

Abstract
Saccharomyces cerevisiae is a robust host for heterologous protein expression. The efficient expression of cellulases in S. cerevisiae is important for the consolidated bioprocess that directly converts lignocellulose into valuable products. However, heterologous proteins are often N-hyperglycosylated in S. cerevisiae, which may affect protein activity. In this study, the expression of three heterologous proteins, β-glucosidase, endoglucanase and cellobiohydrolase, was found to be N-hyperglycosylated in S. cerevisiae. To block the formation of hypermannose glycan, these proteins were expressed in strains with deletions in key Golgi mannosyltransferases (Och1p, Mnn9p and Mnn1p), respectively. Their extracellular activities improved markedly in the OCH1 and MNN9 deletion strains. Interestingly, truncation of the N-hypermannose glycan did not increase the specific activity of these proteins, but improved the secretion yield. Further analysis showed OCH1 and MNN9 deletion up-regulated genes in the secretory pathway, such as protein folding and vesicular trafficking, but did not induce the unfolded protein response. The cell wall integrity was also affected by OCH1 and MNN9 deletion, which contributed to the release of secretory protein extracellularly. This study demonstrated that mannosyltransferases disruption improved protein secretion through up-regulating secretory pathway and affecting cell wall integrity and provided new insights into glycosylation engineering for protein secretion.
The efficient production of recombinant therapeutic and industrial proteins is important for drug discovery and industrial applications1,2. The proteins that do not need post-translational modification can be expressed actively in a prokaryotic expression system, such as Escherichia coli3. However, the majority of heterologous proteins require post-translational modification to ensure appropriate folding and bioactivity and cannot be produced properly in prokaryotic organisms. As a eukaryotic organism, the yeast S. cerevisiae is able to perform post-translational modifications, such as disulfide bond formation, removal of signal peptide and N- and O-glycosylation. Thus, this yeast has been studied widely as a robust host for the expression of recombinant proteins4. However, the expression of heterologous proteins in S. cerevisiae often results in N-hypermannose glycosylation modification.
The glycosylation pathway modifications have been studied extensively in Pichia pastoris5. In S. cerevisiae, the expression of heterologous proteins often results in a hypermannose glycan structure that may result in less activity and changes to their immunogenicity6,7,8,9. Therefore, reducing hypermannose glycans may be one of the key steps for the recombinant protein production. Disruption of the genes involved in the glycosylation modification, such as Mnn2p and Mnn11p, improved the production of recombinant cellulases10,11. Deletion of the α-1,6-mannosyltransferase Och1p increased the production of the active form of human tissue-type plasminogen activator12. Deletion of the enzyme complex M-Pol II component Mnn10p improved secretion of multiple recombinant proteins and endogenous invertase13,14. These previous studies showed that N-glycosylation modification can increase protein secretion.
The S. cerevisiae cell wall composed of glucan polymers, chitin (β-1,4-N-acetylglucosamine polymers) and mannoproteins15. Mannoproteins are important for the cell wall integrity and control the cell wall porosity which allowing substances in and out the cell according to size16,17. Disruption of the mannoprotein network by deletion of cell wall mannoproteins or alteration of protein glycosylation modification can damage the cell wall integrity and increase the cell wall porosity, which often associated with the hyper-production of secretion proteins14,18,19,20. Deletion of KRE6, the gene encoding a β-glucan synthase required for β-1,6-glucan biosynthesis, caused the cell wall defect and enhanced the production of extracellular exo-1,3-β-glucanase which located in the periplasmic space21. The inactivation of Gas1p, a β-1,3-glucanosyltransferase required for cell wall assembly, led to the cell wall defect and increased the secretion of native human insulin-like growth factor 1 significantly22,23. The increase of protein secretion associated with the cell wall defect was also observed in Trichoderma reesei, Hansenula polymorpha and Neurospora crassa24,25,26.
In this study, the impact of N-glycosylation modification on the secretion of heterologous protein was investigated. It was found that three recombinant cellulases, Cel3A, CelA and Cel7A, were N-hyperglycosylated when expressed in S. cerevisiae. Thus, these three heterologous proteins were expressed in strains with deletions in key Golgi mannosyltransferase gene (OCH1, MNN1 and MNN9) to block the formation of hypermannose glycan, and the extracellular activities of the three recombinant proteins were increased significantly. The results revealed that the improvement of extracellular activity was mainly caused by the increase in protein yield, rather than by the specific activity of the proteins. It was also observed that the transcription of key components in the secretory pathway was up-regulated and the cell wall integrity was damaged, which may be responsible for the improvement of protein production.
Results
Recombinant protein secretion was improved by the deletion of mannosyltransferases
We expressed three heterologous proteins, including Saccharomycopsis fibuligera β-glucosidase (Cel3A), Clostridium thermocellum endoglucanase (CelA) and T. reesei cellobiohydrolase I (Cel7A), in S. cerevisiae. Cel3A contains 16 potential N-glycosylation sites (predicted by NetNGlyc) with a theoretical molecular weight of 96.2 KD. Cel7A contains four potential N-glycosylation sites with a theoretical molecular weight of 57 KD. CelA contains four potential N-glycosylation sites with a theoretical molecular weight of 52 KD. When the secreted Cel3A, Cel7A and CelA were treated with peptide-N-glycosidase F (PNGase F), their molecular weights decreased significantly, demonstrating that the secreted Cel3A, Cel7A and CelA were N-hyperglycosylated (Fig. 1A). Cel3A, Cel7A and CelA were then over-expressed in the och1Δ, mnn9Δ and mnn1Δ strains. OCH1 initiates the α-1,6-hypermannose glycan elongation by the first α-1,6-mannose addition, MNN9 determines whether the Man9GlcNAc2 oligosaccharide is elongated with α-1,6-mannose backbone or stopped by the addition of α-1, 2-mannose and MNN1 is responsible for the terminal α-1,3-mannose addition.
Figure 1. Property of recombinant proteins and growth curve of the recombinant strains.

(A) The molecular weights of Cel3A, CelA and Cel7A with (+) or without the PNGase F treatment. For deglycosylation treatment, 48 μL denatured protein with or without 2 μL PNGase F enzyme solution (500 U/mL) was incubated in 50 mM sodium phosphate (pH 7.5) at 37 °C for 2 h. (B) The extracellular activity of Cel3A in recombinant strains. (C) The extracellular activity of CelA in recombinant strains. (D) The extracellular activity of Cel7A in recombinant strains. (E) The molecular weight of secreted Cel3A. (F) The growth curve of wild-type and N-glycosyaltion defective mutants in YPD medium. WT: wild-type strain expressing recombinant protein, och1Δ: OCH1 deletion strain expressing recombinant protein, mnn1Δ: MNN1 deletion strain expressing recombinant protein, mnn9Δ: MNN9 deletion strain expressing recombinant protein. The data are presented as the means ± standard errors from three independent experiments.
The extracellular activities (U/g dry cell weight (U/g DCW)) of Cel3A, CelA and Cel7A in the recombinant strains were shown in Fig. 1B–D. Deletion of N-glycosylation mannosyltransferases in Golgi clearly improved the extracellular activities of the recombinant proteins. Compared with the wild-type strain, the MNN9 deletion strain increased the activity of Cel3A, CelA and Cel7A by 156%, 105% and 230%, respectively, at 72 h. Similarly, the OCH1 deletion strain also yielded higher extracellular activities, with final activities improved by 135%, 102% and 144%, respectively. However, the disruption of MNN1 only enhanced the Cel3A production with a 25% higher activity at 72 h. There was a similar variation trend in enzyme activity at 48 h.
Disruption of the N-glycosylation pathway in Golgi can block the addition of mannose to the protein hypermannose glycan and may affect the molecular weight of the protein. We therefore determined the molecular weight of the extracellular recombinant proteins. Extracellular Cel3A, CelA and Cel7A were collected from the cell culture at 72 h, and the molecular weights were determined by western blot. The molecular weights of Cel3A, CelA and Cel7A in the OCH1 and MNN9 deletion strains were smaller than in the wild-type strain, while the three proteins in the MNN1 deletion strain and the wild-type strain were similar (Fig. 1E and Fig. S1A, S1B). Thus, the N-glycosylation modification such as the OCH1 and MNN9 deletions in S. cerevisiae indeed decreased the hypermannose glycan of recombinant proteins.
We found that the deletion of MNN1 did not affect the growth and that the maximum specific growth rate was similar to the wild-type strain at 0.33 h−1; however, the deletion of OCH1 and MNN9 decreased the growth significantly, with the maximum specific growth rates of 0.17 h−1 (Fig. 1F and Table 1).
Table 1. The maximum specific growth rate of the recombinant protein production strains.
| Strain |
μmaxa (h−1) |
|||
|---|---|---|---|---|
| Wild type | ΔOCH1 | ΔMNN1 | ΔMNN9 | |
| Recombinant strains | 0.32 | 0.17 | 0.31 | 0.17 |
| Rho1p-expressing strains | 0.30 | 0.19 | 0.30 | 0.17 |
| Pkc1p-expressing strains | 0.33 | 0.20 | 0.33 | 0.18 |
aMaximum specific growth rate.
The data are presented as the means ± standard errors from three independent experiments. The standard errors are less than 1%.
A decrease in hypermannose glycan did not improve the activity of the recombinant proteins
To determine if the decrease in hypermannose glycan affected the activity of recombinant protein, the N-glycans of recombinant proteins were removed, after which the changes in enzyme activity and molecular weight were compared with the control before N-glycan removal. PNGase F and the native protein deglycosylation kit containing endoglycosidases F1, F2 and F3 were used for the removal of protein N-glycans. The molecular weights of recombinant proteins Cel3A (Fig. 2A and Fig. S2A), CelA (Fig. 2C and Fig. S2B) and Cel7A (Fig. 2E and Fig. S2C) produced in the wild-type strain and the OCH1, MNN1 and MNN9 deletion strains were decreased to the theoretical molecular weights (approximately 100 KD, 50KD and 60KD, respectively) after the N-glycan removal. According to the decrease in molecular weight, it was observed that the N-glycans of Cel3A proteins could be removed effectively with endoglycosidase F3 treatment but could not be removed with PNGase F, endoglycosidase F1 or endoglycosidase F2 treatment. However, the N-glycans of the CelA and Cel7A proteins could be removed effectively by endoglycosidase F1 treatment. Therefore, we determined the activities of the Cel3A proteins with endoglycosidase F3 treatment, and the activities of the CelA and Cel7A proteins with endoglycosidase F1 treatment (Fig. 2B,D,F). As shown in Fig. 2B, the ratio of extracellular Cel3A activity with and without deglycosyaltion by endoglycosidase F3 treatment from the wild-type (with the complete hypermannose glycan), MNN1 (with a relatively long hypermannose glycan) and MNN9 (with one mannose glycan) deletion strains decreased by 42%, 45% and 28%, respectively. However, the ratio of Cel3A activity with and without deglycosyaltion from the OCH1 deletion strain (without mannose glycan) showed a negligible change after N-glycan removal. This indicated that the truncation of hypermannose glycan did not improve but rather interfered with Cel3A protein activity. Moreover, the removal of the N-glycans with complete or truncated hypermannose glycan did not affect the ratio of CelA and Cel7A activity with and without deglycosyaltion, indicating that the N-glycans were not responsible for their protein activity (Fig. 2D,F). Regardless, the decrease in hypermannose glycan on the recombinant proteins as a result of the deletion of mannosyltransferases did not improve the specific activity of the proteins.
Figure 2. The effect of deglycosylation enzymes to the molecular weight and activity of the recombinant proteins.

(A) The molecular weight of extracellular Cel3A with or without deglycoslylation enzyme treatment. (B) The ratio of extracellular Cel3A activity with or without Endo F3 treatment. Compared with the control, Endo F3 removed the N-glycans efficiently, thus the activity of Cel3A with and without deglycosylation by Endo F3 was measured. (C) The molecular weight of extracellular CelA with or without deglycosylation enzyme treatment. (D) The ratio of extracellular CelA activity with or without Endo F1 treatment. Compared with the control, Endo F1 removed the N-glycans efficiently, thus the activity of CelA with and without deglycosylation by Endo F1 was measured. (E) The molecular weight of extracellular Cel7A activity with or without deglycosylation enzyme treatment. (F) The ratio of extracellular Cel7A activity with or without Endo F1 treatment. Compared with the control, Endo F1 removed the N-glycans efficiently, thus the activity of Cel7A with and without deglycosylation by Endo F1 was measured. Control: samples without deglycosylation enzyme treatment. PNGase F: samples with PNGase F treatment; Endo F 1/2/3: samples with Endo F1, Endo F2 or Endo F3 treatment, respectively. The data are presented as the means ± standard errors from three independent experiments.
Deletion of mannosyltransferases improved the recombinant protein yield
Because the decrease in the hypermannose glycan did not improve the activity of the recombinant proteins, we determined whether the improvement in extracellular activity was attributed to the enhancement of protein yield. Compared with the wild-type strain, the MNN9 deletion strain increased the extracellular protein yields of Cel3A, CelA and Cel7A by 170%, 140% and 160%, respectively, at 72 h. Similarly, the extracellular protein yields of Cel3A, CelA and Cel7A in the OCH1 deletion strain was also improved by 160%, 176% and 102%, respectively (Fig. 3A–C). In addition, the MNN1 deletion strain had a 30% higher Cel3A yield. These results revealed that the enhancement of extracellular protein yield was the main reason for the improvement in extracellular protein activity. Furthermore, the intracellular recombinant protein yield was also determined using Cel3A as an example. Compared with the wild-type strain, intracellular Cel3A production in the OCH1, MNN1 and MNN9 deletion strains increased slightly (Fig. 3D), demonstrating that the total protein production was improved by the deletion of mannosyltransferases.
Figure 3. The yield of recombinant proteins.

(A) The yield of extracellular Cel3A in recombinant strains. (B) The yield of extracellular CelA in recombinant strains. (C) The yield of extracellular Cel7A in recombinant strains. (D) The yield of intracellular Cel3A in recombinant strains. The data are presented as the means ± standard errors from three independent experiments.
The secretory pathway was strengthened, and the UPR was not activated through the IRE1/HAC1 pathway by the OCH1 and MNN9 deletions
Because the efficiency of the protein secretory pathway largely affects recombinant protein production, we therefore determined whether the secretory pathway was affected by the OCH1 and MNN9 deletions. The transcription of key genes in the secretory pathway involved in protein translocation, folding, ER-associated degradation (ERAD) and vesicle trafficking was determined. The results showed that protein folding-related genes, such as KAR2 and SSA1, protein trafficking-related genes, including BOS1, ERV25, SNC2 and SSO1, and genes involved in ERAD, such as DER1 and HRD3 were up-regulated by the OCH1 and MNN9 deletions. However, SEC61, the gene encoding a protein involved in protein translocation, and PDI1, encoding protein disulfide isomerase involved in the formation of disulfide bonds, were unaffected (Fig. 4A). The results indicated that enhancement of the secretory pathway by OCH1 and MNN9 deletions may contribute to improvement in recombinant protein production.
Figure 4. The response of the secretory pathway and the extracellular activity of Cel5A.
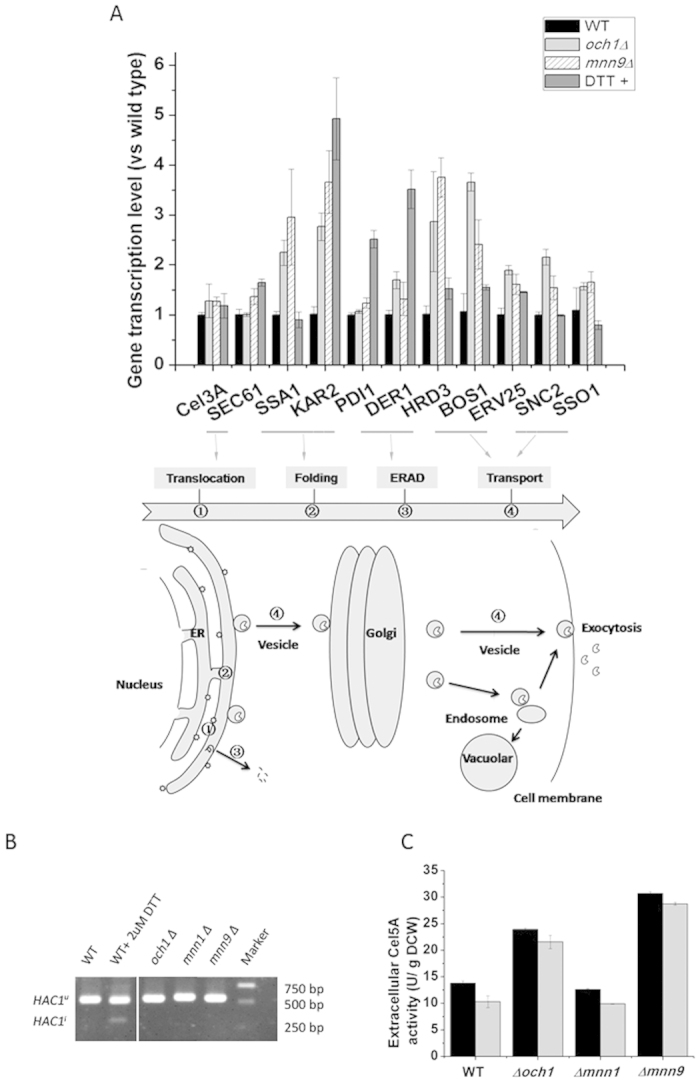
(A) The relative transcription level of components involved in the secretory pathway. (B) HAC1 mRNA splicing levels in wild-type, N-glycosyaltion defective mutants and DTT treated cells (DTT+). DTT+ was used as the positive controls. HAC1u denotes unspliced HAC1 mRNA, and HAC1i denotes induced (spliced) HAC1 mRNA. (C) The activity of extracellular Cel5A (a non-glycosylated protein, a N-glycosylation site mutation rCel5A (rN124D) of Trichoderma reesei endoglucanase Cel5A) in recombinant strains. The data are presented as the means ± standard errors from three independent experiments.
We further investigated whether the up-regulation of the secretory pathway was caused by the activation of the UPR. The accumulation of unfolded or misfolded proteins induces ER stress and triggers the UPR. The transcription factor Hac1p, generating from spliced HAC1 mRNA through the intron removal of unspliced HAC1 mRNA, can regulate UPR by up-regulating the transcription of many genes in the secretory pathway27,28. The HAC1 splicing level was used as an indicator of activation of the UPR. DTT disturbs the formation of protein disulfide bond and is known to induce the UPR. Compared with the OCH1 and MNN9 deletion strains, the DTT-treated wild-type strain up-regulated not only KAR2, BOS1, ERV25, DER1 but also SEC61 and PDI1, while SNC2 and SSO1, involved in vesicle transport from Golgi to membrane, were not affected. The level of spliced HAC1 mRNA was assessed, and accumulation of spliced HAC1 (HAC1i) was observed in the DTT-treated strain, but not in the glycosylation disruption strains (Fig. 4B). This demonstrated that the up-regulation of the secretory pathway was not induced by the UPR through the classic IRE1/HAC1 pathway in OCH1 and MNN9 deletion strains.
To investigate whether the mannosyltransferase deletion also affect the secretion of non-glycosylated protein, we expressed a non-glycosylated protein, a N-glycosylation site mutation rCel5A (rN124D) of Trichoderma reesei endoglucanase Cel5A in the mannosyltransferase deletion strains29. As shown in Fig. 4C, the secretion of rCel5A was also improved significantly in OCH1 and MNN9 deletion strains, indicating that the enhancement of protein secretion was not caused by the change of glycan pattern on the secreted protein, but may be caused by the strengthen of secretory pathway.
OCH1 and MNN9 deletions caused cell wall integrity defects
The cell wall mannoproteins are important components for the synthesis of cell wall. To study whether the cell wall integrity was affected by mannosyltransferase deletion, the transcription of the genes PIR4 and CWP2, encoding cell wall mannoproteins, was determined, which showed an up-regulation in the OCH1 and MNN9 deletion strains, but not in the DTT-treated strain (Fig. 5A). Meanwhile, the OCH1 and MNN9 deletion strains were more sensitive to the cell wall-perturbing materials, such as Congo red and CFW and glucan-digesting enzyme lyticase (Fig. 5B,C and Fig. S3A). Moreover, the carbohydrate composition of cell wall of stationary-phase cells was also determined and the results showed that the percentage of mannan decreased sharply in OCH1 and MNN9 deletion strains (Fig. 5D). The hypermannose glycan of cell surface glycoproteins determines the cell wall porosity, which limits the penetrability of the cell wall19. Thus, the disruption of protein glycosylation may affect the cell wall porosity. The results showed that the relative cell wall porosity of the OCH1 and MNN9 deletion strains was clearly higher than that of the wild-type strain (Table 2). These results demonstrated that the cell wall integrity was disrupted in the OCH1 and MNN9 deletion strains.
Figure 5. The defects of cell wall integrity.

(A) The relative transcription levels of cell wall proteins in recombinant strains and DTT treated strain (DTT+). (B) Congo red sensitivity assay. The cell density was normalized to OD600 of 1.0 and diluted four times in a 10-fold series (100, 10−1, 10−2 and 10−3). Congo red –: cultivation without the addition of Congo red; Congo red+: cultivation with the addition of 5 μg/ml Congo red. (C) The fluorescence intensity of recombinant strains after staining with CFW. (D) Carbohydrate composition of cell wall of wild-type and N-glycosyaltion defective mutants. The data are presented as the means ± standard errors from three independent experiments.
Table 2. The cell wall porosity as determined by the polycation assay.
| Wild type | ΔOCH1 | ΔMNN1 | ΔMNN9 | |
|---|---|---|---|---|
| Porosity | 8.8 ± 0.56 | 49.7 ± 1.15 | 11.1 ± 0.35 | 28.1 ± 2.05 |
Invertase, a yeast endogenous secreted protein, mainly accumulates in the periplasm. In the OCH1 and MNN9 deletion strains, the percentage of extracellular invertase (extracellular activity/extracellular activity and periplasmic activity) increased obviously, while the percentage was not changed in the MNN1 deletion strain (Fig. 6A). It was also observed that the percentage of the extracellular activity of these three recombinant proteins was also increased in the OCH1 and MNN9 deletion strains (Fig. 6B–D). These results demonstrated that the disruption of cell wall integrity contributed to greater protein secretion into the extracellular matrix.
Figure 6. The percentage of the extracellular protein activity (extracellular protein activity/the activity of extracellular and periplasmic protein).

(A) The percentage of the extracellular invertase activity. (B) The percentage of the extracellular Cel3A activity. (C) The percentage of the extracellular CelA activity. (D) The percentage of the extracellular Cel7A activity. The data are presented as the means ± standard errors from three independent experiments.
A defect in the cell wall can lead to a sensitivity to osmotic stress and may be one of the reasons for the growth deficiency of the OCH1 and MNN9 deletion strains. However, growth was not suppressed by sorbitol addition (data not shown). In addition, a cell wall defect can also activate the cell wall integrity signaling pathway to regulate cell wall remodeling for survival. The genes RHO1 and PKC1, involving in cell wall integrity signaling pathway, were over-expressed to strengthen the cell wall integrity30,31. In Rho1p and Pkc1p over-expressing strains, the growth defect of the OCH1 and MNN9 deletion strains was suppressed slightly (Fig. 7 and Table 1). In addition, the sensitivity of Congo red and CFW in Rho1p and Pkc1p over-expressing strains was partially decreased (Fig. S3B,C), indicating that the defect in cell wall integrity was partially suppressed.
Figure 7. The growth curve of the RHO1- and PKC1-expressing strains.

(A) The growth curve of the OCH1 deletion strain expressing RHO1 and PKC1. (B) The growth curve of the MNN9 deletion strain expressing RHO1 and PKC1. The data are presented as the means ± standard errors from three independent experiments.
Discussion
In this study, the recombinant proteins Cel3A, CelA and Cel7A were found to be hyperglycosylated with significant increases in their molecular weights, even though CelA was from a prokaryotic organism. This indicated that heterologous proteins with potential glycosylation sites may be modified by the yeast glycosylation process regardless of their origin. Therefore, Cel3A, CelA and Cel7A were expressed in the Golgi mannosyltransferase deletion strains that blocked the extension of the outer chain to avoid hyperglycosylation. The extracellular activities of the three recombinant proteins were enhanced significantly. Moreover, the molecular weights of the proteins secreted from the OCH1 and MNN9 deletion strains were clearly reduced. These results showed that the disruption of the key mannosyltransferases in the Golgi could successfully block the N-hypermannose glycan extension and increase the extracellular activity of recombinant proteins. This improvement in extracellular activity could result from the increase in protein specific activity or protein yield.
Removal of the N-glycans without the mannose outer chain in the OCH1 deletion strain did not affect the Cel3A activity, while removal of the N-glycans with a complete or truncated mannose outer chain in wild-type, MNN1 and MNN9 deletion strains decreased the Cel3A activity. The results demonstrated that some heterologous proteins required the outer chain of N-hypermannose glycan to ensure their activity and that truncation of the N-hypermannose glycan might not enhance the protein activity but rather have a negative effect. In contrast, the removal of N-glycans from CelA and Cel7A proteins had no effect on their activities, indicating that the hypermannose glycan of some recombinant proteins did not disturb or enhance their protein activity. In conclusion, the truncation of the hypermannose glycan of recombinant proteins in S. cerevisiae did not improve their protein specific activity.
The results demonstrated that improvement in the extracellular protein activity was mainly due to an increase in the protein production. Recombinant protein production is largely affected by the efficiency of the protein secretory pathway. Over-expression of key genes involved in protein folding, such as KAR2 and PDI1, and vesicle trafficking, such as SSO1 and SNC1 can strengthen the secretory pathway and enhance the secretion of heterologous proteins32,33. In the mannosyltransferase deletion strains, many genes in the secretory pathway, including protein folding-related genes such as KAR2 and SSA1, protein trafficking genes such as BOS1, ERV25, SNC2 and SSO1, and the genes involved in ERAD such as DER1 and HRD3, were up-regulated. Thus, improvement in protein yield might be caused by the enhancement of the secretory pathway.
The UPR can regulate the secretory pathway by up-regulating the transcription of many genes in the secretory pathway34. The genes involved in protein translocation, such as SEC61, protein folding, such as KAR2 and PDI1, vesicle trafficking, such as BOS1 and ERV25, and ERAD, such as DER1 and HRD3, were up-regulated in the DTT-treated wild-type strain. However, the genes involved in vesicle trafficking from the Golgi to the membrane, such as SSO1 and SNC2, were not affected. These results were consistent with a previous study, in which the genes in the secretory pathway were up-regulated by the UPR34. The spliced HAC1 mRNA was induced in the DTT-treated strain but not in mannosyltransferase deletion strains. Thus, although the genes up-regulated in the secretory pathway through the OCH1 and MNN9 deletion partially overlapped with the UPR targets, this was induced in an IRE1/HAC1-independent manner.
In a previous study, the OCH1 deletion resulted in a defect in cell wall integrity and triggered the mitogen-activated protein kinase (MAPK) pathway31. Chen et al. reported that MAPK pathway was activated during ER stress and regulated ER stress in an IRE1/HAC1-independent manner35. Scrimale et al. reported that a defect in cell wall integrity caused ER stress and induced the UPR by Hac1p and that this activation was regulated by signaling through the cell wall integrity MAPK pathway36. These reports suggested that a regulatory relationship might exist between cell wall integrity and the secretory pathway. In our study, both OCH1 and MNN9 deletions could up-regulate the components of the secretory pathway. In addition, the OCH1 and MNN9 deletions affected the cell wall integrity. We therefore speculated that the up-regulation of the secretory pathway might correlate with the cell wall defect. However, the relation bewteen cell wall defect and secretory pathway regulation is still not very clear and needs to be investigated further.
A defect in cell wall integrity may be one reason for severe growth deficiency of the OCH1 and MNN9 deletion strains. It is reported that the growth defect of the OCH1 deletion strains can be partially suppressed by an osmotic stabilizer and by the over-expression of Pkc1p, which is involved in the cell wall integrity signaling pathway31. Pkc1p and Rho1p, which is considered to be the master regulator of the cell wall integrity signaling pathway, slightly recovered the growth defect of the OCH1 and MNN9 deletion strains and the defect in cell wall integrity was partially suppressed by the over-expression of Rho1p and Pkc1p30,37. However, the osmotic stabilizer sorbitol did not suppress the growth deficiency of the OCH1 and MNN9 deletion strains. These results indicated that the defect in cell wall integrity might not be the only damaging factor responsible for the growth defect in the OCH1 and MNN9 deletion strains38.
In this study, we found that recombinant proteins with potential glycosylation sites were hyperglycosylated and that disruption of the hypermannose glycan extension in the Golgi significantly improved recombinant protein production. This improvement was mainly due to the enhancement of protein yield rather than to an increase in protein activity. The secretory pathway was strengthened in the OCH1 and MNN9 deletion strains in a HAC1-independent manner, which may contribute to the improved production of recombinant proteins. However, the relationship between glycosylation disruption and secretory pathway regulation is still unclear; further work should be carried out to investigate how glycosylation disruption affects the secretory pathway.
Materials and Methods
Media and growth conditions
Escherichia coli Trans5α (TransGene Biotech) was used for the propagation of plasmids construction. S. cerevisiae CEN.PK102-3A was used as the host for the expression of heterologous protein39. E. coli Trans5α was cultivated in LB medium (5 g/L yeast extract, 10 g/L peptone, 10 g/L glucose). CEN.PK102-3A was grown in YPD medium (10 g/L yeast extract, 20 g/L peptone, 20 g/L glucose). The recombinant strains were cultivated in SC-SCAA medium as previously described40. The cultivation of recombinant strains was performed at 30 °C, 200 rpm in 100-ml flasks with a 40 ml working volume. The initial OD600 (optical density) was 0.2.
Recombinant plasmids and strains construction
The recombinant strains and plasmids involved in this study were listed in Table S1. The primers were shown in Table S2. The ligation was performed using the Gibson assembly41. The Cel3A gene with its native signal sequence and FLAG-tagged sequence was amplified from the plasmid pTH-BGL, the CelA gene with an INU1 signal sequence and myc-tagged sequence was amplified from pTH-CEL (37) and each gene was then ligated into the plasmid PYX242WS42 under the control of the TPI1 promoter and the PGK1 terminator. The gene of Cel5A from T. reesei with a site mutation (rN124D) was amplified from recombinant plasmid pAJ401-cel5A-N124D29 and inserted into plasmid PYX242WS. The yeast centromere plasmid pJFE1 containing the URA3 gene as a marker was used to over-express the genes RHO1 and PKC1. RHO1 and PKC1 were amplified from the genomic DNA of CEN.PK102-3A and ligated into the pJFE1 plasmid43 under the control of the TEF1 promoter and the PGK1 terminator.
The OCH1, MNN9 and MNN1 were each deleted in CEN.PK102-3A as described previously44, resulting in the och1Δ, mnn9Δ and mnn1Δ strains. The plasmids pBGL, pCelA, pCel5A and pYX242WS were used to transform CEN.PK102-3A, och1Δ, mnn9Δ and mnn1Δ. The plasmids pJFE1, pRHO and pPKC were then transformed into the Cel3A-expressing strains.
Protein quantification
The cell culture was centrifuged at 18,000 g for 4 min. The supernatant was collected and the cells were washed and resuspended in 50 mM citrate buffer (pH 5.0) for the measurement of enzyme activity. For extraction of periplasmic and intracellular proteins, the cells were washed with Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7.0). The cells were then resuspended in 1 mL Z buffer with 0.1 mg/mL of lyticase (Sigma, USA) to an OD600 of 5.0 and incubated at 20 °C for 3 hours to remove the cell walls. After incubation with lyticase, the samples were centrifuged at 18,000 g for 4 min and the supernatant were collected to measure the periplasmic enzymes activity. The cellular proteins were extracted from the lysate of the cell pellets, which were broken by a Precellys 24 Homogenizer (Bertin Technologies, France) with glass beads. The protein titer of the recombinant proteins was measured by Flag-tag and myc-tag ELISA detection kits (Biovol, Shanghai, China) in accordance with the manufacturer’s instruction.
β-glucosidase activity was determined as described previously using p-nitrophenyl-β-D-glucopyranoside pNPG (Sigma, USA) as the substrate45. The enzyme was incubated in 50 mM citrate buffer (pH 5.0) with 5 mM pNPG at 50 °C for 30 min. The reaction was stopped by adding 10% sodium carbonate, and p-nitrophenol released from pNPG was determined at 405 nm. The endoglucanase activity assay was quantified using carboxymethylcellulose sodium salt as the substrate (Sigma, USA)46. Reducing sugars were detected at 540 nm after boiling with the dinitrosalicylate (DNS) reagent for 10 min. Cellobiohydrolase activity was determined using p-nitrophenyl-b-D-cellobioside (pNPC) (Sigma, USA) as the substrate as described previously. The enzyme was incubated in 50 mM citrate buffer (pH 5.0) with 1.67 mM pNPG at 50 °C for 30 min. The reaction was stopped by adding 10% sodium carbonate, and the p-nitrophenol released from pNPC was determined at 405 nm47. Invertase activity secreted into the periplasmic space was measured as described previously33. Glucose released from sucrose was quantified using a D-GLUCOSE (GOPOD) kit (Megazyme K-CERA, Wicklow, Ireland).
Analysis of cell wall integrity in recombinant strains
Congo red sensibility was monitored by a qualitative growth assay on plates containing Congo red as described previously with some modification48. Briefly, the cells were grown overnight and harvested by centrifugation. The cell pullets were washed three times and resuspended in sterile H2O. The cell density was normalized to an OD600 of 1.0 and diluted four times in a 10-fold series (OD600 10−1, 10−2, 10−3, 10−4). Four μl of each dilution of the samples was spotted onto the plate with or without 50 ug/mL Congo red.
The lyticase sensibility assay was performed as previously described with some modification48. Cells were harvested by centrifugation, washed three times and resuspended in 10 mM Tris-HCl (pH = 7.4) with an approximate OD600 of 1.5. The cells were then treated at 30 °C with 0.5 U lyticase (Sigma, USA). The residual cells that were not lysed were monitored by OD600.
For cell wall staining, Calcofluor white (CFW) (Sigma, USA) was used as described previously49. Cells at an approximate OD600 of 1.5 were harvested by centrifugation, and washed three times and resuspended in sterile water with 100 μg/mL CFW. The solution was incubated at 30 °C for 2 min, harvested, washed twice with sterile water, and resuspended in 1 ml of water before measuring the fluorescence intensity at 355 nm/460 nm.
The relative cell porosity assay was determined as described previously50. The cells were harvested at 36 h, and washed with 10 mM Tris-HCl (pH = 7.4). The cells were then resuspended in 10 mM Tris-HCl (pH = 7.4) containing 5 ug/mL DEAE-dextran (a large hydrodynamic radius causing only limited cell leakage due to limited passage through the cell wall) or 10 ug/mL poly-L-lysine (a small hydrodynamic radius caused cell leakage independent of cell wall porosity) (Songon Biological Engineering, China) to an density of 10 OD600/mL and incubated at 30 °C for 30 min. Subsequently, the samples were centrifuged at 18,000 g, after which the supernatant was filtered and the UV-absorbing compounds leaking from the cells as a result of by the polycation treatment were measured at 260 nm. The ratio between DEAE-dextran- and poly-L-lysine-induced cell leakage was calculated to determine the relative cell wall porosity.
Isolation of cell wall and analysis of cell wall carbohydrate
The isolation of cell wall and analysis of carbohydrate composition was performed as previously described51. In brief, cells were harvested during stationary-phase at 48 h and washed once with deionized water. The cell pellet was resuspended with 1 ml TE buffer and broken with 1 g acid-washed glass beads. The supernatant was transferred into a 15 ml falcon tube. The glass beads were washed five times and the washing solutions were also collected in 15 ml falcon tube. Samples were centrifuged at 4,800 g for 15 min and the pellets were resuspended in 1 mL TE buffer for centrifugation at 3,000 g for 5 min. The supernatant was dried at 110 °C. Cell walls were hydrolyzed with 72% H2SO4 and the carbohydrate was analyzed by CarboPacTM PA1 anion-exchange column (2 × 250 mm, Dionex). Elution was performed with 18 mM NaOH at a flow rate of 0.5 mL/min.
Extracellular protein concentration and PNGase F treatment
The extracellular protein from the cell cultures were concentrated using the Amicon ultra 15 mL centrifugal filter with an NMWL membrane of 30 KD (Millipore, Germany) and then washed three times with sterilized water. N-linked oligosaccharides of the recombinant proteins were removed using PNGase F or the native protein deglycosylation kit containing endoglycosidase F1, F2 and F3 (Sigma, USA) according to the manufacturer’s instructions. The proteins were then used for the activity assay and western blot analysis.
Western blot
SDS-PAGE and western blots were performed as described previously37. The recombinant proteins were detected using rabbit polyclonal octA-, his- or myc-probe antibodies (Santa Cruz Biotechnology, USA) as the primary antibody and a horseradish peroxidase-labeled goat anti-rabbit IgG as the secondary antibody.
HAC1 mRNA splicing and real-time quantitative PCR (qPCR)
Stains grew in 40 mL of SC-SCAA medium at 30 °C at 200 rpm from OD 0.2 to OD600 0.6, and cells were harvested and washed. Total RNA was extracted using the UNIQ-10 Trizol RNA extraction kit (Sangon Biological Engineering, China). The PrimeScript RT-PCR Kit (Takara, Japan) was used for cDNA synthesis. The relative transcription levels of the genes were quantified by real-time quantitative PCR using the SYBR Green Master Mix Kit (Roche Molecular Biochemicals, Germany). HAC1 mRNA splicing were detected through PCR using cDNA as the template.
Additional Information
How to cite this article: Tang, H. et al. N-hypermannose glycosylation disruption enhances recombinant protein production by regulating secretory pathway and cell wall integrity in Saccharomyces cerevisiae. Sci. Rep. 6, 25654; doi: 10.1038/srep25654 (2016).
Supplementary Material
Acknowledgments
This work was supported by the National High Technology Research and Development Program of China (2012AA022106), the National Natural Science Foundation of China (31300037, 31470163, 31470219 and J1103515), Project of National Energy Administration of China (NY20130402), the National Key Technology R&D Program of China (2014BAD02B07), State Key Laboratory for Microbial Technology (M2013-08).
Footnotes
Author Contributions H.T., X.B. and J.H. designed this work. H.T., S.W., J.W., M.S., M.Z. and M.X. performed the experiment. H.T., S.W. and Y.S. analyzed the data. H.T., X.B. and J.H. wrote and revised the manuscript. All authors read and approved the final manuscript.
References
- Schmidt F. R. Recombinant expression systems in the pharmaceutical industry. Appl. Microbiol. Biotechnol. 65, 363–372 (2004). [DOI] [PubMed] [Google Scholar]
- Lambertz C. et al. Challenges and advances in the heterologous expression of cellulolytic enzymes: a review. Biotechnol. Biofuels. 7, 135 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 72, 211–222 (2006). [DOI] [PubMed] [Google Scholar]
- Idiris A., Tohda H., Kumagai H. & Takegawa K. Engineering of protein secretion in yeast: strategies and impact on protein production. Appl. Microbiol. Biotechnol. 86, 403–417 (2010). [DOI] [PubMed] [Google Scholar]
- Hamilton S. R. et al. Production of complex human glycoproteins in yeast. Science 301, 1244–1246 (2003). [DOI] [PubMed] [Google Scholar]
- Kniskern P. J. et al. Characterization and evaluation of a recombinant hepatitis B vaccine expressed in yeast defective for N-linked hyperglycosylation. Vaccine 12, 1021–1025 (1994). [DOI] [PubMed] [Google Scholar]
- Hoshida H., Fujita T., Cha-aim K. & Akada R. N-glycosylation deficiency enhanced heterologous production of a Bacillus licheniformis thermostable α-amylase in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 97, 5473–5482 (2013). [DOI] [PubMed] [Google Scholar]
- Smith R., Duncan M. & Moir D. Heterologous protein secretion from yeast. Science 229, 1219–1224 (1985). [DOI] [PubMed] [Google Scholar]
- Arico C., Bonnet C. & Javaud C. In Glycosylation Engineering of Biopharmaceuticals Vol. 988 Methods in Molecular Biology (ed Alain Beck ) Ch. 4, 45–57 (Humana Press, 2013). [Google Scholar]
- Matsuoka H. et al. Cell wall structure suitable for surface display of proteins in Saccharomyces cerevisiae. Yeast 31, 67–76 (2014). [DOI] [PubMed] [Google Scholar]
- Suzuki H., Imaeda T., Kitagawa T. & Kohda K. Deglycosylation of cellulosomal enzyme enhances cellulosome assembly in Saccharomyces cerevisiae. J. Biotechnol 157, 64–70 (2012). [DOI] [PubMed] [Google Scholar]
- Nagasu T. et al. Isolation of new temperature-sensitive mutants of Saccharomyces cerevisiae deficient in mannose outer chain elongation. Yeast 8, 535–547 (1992). [DOI] [PubMed] [Google Scholar]
- Wang T.-Y. et al. Systematic screening of glycosylation- and trafficking-associated gene knockouts in Saccharomyces cerevisiae identifies mutants with improved heterologous exocellulase activity and host secretion. BMC. Biotechnol 13, 71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkevičiūtė D. & Sasnauskas K. Disruption of the MNN10 gene enhances protein secretion in Kluyveromyces lactis and Saccharomyces cerevisiae. FEMS. Yeast. Res. 4, 833–840 (2004). [DOI] [PubMed] [Google Scholar]
- Orlean P. Architecture and biosynthesis of the Saccharomyces cerevisiae cell wall. Genetics 192, 775–818 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nobel J. & Barnett J. Passage of molecules through yeast cell walls: A brief essay-review. Yeast 7, 313–323 (1991). [DOI] [PubMed] [Google Scholar]
- Zlotnik H., Fernandez M. P., Bowers B. & Cabib E. Saccharomyces cerevisiae mannoproteins form an external cell wall layer that determines wall porosity. J. Bacteriol 159, 1018–1026 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo M. et al. PST1 and ECM33 encode two yeast cell surface GPI proteins important for cell wall integrity. Microbiology 150, 4157–4170 (2004). [DOI] [PubMed] [Google Scholar]
- De Nobel J. G., Klis F. M., Priem J., Munnik T. & Van Den Ende H. The glucanase-soluble mannoproteins limit cell wall porosity in Saccharomyces cerevisiae. Yeast 6, 491–499 (1990). [DOI] [PubMed] [Google Scholar]
- Gaynor E. C. et al. MCD4 encodes a conserved endoplasmic reticulum membrane protein essential for glycosylphosphatidylinositol anchor synthesis in yeast. Mol Biol Cell 10, 627–648 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. Hyperproduction of β-glucanase Exg1 promotes the bioconversion of mogrosides in Saccharomyces cerevisiae mutants defective in mannoprotein deposition. J Agric Food Chem 63, 10271–10279 (2015). [DOI] [PubMed] [Google Scholar]
- Ram A. F. J. et al. Loss of the plasma membrane-bound protein Gas1p in Saccharomyces cerevisiae results in the release of β-1,3-glucan into the medium and induces a compensation mechanism to ensure cell wall integrity. J Bacteriol 180, 1418–1424 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vai M. et al. Improved secretion of native human insulin-like growth factor 1 from gas1 mutant Saccharomyces cerevisiae cells. Appl Environ Microbiol 66, 5477–5479 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agaphonov M. O., Packeiser A. N., Chechenova M. B., Choi E. S. & Ter-Avanesyan M. D. Mutation of the homologue of GDP-mannose pyrophosphorylase alters cell wall structure, protein glycosylation and secretion in Hansenula polymorpha. Yeast 18, 391–402 (2001). [DOI] [PubMed] [Google Scholar]
- Gratzner H. G. Cell wall alterations associated with the hyperproduction of extracellular enzymes in Neurospora crassa. J Bacteriol 111, 443–446 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlińska-Lenart U. et al. Glycoprotein hypersecretion alters the cell wall in Trichoderma reesei strains expressing the Saccharomyces cerevisiae dolichylphosphate mannose synthase gene. Appl Environ Microbiol 72, 7778–7784 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. S. & Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404 (1996). [DOI] [PubMed] [Google Scholar]
- Ogawa N. & Mori K. Autoregulation of the HAC1 gene is required for sustained activation of the yeast unfolded protein response. Genes to Cells 9, 95–104 (2004). [DOI] [PubMed] [Google Scholar]
- Qin Y. & Qu Y. Asn124 of Cel5A from Hypocrea jecorina not only provides the N-glycosylation site but is also essential in maintaining enzymatic activity. BMB Reports 47, 256–261 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H. et al. Identification of yeast Rho1p GTPase as a regulatory subunit of 1, 3-β-glucan synthase. Science 272, 279–281 (1996). [DOI] [PubMed] [Google Scholar]
- Lee B. N. & Elion E. A. The MAPKKK Ste11 regulates vegetative growth through a kinase cascade of shared signaling components. Proc. Natl. Acad. Sci. USA 96, 12679–12684 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. et al. Engineering protein folding and translocation improves heterologous protein secretion in Saccharomyces cerevisiae. Biotechnol. Bioeng 112, 1872–82 (2015). [DOI] [PubMed] [Google Scholar]
- Ruohonen L., Toikkanen J., Outola M., Soderlund H. & Keranen S. Enhancement of protein secretion in Saccharomyces cerevisiae by overproduction of Sso protein, a late-acting component of the secretory machinery. Yeast 13, 337–351 (1997). [DOI] [PubMed] [Google Scholar]
- Travers K. J. et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 (2000). [DOI] [PubMed] [Google Scholar]
- Chen Y. et al. Identification of mitogen-activated protein kinase signaling pathways that confer resistance to endoplasmic reticulum stress in Saccharomyces cerevisiae. Mol Cancer Res 3, 669–677 (2005). [DOI] [PubMed] [Google Scholar]
- Scrimale T., Didone L., de Mesy Bentley K. L. & Krysan D. J. The unfolded protein response is induced by the cell wall integrity mitogen-activated protein kinase signaling cascade and is required for cell wall integrity in Saccharomyces cerevisiae. Mol. Biol. Cell. 20, 164–175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D. E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics 189, 1145–1175 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen P. J. et al. Defects in protein glycosylation cause SHO1-dependent activation of a STE12 signaling pathway in yeast. Genetics 155, 1005–1018 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entian K.-D. & Kötter P. In Methods in Microbiology Vol. 26 (eds Alistair J. P. Brown & Mick Tuite ) 431–449 (Academic Press, 1998).
- Wittrup K. D. & Benig V. Optimization of amino acid supplements for heterologous protein secretion in Saccharomyces cerevisiae. Biotechnol. Tech 8, 161–166 (1994). [Google Scholar]
- Gibson D. G. Enzymatic assembly of overlapping DNA fragments. Methods. Enzymol 498, 349–361 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. et al. Improvement of L-arabinose fermentation by modifying the metabolic pathway and transport in Saccharomyces cerevisiae. Biomed. Res. Int. 2013, 461204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y. et al. An efficient xylose-fermenting recombinant Saccharomyces cerevisiae strain obtained through adaptive evolution and its global transcription profile. Appl. Microbiol. Biotechnol. 96, 1079–1091 (2012). [DOI] [PubMed] [Google Scholar]
- Xu L., Shen Y., Hou J., Tang H. & Wang C. Promotion of extracellular activity of cellobiohydrolase I from Trichoderma reesei by protein glycosylation engineering in Saccharomyces cerevisiae. Curr. Synthetic. Sys. Biol. 2, 2332–0737 (2014). [Google Scholar]
- Berghem L. E. R. & Pettersson L. G. The Mechanism of enzymatic cellulose degradation. Eur. J. Biochem 46, 295–305 (1974). [DOI] [PubMed] [Google Scholar]
- Bailey M. J., Biely P. & Poutanen K. Interlaboratory testing of methods for assay of xylanase activity. J. Biotechnol 23, 257–270 (1992). [Google Scholar]
- Deshpande M. V., Eriksson K.-E. & Göran Pettersson L. An assay for selective determination of exo-1,4,-β-glucanases in a mixture of cellulolytic enzymes. Anal. Biochem 138, 481–487 (1984). [DOI] [PubMed] [Google Scholar]
- van der Vaart J. M., Caro L. H., Chapman J. W., Klis F. M. & Verrips C. T. Identification of three mannoproteins in the cell wall of Saccharomyces cerevisiae. J. Biotechnol 177, 3104–3110 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan T. C. R., Krömer J. O. & Nielsen L. K. Physiological and transcriptional responses of Saccharomyces cerevisiae to d-Limonene show changes to the cell wall but bot to the plasma membrane. Appl. Environ. Microbiol. 79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nobel J. G., Klis F. M., Munnik T., Priem J. & Van Den Ende H. An assay of relative cell wall porosity in Saccharomyces cerevisiae, Kluyveromyces lactis and Schizosaccharomyces pombe. Yeast 6, 483–490 (1990). [DOI] [PubMed] [Google Scholar]
- Francois J. M. A simple method for quantitative determination of polysaccharides in fungal cell walls. Nat. Protocols 1, 2995–3000 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.