 35.Morecroft I, Dempsie Y, Bader M, Walther DJ, Kotnik K, Loughlin L, Nilsen M, MacLean MR. Effect of tryptophan hydroxylase 1 deficiency on the development of hypoxia-induced pulmonary hypertension. Hypertension 2007;49(1):232–236. [DOI] [PubMed]
- 36.Peng J, Xu X, Mace BE, Vanderveer LA, Workman LR, Slifker MJ, Sullivan PM, Veenstra TD, Clapper ML. Estrogen metabolism within the lung and its modulation by tobacco smoke. Carcinogenesis 2013;34(4):909–915. [DOI] [PMC free article] [PubMed]
- 37.Chen ZH, Hurh YJ, Na HK, Kim JH, Chun YJ, Kim DH, Kang KS, Cho MH, Surh YJ. Resveratrol inhibits TCDD-induced expression of CYP1A1 and CYP1B1 and catechol estrogen-mediated oxidative DNA damage in cultured human mammary epithelial cells. Carcinogenesis 2004;25(10):2005–2013. [DOI] [PubMed]
- 38.Wong CM, Bansal G, Pavlickova L, Marcocci L, Suzuki YJ. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal 2013;18(14):1789–1796. [DOI] [PMC free article] [PubMed]
- 39.Chun YJ, Lee SK, Kim MY. Modulation of human cytochrome P450 1B1 expression by 2,4,3′,5′-tetramethoxystilbene. Drug Metab Dispos 2005;33(12):1771–1776. [DOI] [PubMed]
- 40.Kramer MS, Lane DA. Aminorex, dexfenfluramine, and primary pulmonary hypertension. J Clin Epidemiol 1998;51(4):361–364. [DOI] [PubMed]
- 41.Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International primary pulmonary hypertension study group. N Engl J Med 1996;335(9):609–616. [DOI] [PubMed]
- 42.Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates: implications for primary pulmonary hypertension. Circulation 1999;100(8):869–875. [DOI] [PubMed]
- 43.Wright AF, Ewart M-A, Mair K, Nilsen M, Dempsie Y, Loughlin L, MacLean MR. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc Res 2015;106(2):206–216. [DOI] [PMC free article] [PubMed]
- 44.Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, Ahmad F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol 2010;298(4):H1235–H1248. [DOI] [PMC free article] [PubMed]
- 45.Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor α and β subtypes: insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006;147(9):4132–4150. [DOI] [PubMed]
- 46.Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Weil B, Meldrum DR. Exogenous estrogen rapidly attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction. Shock 2008;30(6):660–667. [DOI] [PubMed]
- 47.Yuan P, Wu WH, Gao L, Zheng ZQ, Liu D, Mei HY, Zhang ZL, Jing ZC. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, prostacyclin and endothelin-1 pathways. Eur Respir J 2013;41(5):1116–1125. [DOI] [PubMed]
- 48.English KM, Jones RD, Jones TH, Morice AH, Channer KS. Gender differences in the vasomotor effects of different steroid hormones in rat pulmonary and coronary arteries. Horm Metab Res 2001;33(11):645–652. [DOI] [PubMed]
- 49.Lahm T, Patel KM, Crisostomo PR, Markel TA, Wang M, Herring C, Meldrum DR. Endogenous estrogen attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction: the effects of sex and menstrual cycle. Am J Physiol Endocrinol Metab 2007;293(3):E865–E871. [DOI] [PubMed]
- 50.DeMarco VG, Whaley-Connell AT, Sowers JR, Habibi J, Dellsperger KC. Contribution of oxidative stress to pulmonary arterial hypertension. World J Cardiol 2010;2(10):316–324. [DOI] [PMC free article] [PubMed]
- 51.Nutter LM, Ngo EO, Abulhajj YJ. Characterization of DNA damage induced by 3,4-estrone-ortho-quinone in human-cells. J Biol Chem 1991;266(25):16380–16386. [PubMed]
- 52.Nutter LM, Wu YY, Ngo EO, Sierra EE, Gutierrez PL, Abulhajj YJ. An o-quinone form of estrogen produces free radicals in human breast cancer cells: correlation with DNA damage. Chem Res Toxicol 1994;7(1):23–28. [DOI] [PubMed]
- 53.Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 2012;302(4):L363–L369. [DOI] [PubMed]
- 54.Jacobs W, van de Veerdonk MlC, Trip P, de Man F, Heymans MW, Marcus JT, Kawut SM, Bogaard HJ, Boonstra A, Vonk Nordegraaf A. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014;145(6):1230–1236. [DOI] [PMC free article] [PubMed]
- 55.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991;115(5):343–349. [DOI] [PubMed]
- 56.Kawut SM, Al-Naamani N, Agerstrand C, Rosenzweig EB, Rowan C, Barst RJ, Bergmann S, Horn EM. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest 2009;135(3):752–759. [DOI] [PMC free article] [PubMed]
- 57.Corey EV, Greyson B, Barr RG, David B, Jason SF, Nicholas SH, James RK, et al. Sex hormones in men with pulmonary arterial hypertension: a case-control study using the multi-ethnic study of atherosclerosis. Am J Respir Crit Care Med 2015;191(Meeting Abstracts, C28):A4093.
- 58.Gribben JG, Ryan DP, Boyajian R, Urban RG, Hedley ML, Beach K, Nealon P, et al. Unexpected association between induction of immunity to the universal tumor antigen CYP1B1 and response to next therapy. Clin Cancer Res 2005;11(12):4430–4436. [DOI] [PubMed]
- 59.Luby TM. Targeting cytochrome P450 CYP1B1 with a therapeutic cancer vaccine. Expert Rev Vaccines 2008;7(7):995–1003. [DOI] [PubMed]