

Abstract
Amorphous films of the anticonvulsant drug carbamazepine are easily accessible by various methods, while the crystallization into specific polymorphs represents a challenging and time-consuming task. In this work, the crystallization of drop cast carbamazepine at silica surfaces is investigated by atomic force microscopy and both in situ and ex situ grazing incidence X-ray diffraction. The pristine films grow with low crystallization rates into a triclinic polymorph, exhibiting poor orientational order within films. However, if iminostilbene, a chemical precursor of carbamazepine, is added to the solution, enhanced crystallization rates result. The individual components crystallize phase-separated upon solvent evaporation without the formation of cocrystals. Iminostilbene reduces the time scale of carbamazepine crystallization from several hours to minutes. Besides the change in crystallization dynamics, iminostilbene induces order to the carbamazepine crystallites, evident as a 110 texture. In situ data of intermixed solutions demonstrate that iminostilbene crystallization occurs first. The iminostilbene crystals then act as templates for carbamazepine growth, whereby fully epitaxial growth is suggested from the results. The findings motivate such an approach for other systems, as this solution-processed, intrinsic epitaxial behavior might be employed in up-scaled manufacturing processes.
Short abstract
Improved crystallization of carbamazepine on the addition of its precursor is investigated by ex situ and in situ experiments.
Introduction
Establishing thin solid films with well-defined morphological and/or crystallographic properties is of key interest in various fields of fundamental and application-directed research, examples including pharmaceutics,1−3 organic electronics,4−6 and colloid science.7,8 In all of these fields, the control over the solid-state properties of the films, such as polymorphism and/or morphology, is established by deliberately altering processing methods and conditions. Deposition techniques mainly include physical9 and chemical vapor deposition,10 deposition from the melt,11 and various preparation techniques from solution (e.g., dip-coating12 and spin-coating13), with each method having its individual advantages and drawbacks.
In this context, solution processing is, in principle, one of the simplest methods at hand for crystallization. There, core deposition parameters like the solute concentration, the solvent type, its evaporation rate and temperature, as well as the substrate nature and its surface properties are known to have the potential to influence both the film morphology and crystal-polymorph formation.14 In the field of pharmaceutical sciences, the deposition of the active pharmaceutical ingredients (typically asymmetric molecules) from solution or melt often results in the formation of initially amorphous layers, reported for ibuprofen,15 paracetamol,16 indomethacine,17,18 clotrimazole,18,19 or carbamazepine,18 among others. While this is desired in terms of easier drug dissolution for practical application, the intrinsic meta-stability of the amorphous form is a major concern; undesired crystalline transition may result in strongly altered therapeutic action.20 Clearly, this demands either for stabilizing its metastable form or, alternatively, for directing crystallization into a specific stable polymorph.
The presence of a substrate during crystallization may already provide a handle for the latter, as it induces severe geometrical constraints. Thus, the entropy of the crystallization system is changed. If the crystallization processes are mainly dominated by the proximity of a surface, this even enables the growth of specific polymorphic forms, so-called surface-induced polymorphs.21 Another important approach for altering film properties is applying additives to the solution. For some systems, the formation of cocrystals has been reported, which can enhance stability and solubility and, eventually, allow for its controlled release.22 These aid substances can further act as seeds for nucleation and thus foster crystal growth into specific polymorphs.23,24 In some cases, even a situation of mutual alignment is achievable by this approach, where one component crystallizes on top of the other, forming ordered layers.25
In the present study, the crystallization behavior of the model drug carbamazepine on oxidized silicon surfaces is investigated. Carbamazepine is an anticonvulsant drug used to treat medical conditions such as epilepsy and trigeminal neuralgia.26 To date, five anhydrous polymorphs,27 various pseudopolymorphs (hydrates and solvates),28 and cocrystals (e.g., with saccharine29 and aspirin30) are reported in literature, mostly on the basis of bulk solution experiments. The aim of the present study is to explore how the presence of a solid surface influences the crystallization process and how its impact is on the polymorphism of carbamazepine. Furthermore, these results are compared to iminostilbene, a precursor in the synthesis of carbamazepine.31 Differences in their behavior provide insight into the role of the carboxamide group during the crystallization process on the surface. Atomic force microscopy (AFM) and both in situ and ex situ grazing incidence X-ray diffraction (GIXD) experiments were performed to acquire information on structure, morphology, and crystallization kinetics of the individual materials. On this basis, their intermixing behavior is explored by comparing films obtained from carbamazepine/iminostilbene solution blends to the pristine materials in the solid state.
Experimental Methods
Carbamazepine (98%) and iminostilbene (97%) powders were purchased from Alfa Aesar (Ward Hill, USA) and Sigma-Aldrich (Munich, Germany), respectively, and used without further treatment (Figure 1). Films of the pristine compounds and blends thereof were prepared by drop casting. Various solutions of different concentrations were prepared in spectral grade tetrahydrofuran (THF) (Fluka, Germany).
Figure 1.

Structural formulas of carbamazepine (5H-dibenzo[b,f]azepine-5-carboxamide) and iminostilbene (5H-dibenzo[b,f]azepine).
As substrates, conventional glass slides (Roth, Germany) or single crystal silicon wafers (Siegert Wafers, Germany) with a native oxide layer were used. Single crystal silicon wafers were mostly employed in the investigation of thin samples in the ex situ grazing incidence X-ray diffraction (GIXD) experiments, as glass slides exhibit a strong amorphous characteristic. Prior to their use, the samples were cut into 2 cm pieces and sonicated in acetone and a 0.1 mol NaOH solution (in that order). Finally, the substrates were rinsed with ultrapure water and dried under a nitrogen stream. Apart from slight chemical and morphological variations, both surfaces are nearly identical. The film forming properties are not affected by the substrate choice, as evidenced by optical microscopy and specular X-ray diffraction (data not shown). However, due to the stronger hydrophilic character of the silicon wafers after NaOH treatment, compared to the glass slides, these samples are more prone to hydrate formation upon storage.
For thin film preparation, a constant solution volume of 25 μL per square centimeter substrate area was dropcast onto the surfaces (leveled precisely horizontally). In addition, the solvent evaporation rate was reduced by covering the samples with Petri dishes.
Ex situ GIXD experiments were performed at Helmholtz-Zentrum Berlin für Materialien und Energie GmbH (HZB, BESSY II) at beamline KMC-2 using a wavelength (λ) of 0.1 nm. The diffracted intensities were recorded with a two-dimensional cross-wire VÅNTEC-2000 detector (Bruker AXS, Germany); a constant argon flow was applied onto the samples during measurement to minimize sample degradation due to radical formation at the surface.32
Time-resolved in situ GIXD experiments were carried out at the Austrian SAXS beamline at the Elettra synchrotron facility (Trieste) using λ = 0.154 nm and a two-dimensional Pilatus3 1M detector. The substrates were housed in a custom-built sample cell equipped with Kapton windows; a top syringe inlet allowed for in situ drop casting and measurements during solvent evaporation.
For both setups, the recorded real space maps of the diffraction signal were transformed into reciprocal space (with qp and qz as in-plane and out-of-plane components of the scattering vector q, respectively) using xrayutilities(33) to allow for the simple comparison of the data from both setups. Two-dimensional pseudocolor plots represent the diffraction intensity on a linear scale (reciprocal space maps), with brighter colors corresponding to higher values.
AFM height micrographs were recorded with a FlexAFM equipped with an Easyscan 2 controller (Nanosurf, Switzerland) in tapping mode. Measurements were performed with Tap 190-Al cantilevers supplied from Budgetsensors (Sofia, Bulgaria). All recorded data were processed with the software package Gwyddion.34
Results and Discussion
The deposition of organic molecular compounds via drop-casting is a standard technique, where the preparation parameters subtly impact the processes of crystallization and film formation. For pristine films of carbamazepine and iminostilbene, respectively, drop cast from THF solution, this is clearly evident. The films exhibit severely altered morphologies and crystallographic properties depending on composition and concentration of the solutions. The concentration, in particular, shows a strong influence on the apparent morphologies, as illustrated by AFM height images in Figure 2 (a,b for carbamazepine and c,d for iminostilbene). At a carbamazepine concentration of 46 mg/mL, drop casting leads to the formation of large, elongated needles of up to several micrometers in length, which lie parallel to the substrate surface. The data suggest that a bunch of individual needles originates from a common center, which, e.g., in Figure 2a, lies slightly off the lower image border; the needle diameters vary between 0.20 and 1.50 μm within this sample. In addition, needles seem to be located on top of others. This points toward carbamazepine crystallization taking place already in the bulk solution, as the concentration of 46 mg/mL is close to saturation. In contrast to a potential growth scenario directly at the substrate–liquid interface, this here results in needles precipitating onto the substrate upon solvent evaporation.
Figure 2.

AFM height images of drop cast films from pure carbamazepine solution (a,b), pure iminostilbene solution (c,d), and of a 1:1 blend thereof (e,f). The respective solute concentrations are 46 (a), 5.5 (b), 5 (c), 0.7 (d), 5.5 (e), and 2.5 (f) mg/mL; elongated structures in (a) could not entirely be captured by AFM due to experimental limitations, see the Supporting Information for larger-area optical microscopy data.
The situation changes drastically if the carbamazepine concentration in THF is decreased. Drop casting from less concentrated solutions (i.e., solute concentrations of 10 mg/mL and less) results in the films being initially amorphous after solvent evaporation. Likely, this is because a system with lower solute concentration will remain less time in the supersaturated state during solvent evaporation, which in turn reduces the formation of nuclei. Importantly, these films then fully crystallize upon storage. In Figure 2b, an example for such an already crystallized film is depicted. This specific sample was prepared from a 5.5 mg/mL solution and the AFM micrograph in Figure 2b was recorded 2 weeks after sample preparation. The morphology of such films consists of a vast number of small, elongated structures with the length of a few micrometers. Additionally, it is noted that about half the structures are radially distributed around common centers; some of these spherulitic structures are highlighted by dashed circles in Figure 2b. Adjacent spherulites often intersect, hindering further growth into larger, fully connected structures. In comparison to the high concentration case, this very structure type also leads to a significantly more homogeneous carbamazepine coverage of the substrate. A power spectral density evaluation of the AFM image yields a root-mean-square roughness of about 170 nm and an autocorrelation length of 3.1 μm. The latter results from common distances in the surface. A comparison with the AFM height image suggests that this 3.1 μm corresponds to the average length of the needle-like structures. This is regarded favorable for pharmaceutical applications of fast therapeutic action because smaller structures generally correlate with faster dissolution rates.3
A strikingly different crystallization behavior is exhibited by iminostilbene, if drop cast onto the silica surface. At a concentration of 5 mg/mL, drop casting results in the formation of bulky structures (Figure 2c), as opposed to the spherulites obtained for carbamazepine at a similar concentration (cf. Figure 2b). Iminostilbene crystals exhibit a preferential growth direction (bright “line” in the image) from which dendritic branches evolve. These structures grow rapidly during solvent evaporation, and the resulting films cover the substrate surface completely with essentially no vacant areas. This indicates that crystal growth now either occurs directly at the substrate surface or, at least, in its vicinity when the drop size reduces on account of solvent evaporation. Likewise, such a dendritic morphology is also characteristic for samples of lower concentration. For instance, at 0.7 mg/mL, iminostilbene films retain their overall dendritic morphology, however, with the peculiar structures being now smaller and somehow more defined in shape, as exemplified in Figure 2d. Branches of constant height (ca. 100 nm) are located off the main growth direction and have a well-defined inclination of approximately 60°. In terms of surface coverage, the formation of dendrites means that extended crystalline networks develop but with a high amount of remaining bare substrate area, i.e., iminostilbene assembles in extended dendritic three-dimensional islands rather than following a layer-by-layer growth with full substrate coverage.35
The chemical similarity between carbamazepine and iminostilbene might suggest a similar growth behavior at the silica surface, which is apparently not the case. The presence (or absence) of the carboxamide group strongly alters the intermolecular interaction. For carbamazepine, this group enables different polymorphic phases through the formation of dimers or catemers via hydrogen bonding.27 In iminostilbene, this motif is absent and intermolecular interactions are mainly dominated by van der Waals forces. Importantly, this (seemingly) small change in chemical composition greatly affects key material parameters like, e.g., the solubility; while the carbamazepine solubility is limited to approximately 46 mg/mL in THF (under ambient conditions), the solubility of iminostilbene is significantly higher, exceeding 100 mg/mL. Conversely, supersaturation is reached delayed during drop casting for iminostilbene, thus altering the crystallization behavior at a surface as the solvent evaporates.
To study the film growth of material blends (carbamazepine–iminostilbene), drop cast films containing the two compounds (in 1:1 molar ratio) were prepared on silica surfaces. Again, a strong dependency of the predominant morphology on the overall solute concentration results. AFM images of films prepared from 5.5 and 2.5 mg/mL solute concentration, respectively, are depicted in Figure 2e,f. Both samples exhibit dendritic structures, as observed for the pristine iminostilbene. However, now the inclination between “main axis” and side branches deviates; initially, 60° were observed (Figure 2c,d) while intermixing results in branches of 90° inclination (Figure 2e,f). Besides the large dendritic structures, small needle-shaped crystallites are evident, located both interstitially and on top of dendrites. These findings suggest that each morphological feature is to be assigned to one molecular species but clarification requires further investigations.
The crystallographic properties of the samples were investigated by grazing incidence X-ray diffraction (GIXD) experiments. In such an experiment, the characteristic Bragg reflections (spots or areas of high diffraction intensity) allow for the identification of the polymorph(s) and texture(s) or, more generally, if crystalline order exists at all in the film. In Figure 3a,b, GIXD results for crystalline carbamazepine films are depicted. Casting from a 46 mg/mL solution, the most prominent diffraction features are spots that connect to rings of constant q. Such rings (“Debye–Scherrer rings”) are characteristic for samples containing a powder-like, random distribution of crystallites in which no preferred orientation with respect to the substrate surface exists. Owing to the macroscopic size of the crystallites (cf. Figure 2a), not every possible orientation is realized within this film so that the rings consist of distinct spots rather than homogeneously distributed intensity as expected for a “perfect” powder. Each spot then corresponds to diffraction with an individual crystallite in a certain orientation. The comparison of the ring diameter (i.e., q-value) with calculated values of various carbamazepine polymorphs known from literature, allows identification of the phase present. The evaluation evidence the occurrence of a trigonal polymorph, with a unit cell of a = 35.454(3) Å, b = 35.454(3) Å, c = 5.253(1) Å, α = β = 90°, and γ = 120° (CSD code CBMZPN03).36
Figure 3.
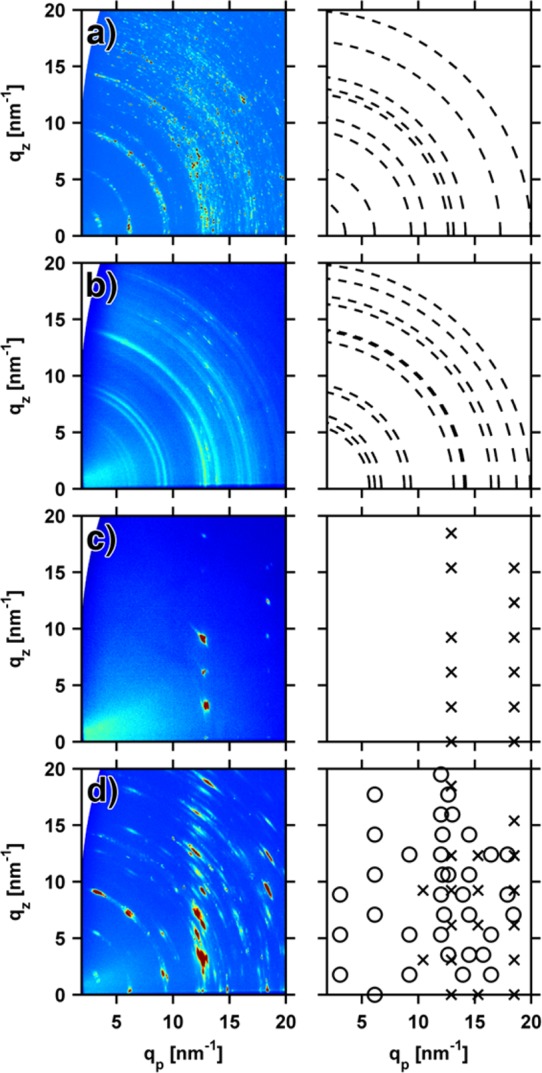
GIXD patterns in reciprocal space map representation (left) and the corresponding assignment (right) for thin films of carbamazepine (a,b), iminostilbene (c), and a 1:1 mixture thereof (d); circles (○) correspond to carbamazepine and crosses (×) to iminostilbene. The films were prepared by drop casting from 46, 5.0, 0.7, and 5.5 mg/mL THF solution, respectively.
At lower solute concentrations of 5 mg/mL, drop cast carbamazepine films behave similarly (see Figure 3b). Powder-like crystallite distribution is evident from an intensity distribution along rings. Owing to the smaller crystallite dimensions obtained from lower solute concentrations (cf. Figure 2b), a more homogeneous intensity distribution results. In some regions stronger intensity (for example at qp = 1.3 nm–1, qz = 0 nm–1) exists, indicating a weak texture within the film. Although being initially amorphous, the samples exhibit only little diffuse scattering so that the amorphous fraction can be expected to be low, i.e., most of the carbamazepine has already crystallized within the 3 weeks between sample preparation and the X-ray experiments. This agrees well with the morphological findings pointing toward defined crystalline fractions of carbamazepine (cf. Figure 2a,b), as the amorphous state typically shows smoother film borders and lower surface roughness. The ring positions evidence the existence of a different polymorph in this sample, with a triclinic unit cell of a = 5.1705(6) Å, b = 20.574(2) Å, c = 22.245(2) Å, α = 84.124(4)°, β = 88.008(4)°, and γ = 85.187(4)° (CSD code CBMZPN11);37 fractions of further polymorphs are not observed within the limits of this experiment. Note that transitions to other polymorphic phases might be feasible by (e.g., thermal) postgrowth treatments but lie beyond the scope of the present study.
In contrast to carbamazepine, the GIXD investigation on iminostilbene reflects a strong texture (Figure 3c). Well-defined Bragg reflections (spots) are found distributed along rods in qz direction at qp = 12.9 and 18.5 nm–1, respectively. These spots are due to iminostilbene grown in an orthorhombic unit cell of lattice constants a = 8.226(3) Å, b = 20.413(6) Å, c = 6.035(2) Å (CSD code BZAZPO).38 This indexes the Bragg reflections as the 1k1 and 2k1 peak series, respectively. The GIXD data evidence that iminostilbene crystallites have a defined contact plane to the substrate surface, i.e., they exhibit a 010 fiber texture at the silica surface. Such a texture can be regarded as two-dimensional powder, as all the crystallites share a common fiber axis while they are azimuthally statistically distributed.
The GIXD experiment of the mixed film containing iminostilbene and carbamazepine shows solely spot-like Bragg features (see Figure 3d) at well-defined locations in the map. This is surprising, as the pristine carbamazepine behaved like a powder (cf. Figure 3b). This means that both materials exhibit fiber-textured growth. There are Bragg spots identical to those found for the pristine iminostilbene, which shows that the polymorphic form of iminostilbene remains the same. Additional Bragg spots, which cannot be ascribed to iminostilbene are, for instance, at qp = 9.1 nm–1, qz = 1.8 nm–1 or qp = 6.2 nm–1, qz = 0 nm–1. Carbamazepine in its trigonal polymorph is able to explain their occurrence.36 The assignment further shows that the carbamazepine crystals have a 110 texture. Overall, this means that iminostilbene addition induces texture within the carbamazepine film portion, which in its pristine form of comparable concentration crystallizes into a different polymorph, exhibiting little orientational order (cf. Figure 3b).
The change in crystallographic order and morphology suggests that iminostilbene acts as a template for the growth of carbamazepine, which, consequently, requires that the former crystallizes prior to the latter. To study in detail the crystallization dynamics, in situ GIXD measurements were performed. The crystallization is monitored as a function of time (t) by continuously recording GIXD patterns with a time resolution of 5 s, starting immediately after drop casting on the substrate surface. For sake of simplicity, the scattering intensities of certain characteristic areas (the respective GIXD maps and areas are given in the Supporting Information) are integrated and plotted as a function of time (Figure 4), allowing the solvent evaporation and the carbamazepine and iminostilbene crystallizations to be followed. The time evolution for a pure 20 mg/mL carbamazepine drop from a THF solution shows several interesting features (Figure 4a). Initially, a rapid decrease in the diffuse scattered intensity is observed (triangles), which, after 40 s, remains constant. This change is due to solvent evaporation, showing that after this time period, no significant amount of solvent is released anymore. The area corresponding to carbamazepine (Figure 4a, circles) requires 5 min until an onset in diffracted intensity can be detected. Over the course of the experiment, a steady increase in intensity is observed for this diffraction signal, which indicates by extrapolation that even after data collection (t ≥ 45 min) crystal growth proceeds; likely, a remaining film portion is still amorphous. The overall time evolution of the carbamazepine diffracted signal (I(t)) exhibits a pronounced “S”-like shape, characteristic for several distinct growth processes. In this context, a well-established formalism for describing such crystallization processes is the Avrami equation.39 In the following, this equation is used with a modification accounting for a variable onset point (parameter d) and a maximum different to one (parameter a), in combination with a linear term (slope s, onset point b) for fitting the experimental data in Figure 4. This leads to
with the Heaviside step function (H) assuring that both terms solely provide positive contributions to the intensity. The addition of a linear term was inferred from the experimental data of the intermixture (Figure 4c), as the Avrami equation cannot account for any interaction within the two-component system. Note that the intensity in the experiment is approximately proportional to the crystallite number and size.40 Therefore, it acts as valuable indicator for the actual crystallization process and, in the framework of this study, for its dynamics. As already mentioned, carbamazepine films are initially unordered and amorphous, but gradually crystallize upon storage. In the above formalism, this is reflected by a low value of the constant k (slowly progressing growth), as well as in the Avrami exponent n. The fits confirm this assumption of the data (Table 1). Usually, the Avrami exponent ranges between 1 and 4 and often relates to a certain growth behavior and/or nucleation type.41 For this experiment, a value of n ≈ 2.5 is obtained, which means growth at constant nucleation rates in all crystal directions takes place (an exponent of 1 may be observed for needle growth). This result further indicates the rather nontextured nature of the pristine carbamazepine crystallization from solution on silica. Supporting ex situ specular X-ray diffraction data as well as optical microscopy suggest that full crystallization within such films may take up to 3 weeks (data not shown). This means that, while the growth is initially initiated shortly after solvent removal (cf. Figure 4a), growth into extended crystals takes several orders of magnitude longer.
Figure 4.

Time evolution of the diffracted intensity from the solvent (△), carbamazepine (○), and iminostilbene (×) in in situ GIXD experiments during drop casting pristine carbamazepine (a), pristine iminostilbene (b), and a 1:1 mixture of both compounds (c); data are normalized to 1, and the number of data points plotted has been reduced for clarity.
Table 1. Fit Parameters of the Intensity vs. Time Data Represented in Figure 4 Following the Modified Avrami Formalism for Carbamazepine (CBZ), Iminostilbene (ISB), and for the Respective Compounds in the Mixturea.
| CBZ | mixture CBZ | ISB | mixture ISB | |
|---|---|---|---|---|
| a [a.u.] | 0.95 ± 0.02 | 0.782 ± 0.006 | 0.88 ± 0.03 | 0.849 ± 0.006 |
| d [s] | 5.1 ± 1.5 | 3.53 ± 0.04 | 1.58 ± 0.03 | 3.38 ± 0.01 |
| k [s–n] | (2.5 ± 2)·10–4 | 0.33 ± 0.02 | 700 ± 1800 | 2.24 ± 0.05 |
| n | 2.5 ± 0.2 | 2.00 ± 0.06 | 2.5 ± 1.3 | 0.86 ± 0.03 |
| s [a.u.] | 0.037 ± 0.001 | 0.015 ± 0.001 | ||
| b [s] | 6.45 ± 0.14 | 2.7 ± 0.2 |
The error is given as the standard error of the fit parameters.
A different time-dependence is observed for pristine iminostilbene (solute concentration 2.5 mg/mL), as shown in Figure 4b. Complete solvent evaporation in this run is observed after 90 s (triangles). On arrival at the background level, i.e., after complete solvent evaporation, it takes a few seconds until diffraction of iminostilbene is detectable (crosses). From this starting point on, the intensity reaches the maximum almost within the time resolution of the experiment (10 s). After this, the intensity remains constant, which indicates that full crystallization has already been achieved in this short period. This is also reflected by the large value of the time constant in the fit according to the Avrami eq (Table 1). As the time resolution was limited by the experimental GIXD setup, both time and Avrami exponents must be treated with some caution; because the time dependence of the intensity (Figure 4b) resembles more a step function than an s-shape curve, their values eventually diverge in the fit (this is also reflected by the large standard error of these parameters).
The in situ investigation of the intermixture of carbamazepine and iminostilbene in 1:1 molar ratio in solution (solute concentration 2.5 mg/mL) demonstrates a deviating behavior in the crystallization behavior for the individual components (Figure 4c). In this experiment, the complete solvent evaporation is observed within 3 min. The onset of iminostilbene diffraction, and thus crystallization, emerges just 10 s thereafter. Eighty-five percent of the maximum intensity is reached after t = 5 min (crosses). After, the curve still has a positive slope up to t = 12 min, i.e., when the experiment was stopped. This is contrary to the pristine film, which did not show a further increase in intensity after a couple of seconds (Figure 4b). Likewise, film preparation from the blend delays the onset of carbamazepine crystallization with respect to that of iminostilbene, as seen, e.g., by the 30 s delay, at which point the latter already exhibits 60% of the normalized intensity (cf. Figure 4c, data points marked with circles and crosses). After the initial growth, a less steep increase in intensity is observed, although crystal growth continues until the end of the experiment (t = 12 min); a positive slope at the end indicates that complete carbamazepine crystallization has not been achieved, and crystal growth further proceeds after the experiment. This linear behavior is just present in the case of the intermixture. A likely explanation is found in interactions between the two compounds, which, at a certain point, start to dominate the crystallization process, resulting in only specific crystallites and/or facets to grow further.
The altered crystallization behavior upon blending is also reflected by the Avrami fit parameters (Table 1). The constant k remains large for iminostilbene but the Avrami exponent is reduced significantly to n = 0.86, as compared to the pristine material. Carbamazepine shows a significant enhancement both in order and in crystallization rate compared to pristine samples, which is also represented by the constant k and the Avrami exponent; crystal growth proceeds much faster and the decreased value of n = 2 indicates a more oriented growth, as compared to the pristine compound. It is noteworthy to mention that all these results are specific to the conditions used to cast the films. A change in the process parameters (e.g., utilized solvent) may lead to a very different behavior, altering crystallization rates and morphology, or may even result in a different polymorphic form.
The ex situ and in situ experiments show, overall, that blending carbamazepine and iminostilbene in solution affects both their crystallization simultaneously. Comparing blended to pristine samples suggests that demixing occurs, i.e., each component crystallizes in its own crystals without the incorporation of the other species, thus cocrystal formation is not observed. However, the morphology is altered. The inclination of the dendritic branches in iminostilbene change from 60° to 90°, suggesting a change of the crystal facet being facilitating during growth, a clear indication of altered interactions in the system. The solvent needs to solvate both types of molecules simultaneously, which, in turn, means that a lower number of solvent molecules is available per solute moiety, and thus, an altered solvent quality results. Such changes in the solvent quality often explains a variation of the growing facets.
In reverse, iminostilbene addition likewise results in an alteration of the carbamazepine growth behavior. In its pristine form, carbamazepine crystallizes in a random fashion on silica surfaces, i.e., a powder-like behavior is present, which takes a long time for crystallization (see above).42 Upon adding iminostilbene to the solution, carbamazepine retains its needle-type character in the drop cast films. The needles are now located in voids between or on top of the dendrites; however, they are always in contact to the iminostilbene morphology. No indications of pure isolated carbamazepine located directly on the surface are found, suggesting that the hydrophobic interaction between the two organic molecules dominates over the interactions with the more hydrophilic silica surface. A closer inspection of the morphology reveals additional order, as needles located on top (exemplary areas marked by dashed lines in Figure 2f) exhibit a characteristic inclination of 45° with respect to the underlying dendritic structures. While this behavior is less perceptible for the higher concentration case where the needles are mostly located interstitially (exemplary areas are marked by arrows in Figure 2e), such an inclination is, however, still present to a certain extent. This indicates that carbamazepine aligns epitaxially on iminostilbene.
As a requirement for epitaxy, iminostilbene growth has to be prior to that of carbamazepine. In fact, the time required to “assemble” such a template by means of dendritic growth is experimentally evident, as the onset of carbamazepine growth is observed significantly later on intermixing. Furthermore, the measurements show that such a template allows inducing a certain degree of alignment to carbamazepine. On the one hand, there is a common contact plane, [110], as a significant fraction of needles show azimuthal alignment with respect to the underlying substrate. This in general is referred to as epitaxy. In addition, this epitaxial-like growth obviously reduces the time required to initiate crystallization for carbamazepine. The reason for this behavior, however, cannot be unambiguously identified as the full epitaxial relationship remains unknown. Nevertheless, in general, faster crystallization occurs as nuclei seem to form faster and eventually evolve into crystals over time. The presence of a solid surface, e.g., that of the dendritic iminostilbene structures, minimizes the entropy of the system. As a consequence, nucleation is facilitated and thus promotes faster crystallization. The mutual epitaxial alignment of two crystals can, in general, be described by various approaches. This includes energy minimization or lattice matching,43 and reports exist that suggest surface corrugation to be responsible for crystal alignment.44 Sketching the respective molecular assemblies in the carbamazepine and iminostilbene unit cells, in consideration of their experimentally determined contact planes, allows to illustrate their mutual molecular orientation at the crystals interface; a structural model is depicted in Figure 5. By nature, the relative azimuth of the respective crystals is not directly accessible from the present GIXD data. However, a closer inspection of the crystal structures reveals that both lattices exhibit regularly spaced and linear hydrogen rows on their contact planes (oriented into the plane of the page in Figure 5). Therefore, we expect the epitaxial order to be influenced by this very corrugation and that these hydrogen rows align parallel in the film, as indicated in Figure 5.
Figure 5.
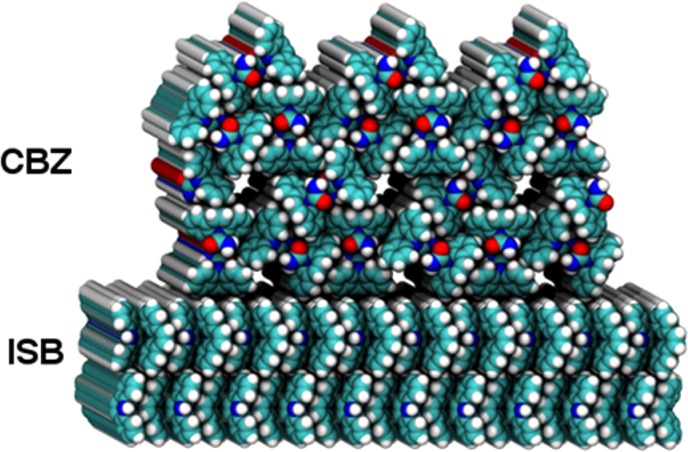
Sketch of the suggested epitaxial alignment of carbamazepine (CBZ) on iminostilbene (ISB) on the basis of the experimental GIXD data; layers are illustrated using the software package VMD.45
Conclusion
The order of carbamazepine and the time frame for crystal growth (dynamic) is demonstrated to change on the addition of iminostilbene during the production process. Ex situ and in situ experiments reveal poor ordering and the long time scales necessary for full crystallization within the pristine carbamazepine films, when drop cast onto silica surfaces. On intermixing with iminostilbene, the order with respect to the substrate surface is drastically changed, and the time required for crystallization is reduced as well. Iminostilbene exhibits well-defined dendritic structures of crystallographic 010 texture, which develop rapidly after solvent evaporation. This results in demixing of the two compounds rather than cocrystallization. The earlier appearance of iminostilbene crystals, however, then acts as a template for carbamazepine, which crystallized much faster upon solvent evaporation. In situ experiments evidence that the logical requirement of the template to be established prior to initial carbamazepine crystallization is indeed fulfilled, allowing epitaxial growth. Such an experimental approach may be of high interest for systems exhibiting slow crystallization, often present for pharmaceutics. In particular, using precursors and their successors may generally facilitate epitaxial growth due to their chemical and thus often structural similarities. Employing intrinsic epitaxy/alignment via a simple solution-casting process, as demonstrated here, could be of interest for various practical applications, including pharmaceutical formulation. This may allow for the crystallization of active drugs into a preferential alignment, enhancing dissolution properties, at once, while still maintaining a low-cost and high-throughput manufacturing process.
Acknowledgments
The work was funded by the Austrian Science Fund (FWF): [P25541-N19]. The authors want to thank the NAWI-Graz. We thank Helmholtz-Zentrum Berlin (HZB) for the allocation of synchrotron radiation beamtime and Daniel Többens (HZB) for experimental support in using beamline KMC-2. The in situ GIXD experiments were carried out at the Austro SAXS beamline at Elettra, Trieste, Italy. The authors thank Heinz Amenitsch for the support during the beamtime.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.cgd.6b00090.
The authors declare no competing financial interest.
Supplementary Material
References
- Newman A. W.; Byrn S. R. Drug Discovery Today 2003, 8, 898–905. 10.1016/S1359-6446(03)02832-0. [DOI] [PubMed] [Google Scholar]
- Yan W.; Hsiao V. K. S.; Zheng Y. B.; Shariff Y. M.; Gao T.; Huang T. J. Thin Solid Films 2009, 517, 1794–1798. 10.1016/j.tsf.2008.09.080. [DOI] [Google Scholar]
- Werzer O.; Baumgartner R.; Zawodzki M.; Roblegg E. Mol. Pharmaceutics 2014, 11, 610–616. 10.1021/mp4006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipp D.; Street R. A.; Völkel A.; Ho J. J. Appl. Phys. 2003, 93, 347–355. 10.1063/1.1525068. [DOI] [Google Scholar]
- Chang P. C.; Lee J.; Huang D.; Subramanian V.; Murphy A. R.; Fréchet J. M. J. Chem. Mater. 2004, 16, 4783–4789. 10.1021/cm0496570. [DOI] [Google Scholar]
- Virkar A. A.; Mannsfeld S.; Bao Z.; Stingelin N. Adv. Mater. 2010, 22, 3857–3875. 10.1002/adma.200903193. [DOI] [PubMed] [Google Scholar]
- Li Y.; Cai W.; Duan G. Chem. Mater. 2008, 20, 615–624. 10.1021/cm701977g. [DOI] [Google Scholar]
- Newbloom G. M.; Kim F. S.; Jenekhe S. A.; Pozzo D. C. Macromolecules 2011, 44, 3801–3809. 10.1021/ma2000515. [DOI] [Google Scholar]
- Mahan J. E.Physical Vapor Deposition of Thin Films; Wiley, 2000. [Google Scholar]
- Coclite A. M.; Howden R. M.; Borrelli D. C.; Petruczok C. D.; Yang R.; Yagüe J. L.; Ugur A.; Chen N.; Lee S.; Jo W. J.; Liu A.; Wang X.; Gleason K. K. Adv. Mater. 2013, 25, 5392–5423. 10.1002/adma.201301878. [DOI] [PubMed] [Google Scholar]
- Wieringa R. H.; Schouten A. J. Macromolecules 1996, 29, 3032–3034. 10.1021/ma9508358. [DOI] [Google Scholar]
- Tsao H. N.; Cho D.; Andreasen J. W.; Rouhanipour A.; Breiby D. W.; Pisula W.; Müllen K. Adv. Mater. 2009, 21, 209–212. 10.1002/adma.200802032. [DOI] [Google Scholar]
- DeLongchamp D. M.; Vogel B. M.; Jung Y.; Gurau M. C.; Richter C. A.; Kirillov O. A.; Obrzut J.; Fischer D. A.; Sambasivan S.; Richter L. J.; Lin E. K. Chem. Mater. 2005, 17, 5610–5612. 10.1021/cm0513637. [DOI] [Google Scholar]
- Erdemir D.; Lee A. Y.; Myerson A. S. Acc. Chem. Res. 2009, 42, 621–629. 10.1021/ar800217x. [DOI] [PubMed] [Google Scholar]
- Kellner T.; Ehmann H. M. A.; Schrank S.; Kunert B.; Zimmer A.; Roblegg E.; Werzer O. Mol. Pharmaceutics 2014, 11, 4084–4091. 10.1021/mp500264e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann H. M. A.; Werzer O. Cryst. Growth Des. 2014, 14, 3680–3684. 10.1021/cg500573e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka E.; Makita M.; Yamamura S. Chem. Pharm. Bull. 1986, 34, 4314–4321. 10.1248/cpb.34.4314. [DOI] [PubMed] [Google Scholar]
- Van Eerdenbrugh B.; Baird J. A.; Taylor L. S. J. Pharm. Sci. 2010, 99, 3826–3838. 10.1002/jps.22214. [DOI] [PubMed] [Google Scholar]
- Ehmann H. M. A.; Zimmer A.; Roblegg E.; Werzer O. Cryst. Growth Des. 2014, 14, 1386–1391. 10.1021/cg401859p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L. Adv. Drug Delivery Rev. 2001, 48, 27–42. 10.1016/S0169-409X(01)00098-9. [DOI] [PubMed] [Google Scholar]
- Jones A. O. F.; Chattopadhyay B.; Geerts Y. H.; Resel R. Adv. Funct. Mater. 2016, 3169. 10.1002/adfm.201503169. [DOI] [Google Scholar]
- McNamara D. P.; Childs S. L.; Giordano J.; Iarriccio A.; Cassidy J.; Shet M. S.; Mannion R.; O’Donnell E.; Park A. Pharm. Res. 2006, 23, 1888–1897. 10.1007/s11095-006-9032-3. [DOI] [PubMed] [Google Scholar]
- Kline B. J.; Saenz J.; Stanković N.; Mitchell M. B. Org. Process Res. Dev. 2006, 10, 203–211. 10.1021/op050176r. [DOI] [Google Scholar]
- Chadwick K.; Myerson A.; Trout B. CrystEngComm 2011, 13, 6625. 10.1039/c1ce05871a. [DOI] [Google Scholar]
- Brinkmann M.; Wittmann J.-C. Adv. Mater. 2006, 18, 860–863. 10.1002/adma.200501838. [DOI] [Google Scholar]
- Bertilsson D. L.; Tomson T. Clin. Pharmacokinet. 2012, 11, 177–198. 10.2165/00003088-198611030-00001. [DOI] [PubMed] [Google Scholar]
- Arlin J.-B.; Price L. S.; Price S. L.; Florence A. J. Chem. Commun. 2011, 47, 7074–7076. 10.1039/c1cc11634g. [DOI] [PubMed] [Google Scholar]
- Harris R. K.; Ghi P. Y.; Puschmann H.; Apperley D. C.; Griesser U. J.; Hammond R. B.; Ma C.; Roberts K. J.; Pearce G. J.; Yates J. R.; Pickard C. J. Org. Process Res. Dev. 2005, 9, 902–910. 10.1021/op0500990. [DOI] [Google Scholar]
- Fleischman S. G.; Kuduva S. S.; McMahon J. A.; Moulton B.; Bailey Walsh R. D.; Rodríguez-Hornedo N.; Zaworotko M. J. Cryst. Growth Des. 2003, 3, 909–919. 10.1021/cg034035x. [DOI] [Google Scholar]
- Vishweshwar P.; McMahon J. A.; Oliveira M.; Peterson M. L.; Zaworotko M. J. J. Am. Chem. Soc. 2005, 127, 16802–16803. 10.1021/ja056455b. [DOI] [PubMed] [Google Scholar]
- Vardanyan R.; Hruby V.. Synthesis of Essential Drugs; Elsevier, 2006. [Google Scholar]
- Neuhold A.; Novák J.; Flesch H.-G.; Moser A.; Djuric T.; Grodd L.; Grigorian S.; Pietsch U.; Resel R. Nucl. Instrum. Methods Phys. Res., Sect. B 2012, 284, 64–68. 10.1016/j.nimb.2011.07.105. [DOI] [Google Scholar]
- Kriegner D.; Wintersberger E.; Stangl J. J. Appl. Crystallogr. 2013, 46, 1162–1170. 10.1107/S0021889813017214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nečas D.; Klapetek P. Cent. Eur. J. Phys. 2011, 10, 181–188. [Google Scholar]
- Venables J. A.; Spiller G. D. T.; Hanbucken M. Rep. Prog. Phys. 1984, 47, 399–459. 10.1088/0034-4885/47/4/002. [DOI] [Google Scholar]
- Lowes M. M.; Caira M. R.; Lötter A. P.; Van der Watt J. G. J. Pharm. Sci. 1987, 76, 744–752. 10.1002/jps.2600760914. [DOI] [PubMed] [Google Scholar]
- Grzesiak A. L.; Lang M.; Kim K.; Matzger A. J. J. Pharm. Sci. 2003, 92, 2260–2271. 10.1002/jps.10455. [DOI] [PubMed] [Google Scholar]
- Reboul J. P.; Cristau B.; Soyfer J. C.; Astier J. P. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1981, 37, 1844–1848. 10.1107/S0567740881007383. [DOI] [Google Scholar]
- Avrami M. J. Chem. Phys. 1939, 7, 1103–1112. 10.1063/1.1750380. [DOI] [Google Scholar]
- Birkholz M.Thin Film Analysis by X-Ray Scattering; Wiley-VCH Verlag GmbH & Co. KGaA, 2005. [Google Scholar]
- Christian J. W.The Theory of Transformations in Metals and Alloys; Pergamon: Oxford, 2002. [Google Scholar]
- Gunn E. M.; Guzei I. A.; Yu L. Cryst. Growth Des. 2011, 11, 3979–3984. 10.1021/cg2005503. [DOI] [Google Scholar]
- Mitchell C. A.; Yu L.; Ward M. D. J. Am. Chem. Soc. 2001, 123, 10830–10839. 10.1021/ja004085f. [DOI] [PubMed] [Google Scholar]
- Koini M.; Haber T.; Werzer O.; Berkebile S.; Koller G.; Oehzelt M.; Ramsey M. G.; Resel R. Thin Solid Films 2008, 517, 483–487. 10.1016/j.tsf.2008.06.053. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.