

Abstract
The thermal decomposition of fulvene endoperoxides ordinarily proceeds via an allene oxide intermediate affording oxepin-2(3H)-one derivatives. We have now uncovered new, unusual pathways in these decompositions where the presence of a hydroxyl group on the alkyl or aryl attached to the fulvene exocyclic double bond has a profound effect on the fate of the reactive intermediates derived from the unstable endoperoxides. Computational work supports the proposed mechanistic pathways.
Keywords: fulvenes, endoperoxides, allene oxides, cyclopropanones, oxy-allyl
Graphical Abstract

1. Introduction
Fulvene-singlet oxygen (1O2) adducts are among the most strained and least stable bicyclic endoperoxides known to date.1 It is not surprising that they are also the most intensely studied organic peroxides owing to the unusual pathways encountered during their decomposition. The most notable feature common to fulvene endoperoxides is the fact that the rupture of the labile O-O bond leads to reactive intermediates such as allene oxides and cyclopropanones (Scheme 1).2,3
Scheme 1.

Normal course of fulvene endoperoxide isomerizations
Though the latter had been implicated as likely precursors of the oxepin-3(2H)-ones (e.g., 4) via electrocyclization of the transient cyclopropanone derivative, we have been able to verify the proposed mechanism by isolating a stable allene oxide from the photooxygenation of a saturated analog of 1 (t-butyl substitution at the exocyclic double bond).4 Moreover, we trapped the cyclopropanone intermediates derived from the saturated fulvene endoperoxides by Diels-Alder reactions,5 intramolecularly via vinylcyclopropanone-cyclopentenone cyclizations,6 and reported the first aliphatic examples of [3.4] sigmatropic shifts. (Scheme 2).7
Scheme 2.

Trapping experiments of cyclopropanones derived from saturated fulvene endoperoxides
More recently we reported that endoperoxides derived from bicyclic fulvenes (i.e., 1,2-dihydropentalenes) undergo decomposition in an entirely different manner; the cyclopropanone intermediate in this case undergoes decarbonylation or intramolecular 1,3-acyl shift in a temperature-dependent reaction.8
We now report that the course of the thermal isomerizations of fulvene endoperoxides is dramatically altered by incorporating a hydroxyl group into the substituents at the exocyclic double bond of the starting fulvene. The systems we studied, 11, 12 and 13, are shown in Figure 1.
Figure 1.

Fulvenes 11, 12, and 13 used in this study carrying OH groups.
2. Results and Discussion
All three substrates are readily available in high yields by the catalytic method we reported recently, or the stoichiometric method by Stone and Little.9
Photooxygenation of 11 in CH2Cl2 at −78 °C, using TPP as sensitizer and allowing the photolysate to warm up to room temperature gave a mixture of two products, formed in a ratio of 3:1, respectively, which were separated from one another by flash chromatography and identified as 14 and 15 (combined yield 72%, Scheme 3).
Scheme 3.

Photooxygenation of 11 leading to 14 and 15.
Whereas the formation of 14 was expected, furan 15 is an unusual product, and its formation is unprecedented. A favorable shift-often observed in oxygen centered radicals- is a 1,2-H shift to the oxygen radical10 (transition state enthalpy = −11.8 kcal/mol relative to 16), giving an allenyl enol, 19. Rapid tautomerization would also lead to 20, and eventually to furan 15 via the corresponding γ-lactol followed by dehydration (Scheme 4).
Scheme 4.

Mechanism for furan 15 formation
Alternatively, a 1,2-hydrogen atom transfer (HC→C·)in the oxygen-centered radical 18, ensuing a common β-fission of the initial diradical might also account for the furan precursor (18→20→15), however, we consider a 1,2-H transfer to the adjacent vinyl highly unlikely due to a calculated activation enthalpy (M062x/6-311+G** with a PCM CH2Cl2 solvent model) that is 8.8 kcal/mol above the energy of 16.11
In the second system we studied (12) the hydroxyl group is one more carbon further down the alkyl chain than in 11, and this subtle variation altered the course of the endoperoxide decomposition dramatically. Upon singlet oxygen addition at 78 °C in CH2Cl2, and subsequent warming the solution to room temperature, 12 gave a 2:1 mixture of 2212 and 23 in a combined yield of 68% (Scheme 5).
Scheme 5.

Photooxygenation products from 12
Again, the formation of the expected product 23 deserves no further comment. Compound 23, on the other hand, apparently stems from a pathway wherein a fragmentation must have occurred since 23 lacks a CH2O unit (formaldehyde) as compared to the endoperoxide 21 derived from 12. A mechanism, consistent with the results is outlined in Scheme 6.
Scheme 6.

Mechanism for the formation of 23 from 21
A reasonable pathway rationalizing the formation of 23 involves a rare 1,7-hydrogen atom transfer (HAT)13 from the hydroxyl group to the proximal oxygen-centered radical in 25. Though rarely observed, it has been found that if the proper spatial orientation of the relevant hydrogen atom toward the oxygen- or carbon-centered radical is provided (an 8-membered transition state obviously suffers from a considerably unfavorable entropy of activation), 1,7-hydrogen abstractions can effectively compete with the more ubiquitous 1,5-H shifts. Molecular modeling at B3LYP/6-31G* level of theory indicates a favorable geometry for the 1,7-H abstraction with a H—O distance of 1.83 Å. The ensuing fragmentation in 26 leading to 23 and formaldehyde (27) is akin to the retro-Paterno-Buchi reaction, though carbon-centered 1,4-diradical intermediates have been implicated in the latter reactions.14 Although 22 and 23 could be separated from one another by flash chromatography, longer exposure of 23 to silica gel resulted in acid-catalyzed epoxide ring opening followed by a 1,2-hydride shift to give cis-4-hydroxyl-5-isopropenylcyclopent-2-enone (29). Moreover, compound 22 quantitatively isomerized to 28 during SiO2 chromatography via acid-catalyzed translactonization (Scheme 7).
Scheme 7.

SiO2 catalyzed isomerizations of 22 and 23
In the third system of our study, the hydroxyl group is placed in the ortho position of a phenyl group at C6 in the fulvene derived from salicyl aldehyde (13). Singlet oxygen addition was conducted under the same conditions as before at −78 °C, however, CD2Cl2 was used as solvent in order to monitor the progress of the reaction by 1H NMR and to avoid the loss of volatile products during the solvent removal by rotary evaporation. Indeed, upon warming the photolysate to room temperature, the 1H NMR of the crude product mixture revealed that two products, furan (30) and 2-coumaranone (31, benzofuran-2(3H)-one) were formed in a 1:1 ratio (Scheme 8).
Scheme 8.

Products 30 and 31 from the singlet oxygenation of 13
The formation of 30 and 31 from the singlet oxygenation of 13 is remarkable in that the endoperoxide appears to have undergone a major fragmentation during one phase of decomposition. At first glance it is not obvious how the phenolic OH would affect the endoperoxide isomerization to the extent that two aromatic compounds, 30 and 31 would be split from one reactive intermediate. A plausible mechanism consistent with the experimental results is depicted in Scheme 9.
Scheme 9.

Photooxidative fragmentation of fulvene 13 into furan (30) and 2-coumaranone (31)
The key impact of the phenol is stabilization of the oxy-allyl intermediate, 34a. Strong hydrogen bonding is possible with the phenolic OH and computational modeling (M062x/6-311+G** with a CH2Cl2 solvent model) suggests that it might be better described as a quinone methide, 34b. In any case, the interaction provides over 20 kcal/mol of stabilization of the oxy-allyl intermediate relative to the allene oxide, 33.
This creates a situation where the oxy-allyl intermediate is more stable than the allene oxide and cyclopropanone isomers by 18.5 and 9.3 kcal/mol, respectively, predisposing the system to follow the path in Scheme 10 (ring-expansion to the lactone must pass through a cyclopropanone intermediate)15. In the absence of the hydroxyl substituent, the cyclopropanone is the preferred isomer on the potential energy surface by nearly 9 kcal/mol. In addition, the barrier to furan cyclization increases by 12 kcal/mol relative to the allene oxide (Supplementary Material, Table S1).
Scheme 10.

Thermal isomerization of the saturated endoperoxide 37 to 38
Next, we studied the thermal behavior of the saturated analog of endoperoxide 32. Selective diazene reduction of 32 at low temperature (−78–0 °C) gave the saturated endoperoxide 37. An aliquote of the endoperoxide solution in CH2Cl2was carefully concentrated under vacuum and a 1H NMR spectrum was taken at 0 °C confirming the structure of 37. It was left at room temperature overnight to decompose, and the crude product analyzed by 1H NMR. It showed the presence of one single product, the bicyclic acetal 38 (Scheme 10).
During the SiO2 chromatography, 38 partially isomerized to the hemiacetal 42 by way of endo-3-(2-hydroxyphenyl)-7-oxabicyclo[2.2.1]heptan-2-one (41). The endo-stereochemistry in 42 was assigned based on the coupling constants (3J) of C1-H, C3-H and C4-H (3J3,4= 6.0 Hz).16 This assignment is also consistent with the Z configuration we postulated for the allene oxide 33 derived from the unsaturated analog (Scheme 11).
Scheme 11.

Formation of 38 from allene oxide 39; SiO2 catalyzed isomerization of 38 to 42
Whereas the C-C cleavage in 35 leading to 36 and furan (30), an aromatic compound, is favored, this pathway is impeded in the saturated analog 39. Intramolecular cyclization of 40 leads to the bicyclic acetal 38. We previously reported similar intramolecular allene oxide-bicyclic acetal cyclizations during thermal isomerizations of saturated fulvene endoperoxides.4-6
It is noteworthy that the hydroxyl group indeed alters the course of fulvene endoperoxide decompositions since the methoxy derivative of 13, the fulvene derived from o-anisaldehyde (43)17 gave upon photooxygenation the expected oxepin-3(2H)-one 44 (Scheme 12). During chromatography on SiO2 partial double bond isomerization to the conjugated oxepin-2(7H)-one isomer 45 occurred due to monosubstitution at C3.18
Scheme 12.
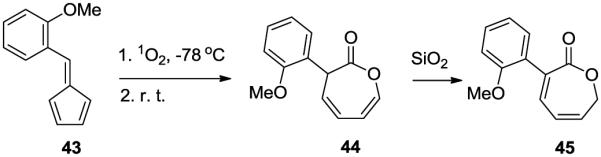
Photooxidative isomerization of 43 to the oxepinone derivatives.
3. Conclusions
In conclusion, we report here some unusual pathways in fulvene endoperoxide decompositions brought on by the presence of substituents at C6 carrying hydroxyl groups: furan formation, 1,2- or 1,7-hydrogen shifts, and fragmentations represent significant deviations from the previously reported decomposition mechanisms observed in these reactions. Moreover, the proposed pathways have been supported by computational modeling in several cases.
Supplementary Material
Fulvene endoperoxides ordinarily decompose via allene oxide intermediates
A hydroxyl substituent has a significant effect on the decomposition mechanism
In some cases the hydroxyl group causes intramolecular 1,7-H shifts
Allene oxide/oxy-allyl species have been implicated in some of the isomerizations
Molecular modeling studies support the proposed pathways
Acknowledgments
I. Erden acknowledges financial support of this work by funds from the National Institutes of Health (Grant No. SC1 GM082340). D.P. is grateful to University of Florence, Italy, for financial support for a study visit at SFSU. The mass spectrometry work at SFSU was in part supported by a grant from the National Science Foundation (CHE-1228656) and is gratefully acknowledged. S. Gronert acknowledges support from National Science Foundation (CHE-1300817). We also thank Dr. Robert Yen, SFSU, for the HRMS spectra and Mr. Krish Kumar for technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary material (detailed experimental procedures and characterization data for all compounds; 1H and 13C NMR and FT-IR and HRMS data; full citation for reference (10); Table S1 contains calculated reaction enthalpies; and energies and geometries (xyz format) for all computed species) associated with this article can be found in the online version at
References and notes
- 1.Saito I, Matsuura T. The Oxidation of Electron-Rich Aromatic Compounds. In: Wasserman HH, Murray RW, editors. Singlet Oxygen. Vol. 40. Organic Chemistry Series; Academic Press Inc.; New York, NY: 1979. pp. 511–574. [Google Scholar]
- 2.(a) Harada N, Suzuki S, Uda H, Ueno H. J. Am. Chem. Soc. 1972;94:1777–1778. [Google Scholar]; (b) Skorianetz W, Schulte-Elte KH, Ohloff G. Angew. Chem. Int. Ed. Engl. 1972;11:304–305. [Google Scholar]; (c) Skorianetz W, Schulte-Elte KH, Ohloff G. Helv. Chim. Acta. 1971;54:1913–1922. [Google Scholar]; (d) Zhang X, Lin F, Foote CS. J. Org. Chem. 1995;60:1333–1338. [Google Scholar]; (e) Erden I, Ocal N, Song J, Gleason C, Gärtner C. Tetrahedron. 2006;62:10676–10682. doi: 10.1016/j.tet.2006.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adam W, Erden I. Angew. Chem. Int. Ed. Engl. 1978;17:210–211. [Google Scholar]
- 4.(a) Erden I, Drummond J, Alstad R, Xu F. Tetrahedron Lett. 1993;34:1255–1258. [Google Scholar]; (b) Erden I, Drummond J, Alstad R, Xu F. Tetrahedron Lett. 1993;34:1255–1258. [Google Scholar]
- 5.Erden I, Amputch M. Tetrahedron Lett. 1987;28:3779–3782. [Google Scholar]
- 6.(a) Erden I, Drummond J, Alstad R, Xu F. J. Org. Chem. 1993;58:3611–3612. [Google Scholar]; (b) Erden I, Gärtner C. Tetrahedron Lett. 2009;50:2381–2383. doi: 10.1016/j.tetlet.2009.02.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erden I, Xu F, Cao W. Angew. Chem. Int. Ed. Engl. 1997;36:1516–1518. [Google Scholar]
- 8.Erden I, Ma J, Gärtner C, Azimi S, Gronert S. Tetrahedron. 2013;69:5044–5047. doi: 10.1016/j.tet.2013.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Coskun N, Erden I. Tetrahedron. 2011;67:8607–8614. doi: 10.1016/j.tet.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Erden I, Xu FP, Sadoun A, Smith W, Sheff G, Ossun M. J. Org. Chem. 1995;60:813–820. [Google Scholar]; (c) Stone K, Little RD. J. Org. Chem. 1984;49:1849–1853. [Google Scholar]
- 10.Von Sontag C. Free-Radical-Induced DNA Damage and its Repair: A Chemical Perspective. Springer; Heidelberg: 2006. p. 138. 7. [Google Scholar]
- 11.Frisch MJ, et al. Gaussian09, Revision C.01. Gaussian Inc.; Wallingford CT: 2009. see Supplementary Material for full reference. [Google Scholar]
- 12. Though compound 23 was identified in the crude 1H NMR spectrum based on its characteristic signals, it was unstable on the silica gel column, in spite of pre-treatment with NEt3. It quantitatively underwent translactonization to 28 during chromatography (see Scheme 8)
- 13.Attouche A, Urban D, Beau J-M. Angew. Chem. Int. Ed. Engl. 2013;52:9572–9575. doi: 10.1002/anie.201301783. To the best of our knowledge, a 1,7-H transfer from ROH to R’O·has not previously been reported. For a 1,7-H shift from a benzylic methylene to an alkoxy radical, see. references cited therein. [DOI] [PubMed] [Google Scholar]
- 14.(a) Paterno E, Chieffi G. Gazz. Chim. Ital. 1909;39:341. [Google Scholar]; (b) Buchi G, Inman CG, Lipinsky ES. J. Amer. Chem. Soc. 1954;76:4327–4331. [Google Scholar]
- 15. Cyclopropanone formation is computed to be rate-limiting in the path to the ring-expanded lactone. The computed barrier to furan cyclization is 3 kcal/mol higher than the barrier to cyclopropanone formation. Although the polarized continuum model calculations are not able to reproduce the preference for the furan cyclization, they clearly highlight the dramatic impact of the phenol on the stability of the oxy-allyl intermediate and on the relative reaction barriers.
- 16. Numbering based on the uncyclized bicyclic ketone 41.
- 17.Cini M, Bradshaw TD, Woodward S, Lewis W. Angew. Chem. Int. Ed. Engl. 2015;54:14179–14182. doi: 10.1002/anie.201508034. [DOI] [PubMed] [Google Scholar]
- 18. See Supplementary Material for experimental details and spectral characterization of 44 and 45.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.