

Abstract
Transfer RNA (tRNA) molecules contain many chemical modifications that are introduced after transcription. A major form of these modifications is methyl transfer to bases and backbone groups, using S-adenosyl methionine (AdoMet) as the methyl donor. Each methylation confers a specific advantage to tRNA in structure or in function. A remarkable methylation is to the G37 base on the 3' side of the anticodon to generate m1G37-tRNA, which suppresses frameshift errors during protein synthesis and is therefore essential for cell growth in all three domains of life. This methylation is catalyzed by TrmD in bacteria and by Trm5 in eukaryotes and archaea. Although TrmD and Trm5 catalyze the same methylation reaction, kinetic analysis reveal that these two enzymes are unrelated to each other and are distinct in their reaction mechanism. This chapter summarizes the kinetic assays that are used to reveal the distinction between TrmD and Trm5. Three types of assays are described, the steady-state, the pre-steady-state, and the single turnover assays, which collectively provide the basis for mechanistic investigation of AdoMet-dependent methyl transfer reactions.
Keywords: AdoMet-dependent methyl transfer, rapid equilibrium binding
1. Introduction
Naturally occurring tRNA molecules contain many post-transcriptionally modified bases and backbones, where the standard nucleotides (A, C, G, and U) are modified with the addition of various chemical moieties. Each post-transcriptional modification is synthesized in a specific enzymatic pathway on the primary tRNA transcripts. Modifications that occur in the tRNA elbow region mainly contribute to the tertiary folding of the nucleic acid and those in the anticodon region mainly contribute to the protein synthesis activity on the ribosome. These and other post-transcriptional modifications can also play a role in the cellular response to stress (Yi and Pan, 2011). Although other types of cellular RNA (e.g., mRNA, rRNA, microRNA, piwi-interacting RNA, and small interfering RNA) also contain post-transcriptional modifications (Yi and Pan, 2011), tRNA is distinguished with the most diversity and abundance. A major form of post-transcriptional modifications in tRNA is methylation. For example, of the 76 nucleotides in the standard sequence framework of E. coli tRNA, up to 9 different types of methylation can occur (Figure 1A, Table 1), whereas other types of modifications (pseudouridynation, thiolation, dihydrouridylation, etc) are much less frequent. Given the dominance of methylation, the question of how the addition of a single methyl group to a nucleotide can modulate tRNA structure and activity becomes increasingly important.
Figure 1.

AdoMet-dependent methyl transfer to E. coli tRNA. (A) The cloverleaf structure of tRNA is made up of nucleotides as circles. Conserved nucleotides are shown in letters, non-conserved nucleotides are shown as closed circles, and the anticodon nucleotides are shown as open circles. The position of each AdoMet-dependent methylation (catalyzed by the E. coli enzyme in parentheses) is indicated. The nucleotide numbering is based on the standard sequence framework of tRNA (Sprinzl et al., 1998). (B) The chemical structure of AdoMet.
Table 1. AdoMet-dependent tRNA methyl transferases in E. coli.
References for each enzyme are as follows: EcTrmA (Ny and Bjork, 1980), EcTrmB (De Bie et al., 2003), EcTrmC (Taya and Nishimura, 1973), EcTrmD (Bystrom and Bjork, 1982b), EcTrmH (Persson et al., 1997), EcTrmJ (Purta et al., 2006), EcTrmL (Benitez-Paez et al., 2010), EcTrmM (Golovina et al., 2009), and EcTrmO (Kimura et al., 2014). Note that EcTrmO has a unique single-sheeted β-barrel structure that does not belong to any of the 5 known classes of methyl transferases and is designated as class VIII (Kimura et al., 2014). Only the first reference is given for each enzyme.
| Enzyme (Synonyms) |
Modification | Chemical structure | Class | Enzyme (Synonyms) |
Modification | Chemical structure | Class |
|---|---|---|---|---|---|---|---|
| TrmA (rT) |
m5U54 (T54) | 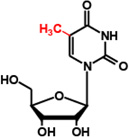 |
I | TrmJ (YfhQ) |
Cm32/Um32 | 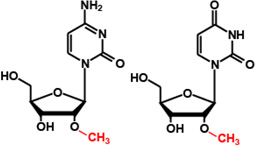 |
IV |
| TrmB (TrmI, YggH) |
m7G46 | 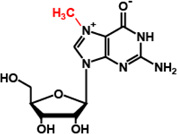 |
I | TrmL (YibK) |
Cm34/ cmnm5Um34 |
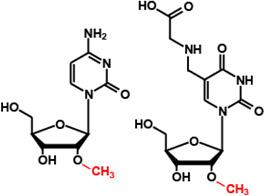 |
IV |
| TrmC (MnmC, YfcK) |
mnm5s2U34 | 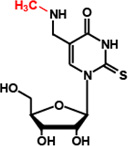 |
I | ||||
| TrmD | m1G37 | 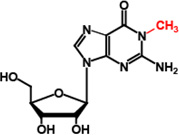 |
IV | TrmM (YfiC) |
m6A37 | 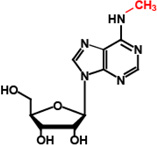 |
I |
| TrmH (SpoU) |
Gm18 | 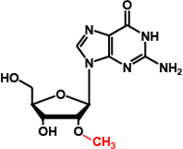 |
IV | TrmO (TsaA, YaeB) |
m6t6A37 | 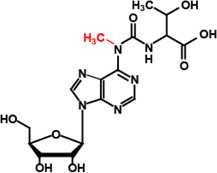 |
VIII |
To address the role of each methylation to tRNA, it is necessary to understand the reaction mechanism of the methyl transferase and to relate it to biology. One consideration is the methyl donor of the methyl transferase. The greatest majority of methyl transferases use AdoMet as the methyl donor. The preference for AdoMet over other methyl donors, such as folate, reflects the favorable energetics resulting from a nucleophilic attack on the positively charged methyl group of the sulfonium center (Figure 1B). The energy release upon methyl transfer from AdoMet is more than twice of the energy release upon hydrolysis of ATP to ADP and Pi (Cantoni, 1975). The target atoms performing the nucleophilic attack on the methyl group of AdoMet are diverse, including nitrogen, oxygen, carbon, and sulfur. AdoMet itself is synthesized by condensation of methionine with ATP by methionine adenosyl transferase (or SAM synthetase) (Markham et al., 1980), and the product of methyl transfer is S-adenosyl homocysteine (AdoHcy). The ratio of AdoMet versus AdoHcy is defined as the methylation potential and it is subject to change according to the energy state of a cell. In mammals, the reduction of the methylation potential lengthens the circadian rhythm (Fustin et al., 2013), indicating a broad impact on cell physiology. This observation then raises the question of whether the circadian lengthening is due to the loss of methylation at specific sites in a specific subset of tRNA. It is possible that, upon the reduction of the methylation potential, certain types of tRNA methylation remain while others decrease or disappear, leading to changes in the tRNA structure and activity. To address the dynamics of such changes in cells, a pre-requisite is an understanding of the sensitivity of each tRNA methyl transferase to alterations of the AdoMet level in kinetic analysis
A second consideration is the structural conformation of AdoMet when bound to a tRNA methyl transferase. To date, at least 5 classes (class I–V) of structurally distinct AdoMet-dependent methyl transferases have been identified (Schubert et al., 2003). The distinction among the 5 classes is the topological structural fold that binds AdoMet and as such the conformation of AdoMet in each fold. This wide diversity among AdoMet-dependent methyl transferases is paralleled only by the diversity among ATP-dependent protein kinases and phosphoryl transferases (Schubert et al., 2003). The discovery of different AdoMet conformations among methyl transferases suggests the existence of distinct reaction mechanisms. For example, the class I fold is the most common of the 5 classes and it includes the greatest majority of tRNA methyl transferases, including TrmA for synthesis of m5U54 and TrmB for synthesis of m7G46 in E. coli (Table 1) (Hou and Perona, 2010). The class I fold binds AdoMet in the open space of a dinucleotide-fold (also known as the Rossmann-fold) (Schubert et al., 2003). A signature of the class I fold is that AdoMet adopts a straight conformation, where the adenosine and methionine moieties are extended in opposite direction from each other (Schubert et al., 2003). The other type of tRNA methyl transferases has the class IV fold, which is characterized by binding AdoMet to the bottom of a deep cleft in a topologically knotted protein fold. The class IV fold is rare among protein structures and it is made up of three passages of the protein backbone in and out of a loop in a structure known as the trefoil-knot fold (Nureki et al., 2002; Nureki et al., 2004). A signature of the class IV fold is that AdoMet adopts a sharp bent conformation, where the two component moieties are spatially facing each other almost at a right angle (Schubert et al., 2003). Only a limited number of E. coli tRNA methyl transferases have the class IV fold and these include TrmD for synthesis of m1G37, TrmH for synthesis of Gm18 (Gm = 2’-O-methyl), TrmJ for synthesis of Cm32/Um32, and TrmL for synthesis of Cm34/Um34 (Table 1).
TrmD presents an important case for why kinetic analysis of methyl transfer is critical for understanding the reaction mechanism. TrmD is broadly conserved in the bacterial domain (Bystrom and Bjork, 1982a, b), while its counterpart in the eukaryotic and archaeal domain is Trm5 (Christian et al., 2004; Christian et al., 2013). Both TrmD and Trm5 are essential for cell growth (Baba et al., 2006; Bjork et al., 2001), because their reaction product m1G37 occurring on the 3' side of the tRNA anticodon is necessary to suppress +1-frameshift errors on the ribosome. Unlike mis-sense errors, +1-frameshift errors are deleterious, resulting in pre-mature termination of protein synthesis and leading to cell death. However, while TrmD and Trm5 catalyze the same reaction, they are fundamentally distinct (Ahn et al., 2003; Christian et al., 2004; Elkins et al., 2003; Goto-Ito et al., 2008; Goto-Ito et al., 2009). The distinction is manifested in their structure: while TrmD is an obligated dimer that uses the class IV-fold for AdoMet binding, Trm5 is an active monomer that uses the class I-fold (Christian et al., 2004). More importantly, the distinction is manifested in their kinetics: while the slow step of the TrmD reaction is the chemistry of methyl transfer, that of the Trm5 reaction is after methyl transfer and is associated with release of the m1G37-tRNA product (Christian et al., 2010b). Indeed, it is with the understanding of their kinetic distinction that we now gain insight into how their structural distinction affects methyl transfer. For example, while TrmD recognizes AdoMet using a rigid lock-and-key mode, Trm5 uses an induced-fit mode (Lahoud et al., 2011). While TrmD binds tRNA by recognizing only the anticodon stem-loop, Trm5 recognizes the entire tRNA L-shape with an emphasis on the elbow region (Christian and Hou, 2007; Goto-Ito et al., 2009). While TrmD discriminates the target G37 base by carefully reading all three functional groups on the Watson-Crick face, Trm5 is more relaxed and reads only two of the three functional groups (Sakaguchi et al., 2012). Finally, while both enzymes involve a general base to abstract the N1 proton of G37 to activate the nucleophile, TrmD differs from Trm5 by engaging a catalytic Mg2+ ion for the proton abstraction (Christian et al., 2010a; Sakaguchi et al., 2014). Together, these mechanistic distinctions show that the structural difference between TrmD and Trm5 has resulted in the kinetic distinction of their methyl transfer. Importantly, this correlation between structure and mechanism would not have been realized without the development of kinetic assays to monitor the methyl transfer reaction of each enzyme.
Here we describe three kinetic assays that were used to distinguish TrmD from Trm5. Combined, these assays provide information on the rate constant of each catalytic turnover (kcat), the substrate concentration (Km) that permits methyl transfer proceeding at the half-maximum turnover rate, the rate constant of methyl transfer in a single turnover (kchem), the substrate binding affinity (Kd) for the methyl transferase in a single turnover, and the rate-limiting step in multiple rounds of turnover. A summary of these kinetic parameters for E. coli TrmD (EcTrmD) and the archaeal Methanococcus jannaschii Trm5 (MjTrm5) is shown in Table 2. The central component of these assays is the radioactive [3H-methyl]-AdoMet as the methyl donor (abbreviated as 3H-AdoMet hereafter), which permits the incorporation of [3H-methyl] to synthesize the product m1G37-tRNA. While the radioactivity in the product tRNA is an integral part of the nucleic acid and therefore is acid precipitable, that in the free AdoMet substrate is not acid precipitable and can be washed away. Measuring the radioactivity of the product tRNA, usually collected on filter pads, then quantifies the amount of product synthesis. This quantitative information is valuable for insight into the kinetic and catalytic mechanism. An advantage of this method is that because the radiolabel is placed on AdoMet, the substrate G37-tRNA can be prepared without label in one transcription reaction.
Table 2.
Kinetic parameters of EcTrmD and MjTrm5.
| Parameter | EcTrmD | MjTrm5 | Assays | References |
|---|---|---|---|---|
| Km (tRNA), µM | 3.1 ± 0.1 | 0.70 ± 0.03 | steady-state | (Elkins et al., 2003) (Masuda et al., 2013) |
| kcat, s−1 | 0.09 ± 0.01 | 0.017 ± 0.002 | steady-state | (Christian and Hou, 2007) |
| 0.020 ± 0.007 | pre-steady-state | (Christian et al., 2010b) | ||
| Kd (tRNA), µM | 0.44 ± 0.04 | 1.4 ± 0.1 | single turnover | (Christian et al., 2010b) |
| Kd (AdoMet), µM | 0.8 ± 0.1 | single turnover | (Christian et al., 2010b) | |
| Kd (m1G37-tRNA), µM | 0.2 ± 0.1 | pre-steady-state | (Christian et al., 2010b) | |
| Kd (AdoHcy), µM | 0.020 ± 0.005 | pre-steady-state | (Christian et al., 2010b) | |
| kchem, s−1 | 0.09 ± 0.01 | 0.12 ± 0.03 | pre-steady-state | (Christian et al., 2010b) |
| 0.12 ± 0.01 | single turnover | (Christian et al., 2010b) |
In contrast, there exists an alternative and also quantitative assay (Swinehart et al., 2013), which places the label on the substrate G37-tRNA while using unlabeled AdoMet. In this alternative method, the G37-tRNA substrate is prepared with a site-specifically placed 32P at the 5' end of G37, so that the label is associated only with G37 in the substrate or m1G37 in the product (Jackman et al., 2003). After methyl transfer, the substrate and product tRNAs are digested to single 5'-monophosphate nucleotides, and G37 and m1G37 are separated by one-dimensional thin layer chromatography (TLC) and quantified individually on a phosphorimager screen. A major challenge of this alternative assay, however, is that the site-specific labeling of 5'-32P-G37 in tRNA is a multi-step process, involving synthesis of a tRNA fragment starting with G37, 5'-32P-labeling of this fragment, and joining this labeled fragment with another tRNA fragment to reconstitute the full-length molecule. While this multi-step labeling procedure was successful for the G37-tRNA substrate, it may be difficult for other tRNA substrates, possibly involving ligation of three or more fragments (Sakaguchi et al., 2012).
2. Methodology
Steady state assays
Pre-steady-state assays
Single turnover assays
3. Steady state assays
For decades, kinetic analysis of methyl transfer to tRNA substrates has been dominated by steady state assays. In these assays, the enzyme is in catalytic amounts, the tRNA substrate is in excess, the AdoMet concentration is saturating, and the reaction proceeds over multiple rounds of turnover. Fitting the data of the initial rate of methyl transfer (V0) as a function of the tRNA substrate concentration to the Michaelis-Menten equation yields the catalytic turnover kcat and the Michaelis constant for the tRNA substrate Km (tRNA). Conversely, analysis of the initial rate (V0) as a function of the AdoMet concentration, while tRNA is saturating, yields the catalytic turnover kcat and the Michaelis constant Km (AdoMet). In both cases, the kcat value should be similar, which is a validation for the reliability of the assay. Using such steady state assays, the kinetic parameters of EcTrmD were determined and used to evaluate the corresponding parameters of mutant enzymes harboring amino acid substitutions (Elkins et al., 2003). This analysis revealed the broad landscape of the enzyme, showing the residues important for synthesis of m1G37-tRNA. Because steady state assays require only minimal amounts of the enzyme, they are easy to prepare and are usually the first assays to perform for a methyl transfer reaction.
3.1. Preparation of a target tRNA transcript
The substrate tRNA is prepared as a transcript from run-off transcription of a DNA template encoding a native or mutant sequence. The DNA template sequence can be cloned into a plasmid and made available for run-off transcription by digestion with a restriction enzyme to expose the 3' end. Alternatively, the DNA template can be constructed from overlapping oligonucleotides (Hou, 2012; Zhang et al., 2008). The template sequence is usually placed behind the promoter of the highly processive T7 RNA polymerase and the transcription by this enzyme gives the best yield for the range of tRNA lengths (70–90 nucleotides). An over-producer strain of T7 RNA polymerase is available (Tabor et al., 1987) and the enzyme can be purified in house and titrated to the level where 10 µL of the enzyme gives visible precipitation of pyrophosphate-Mg2+ conjugates in less than one hour at 37 °C. The pyrophosphate is released from NTP upon incorporation of NMP during active transcription. The observation of such precipitation is usually a good indication of strong transcription. If no precipitation is observed in more than one hour, then add more T7 RNA polymerase to the transcription reaction.
An important note of T7 RNA polymerase is that it initiates transcription only with G (guanosine, GMP, or GTP). Therefore, substrate sequences that do not start with a G are prepared with a hammerhead ribozyme sequence between the promoter and the first nucleotide of the tRNA gene (Fechter et al., 1998; Liu et al., 2012; Pham et al., 2014). Transcription of this hybrid construct gives rise to a fusion, where the synthesized ribozyme in the upstream portion will self-cleave to liberate the downstream substrate tRNA with a 5'-OH end. Another consideration is the propensity of T7 RNA polymerase to generate heterogeneous 3' ends, which can affect some post-transcriptional modification reactions (Roovers et al., 2006). This problem can be reduced, but not completely eliminated, by two methods. One is to use the “foot” mutant of T7 RNA polymerase, which lacks the C-terminal F882-A883 residues of the wild-type enzyme and as a result exhibits reduced processivity (Mookhtiar et al., 1991). The other is to introduce two consecutive 2'-O-methyl backbone modifications to the 5'-terminus of the non-coding strand of the DNA template to reduce the processivity of T7 RNA polymerase (Kao et al., 1999). Notably, introduction of the 2'-O-methyl backbone modifications are not easily achieved with plasmid DNA templates, but are readily accommodated by chemical synthesis of oligonucleotides for constructing DNA templates. The synthesized tRNA transcripts are separated from DNA templates by denaturing 12% polyacrylamide/7M urea (12%PAGE/7M urea) gels, identified by UV shadowing, extracted from gel materials into the TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA), collected by 3 volumes of ethanol precipitation, washed by 70% ethanol, and resuspended and stored in TE.
3.2. Methyl transfer in steady-state conditions
The representative enzymes are EcTrmD and MjTrm5, each of which is well characterized and has a high-resolution crystal structure in complex with AdoMet or the product AdoHcy (Elkins et al., 2003; Goto-Ito et al., 2008; Goto-Ito et al., 2009). Additionally, EcTrmD is homologous to Haemophilus influenza TrmD, which has crystal structures in the apo form and in the form with AdoMet or AdoHcy (Ahn et al., 2003), while MjTrm5 is homologous to human Trm5 (Homo sapiens Trm5, HsTrm5) with similar kinetic parameters (Christian et al., 2013). The chosen assay temperature is 37 °C for EcTrmD and HsTrm5 and 55 °C for MjTrm5. The G37-tRNA substrate for each enzyme is the transcript of EctRNALeu, HstRNACys, and MjtRNACys, respectively. The reaction buffer for EcTrmD is 0.1 M Tris-HCl (pH 8.0), 24 mM NH4Cl2, 6 mM MgCl2, 4 mM dithiothreitol (DTT), 0.1 mM EDTA, and 0.024 mg/mL bovine serum albumin (BSA). The same reaction buffer applies to MjTrm5 and HsTrm5, except that NH4Cl2 is increased to 100 mM. To determine the kinetic parameters for the tRNA substrate, the substrate concentration varies while AdoMet is saturating; conversely, to determine the kinetic parameters for AdoMet, the concentration of the methyl donor varies, while tRNA is saturating. In either case, the substrate must exist in excess of the enzyme (preferably more than 50-fold), so that the enzyme can catalyze multiple turnovers while the reaction consumes no more than 5% of the substrate.
-
Prepare the working stock of 3H-AdoMet by mixing as follows:
Purchase 3H-AdoMet commercial solution (Perkin Elmer, NET155H, 60 Ci/mmol, 6.6 µM, 0.55 µCi/µL).
Purchase unlabeled AdoMet (1 mM): dissolve 0.57 mg of AdoMet in 1.0 mL water with 1 µL concentrated H2SO4 (final concentration of the acid = 12 mM). Store the solution at −20 °C.
Mix 200 µL 3H-AdoMet commercial solution with 90 µL unlabeled AdoMet (1 mM) to give a final concentration of ~300 µM AdoMet with a specific activity of 2,650 dpm/pmole.
- Calculate the specific activity of the working stock 3H-AdoMet from step 1.
- The working stock consists of 200 µL of the commercial 3H-AdoMet (6.6 µM, 0.55 µCi/µL) and 90 µL of unlabeled AdoMet (1 mM)
- Total concentration of the commercial 3H-AdoMet in the mixture: 4.55 µM
- Total concentration of unlabeled AdoMet in the mixture: 310.34 µM
- Combined concentration of AdoMet: 314.89 µM
- Total µCi in the mixture: 0.55 µCi/µL × 200 µL = 110 µCi
- Specific activity in dpm/pmole: [110 µCi × (2.2 × 106 dpm/µCi)] / [314.89 pmole/µL × 290 µL)] = 2,650 dpm/pmole.
As an initial determination of the kinetic parameters for the G37-tRNA substrate for EcTrmD or MjTrm5, set up a series of reactions, where the concentration of G37-tRNA increases by 2-fold from 0.1 to 25 µM. This range of concentration usually covers the Km (tRNA) (5 µM for EcTrmD and 0.5 µM for MjTrm5) (Christian et al., 2013; Elkins et al., 2003). Include in the series a reaction without tRNA as a control. Once an initial Km (tRNA) is determined, one should repeat the Km (tRNA) analysis by ranging the G37-tRNA concentration between 1/5 and 5× of the initial Km (tRNA) value.
- Each reaction in the series above should have the following components:
Reaction components Volume to add (µL) Final concentration (µM) tRNA/TE 10.0 0.1–25 Heat-cool (HC buffer) 2.0 5× buffer 5.0 1.0× 3H-AdoMet (310 µM, 2,650 dpm/pmol) 3.0 37.2 EcTrmD or MjTrm5 (25 nM) 5.0 0.005 Total 25.0 Add the 5× buffer, the working stock 3H-AdoMet, and initiate the reaction by adding EcTrmD or MjTrm5. At the specified time points (e.g., 2, 4, 6, 8, and 10 min), remove 5 µL from the reaction, spot it onto a 1 cm2 Whatman 3MM filter pad, and place the filter pad into a beaker containing 5% trichloroacetic acid (TCA).
After all pads are in TCA, shake the solution for 10 min at 4 °C to wash off un-incorporated 3H-AdoMet, while allowing the synthesized m1G37-tRNA to precipitate in TCA. Decant and repeat the 5% TCA wash.
Wash all filter pads with 95% ethanol by shaking for 10 min at 4 °C in the beaker. Repeat the ethanol wash one more time.
Wash all filter pads with ether. Agitate gently by hand and let the ether solution sit at room temperature for 5 min under a fume hood. Decant ether and dry the filter pads under the fume hood for 15 min.
Transfer each filter pad to a scintillation solution in a vial and measure the amount of radioactivity using a liquid scintillation counter.
Calculate the amount of m1G37-tRNA synthesis based on the specific activity of the working stock of 3H-AdoMet.
Correct the 3H counting by measuring the quenching factor using the following procedure: Take a 5 µL aliquot at the final time point of a reaction and pass it through a quick spin column to remove unincorporated 3H-AdoMet, which stays with the column. Directly transfer the eluate 5 µL (which contains counts only associated with the methylated tRNA) into the liquid scintillation fluid and measure the counts. The ratio of the direct measurement of this count over the count on the TCA precipitated filter pad at the same time point reveals the quenching factor, which should be used to correct the fraction of methylation. For the protocol described here, the quenching factor is usually 4.
- For each enzyme concentration, fit the data of the time course to the linear equation:
where y is the pmoles of synthesis of m1G37-tRNA, x is the time, a is the initial rate (V0) of synthesis in pmoles/sec, and b is the intercept on the y axis, which arises from background counts of the reaction due to non-specific binding of 3H-AdoMet to filter pads. The value of b should be closely similar to the value obtained from the control reaction containing no tRNA.(equation 1) - Plot the initial rate V0 as a function of the G37-tRNA concentration by fitting the data to the Michaelis-Menten equation:
where V0 is the initial rate of synthesis of m1G37-tRNA (in pmole/sec), S is the substrate G37-tRNA concentration (in µM), Vmax is the maximum rate (in pmole/sec), kcat is the catalytic turnover (in sec−1), E0 is the enzyme quantity in the reaction (in pmole), and Km is the Michaelis constant for the G37-tRNA substrate (in µM).(equation 2)
3.3. Experimental considerations
To determine the kinetic parameters of AdoMet, one should design a series of reactions similar to steps 3–4 in Section 3.2 but vary the AdoMet concentration in 0.1–25 µM, while the tRNA substrate is saturating (~10× of Km (tRNA)). The suggested range of AdoMet concentration covers the Km (AdoMet) for EcTrmD (5–8 µM) and MjTrm5 (0.5 µM) (Christian et al., 2013; Lahoud et al., 2011). By having a working stock of 3H-AdoMet (step 2), one can use this stock to make appropriate dilution to the desired concentration. However, an important difference here from the determination of Km (tRNA) is that the radioactivity of AdoMet varies with each dilution. To address this difference, one needs to have a no-tRNA control reaction for each AdoMet concentration to provide the background counts for correction of the non-specific radioactivity bound to filter pads at each concentration. The counts after correction of the no-tRNA control for each AdoMet concentration, followed by correction for the filter-quenching factor, are then converted to pmoles of product synthesis. This information is then used to determine the Km (AdoMet) as described in steps 11–13.
The steady state parameters are each a composite term of multiple turnovers. Therefore, kcat does not mean the rate constant of methyl transfer and Km does not mean the binding affinity of the substrate to the enzyme. The kinetic assays described below, pre-steady-state and single turnover assays, are designed to complement steady state assays.
4. Pre-steady-state assays
The recent development of pre-steady-state assays has shed new light on the distinction between TrmD and Trm5. In pre-steady-state assays, the G37-tRNA substrate is maintained in 10-fold molar excess of the enzyme and both the substrate and the enzyme are at the µM level to permit one turnover of methyl transfer on the enzyme, followed by multiple rounds of steady-state turnover. It was in fact in pre-steady-state assays that the kinetics of TrmD was distinguished from Trm5. Specifically, while the TrmD reaction occurred linearly over time, the Trm5 reaction occurred in a rapid burst phase followed by a slower and linear phase (Fig. 2) (Christian et al., 2010b). Fitting the TrmD data to a linear equation revealed a slope of 0.09 ± 0.01 s−1, similar to the value of kcat in steady state. Fitting the Trm5 data to a burst equation revealed a rate constant of 0.12 ± 0.03 s−1 for the first turnover and a rate constant of 0.020 ± 0.007 s−1 for steady-state turnover kcat. The rate constant for the first turnover is associated with the chemistry of methyl transfer (kchem) under conditions of rapid equilibrium binding and it can report either the chemical step or the pre-chemistry active-site rearrangement step of the enzyme. Because TrmD maintains the same rate constant in the first turnover as in steady-state turnover, its catalytic cycle is limited by kchem. In contrast, because Trm5 exhibits a faster kchem in the first turnover relative to the steady-state kcat, its catalytic cycle is limited by kcat, which is associated with the release of the m1G37-tRNA product. Indeed, by treating m1G37-tRNA as an inhibitor of Trm5, pre-steady-state assays showed that increasing concentration of the inhibitor progressively decreased the burst amplitude, a parameter that indicates the active fraction of the enzyme (Christian et al., 2010b). Similarly, increasing concentration of AdoHcy also decreased the burst amplitude (Christian et al., 2010b). The analysis of the Kd of each inhibitor for Trm5 then revealed that the enzyme has a tighter binding affinity to AdoHcy relative to AdoMet and a tighter binding affinity to m1G37- relative to G37-tRNA. These results support the notion that the tight binding of Trm5 to either product contributes to the slow product release in catalytic turnover.
Figure 2.

Pre-steady-state kinetics of m1G37-tRNA synthesis. (A) Monitoring the time course of synthesis upon mixing EcTrmD (1 µM) with EctRNALeu (10 µM) and AdoMet (30 µM) at 37 °C. The time-dependent synthesis is calculated as the amount of synthesis per active site of the enzyme (%) and is fit to equation 1 to determine the slope (a = slope and b = 0). (B) Monitoring the time course of synthesis upon mixing MjTrm5 (1 µM), MjtRNACys (10 µM), and AdoMet (25 µM). The time-dependent synthesis is calculated as the amount of synthesis per active site of the enzyme (%) and is fit to equation 3 to determine kchem and kcat. This figure is adapted from Figure 1 in (Christian et al., 2010b).
4.1. Methyl transfer in pre-steady-state conditions
Using EcTrmD and MjTrm5 as the examples, the reaction buffer for each is as described in section 3.2. Because methyl transfer of the first turnover for EcTrmD (kobs = 0.09 ± 0.01 s−1) or MjTrm5 (kobs = 0.12 ± 0.03 s−1) occurs on the time scale of 0.1 s−1 or faster (Christian et al., 2010b), meaning that it takes less than 10–11 sec to complete the first turnover, the pre-steady-state measurement should be carried out on a rapid mixing and quench instrument. Our lab uses the KinTek RQF-3 model (KinTek Corp, Texas, www.kintek-corp.com), which operates with a computer panel to control rapid mixing of the contents of two syringes and time-dependent quenching of the reaction. For methyl transfer that proceeds on a slower time scale, the mixing and quenching can be performed without the instrument. We have found that data obtained from the same protocol performed with or without the instrument are similar, with the standard deviation less than 20%.
Take an aliquot of the unmodified transcript of G37-tRNA (200 pmole) and adjust the volume with TE to 15 µL. Heat the tRNA solution for 3 min at 80 °C, which is above the estimated melting temperature of the tRNA transcript. Quickly spin the solution and add 5 µL of the HC buffer. Anneal the G37-tRNA at 37 °C for 15 min. The resulting stock concentration of G37-tRNA is 100 µM.
- Prepare the G37-tRNA solution for syringe #1 of the RQF-3 instrument:
Syringe #1 Volume 2× Concentration 1× Concentration G37-tRNA transcript (100 µM) 60.0 µL 20.0 µM 10.0 µM ddH2O 151.0 µL 5× buffer 60.0 µL 1× Working stock of 3H-AdoMet 29.0 µL 30.0 µM 15.0 µM Total 300.0 µL - Prepare the enzyme solution for syringe #2 of the RQF-3 instrument:
Syringe #2 Volume 2× Concentration 1× Concentration EcTrmD or MjTrm5 (400 µM) 1.5 µL 2.0 µM 1.0 µM ddH2O 238.5 µL 5× buffer 60.0 µL 1× Total 300.0 µL On the RQF-3 instrument, fill the large syringe on each side with 1× buffer and the middle syringe with 5% TCA. Fill syringe #1 with 300 µL of the tRNA solution to one sample loop and syringe #2 with 300 µL of the enzyme solution to the second sample loop. Enter time points on the control panel. Upon hitting the start button, the 1× buffer pushes 15 µL from syringe #1 and 15 µL from syringe #2 into the reaction loop. After the specified time lapse (e.g., 0, 1, 3, 5, 7, 9, 12, 15, 20, 25, 30, 40, 50, 60, 80, 100, and 120 sec), the reaction is quenched with 54 µL of 5% TCA.
Collect the quenched solution of each time point in an Eppendorf tube. Spot 20 µL of the quenched solution onto a 1 cm2 Whatman 3 MM filter pad. Wash all filter pads with 5% TCA twice, followed by 95% ethanol twice, and followed by ether once. Dry filter pads in hood and measure radioactivity of each filter pad in a scintillation counter. Correct the counts for the quenching factor, and calculate the synthesis of m1G37-tRNA based on the corrected counts. Calculate the fractional conversion from G37 to m1G37-tRNA and plot the data vs. time. These procedures are as described in steps 6–12 of section 3.2.
- Fit the data to the burst equation as shown below:
where yo is the y intercept, A is the amplitude of the initial exponential phase (representing the active fraction of the enzyme), k1 is the observed rate constant of the initial exponential regression, k2 is the apparent rate constant of the steady-state phase, and t is the time in seconds (Dupasquier et al., 2008; Zhang et al., 2006).(equation 3)
4.2. Experimental considerations
The appearance of burst kinetics implies slow product release after methyl transfer. To test this hypothesis, one can use the burst phase to determine the enzyme affinity for the product. For example, to determine the enzyme affinity for the m1G37-tRNA product, we purified the product from the methyl transfer reaction by using RNase H to selectively cleave the substrate G37-tRNA (Hou, 2012; Hou et al., 2006). The purified m1G37-tRNA was used to form a series of Trm5-product complexes, which was tested for the forward methyl transfer reaction in pre-steady-state assays. We showed that while increasing concentration of the product had little effect on the rate of the burst or steady-state phase, it progressively decreased the amplitude of the burst phase (Christian et al., 2010b), indicating that the dissociation of the product from Trm5 was slow and it limited the availability of the enzyme for the forward reaction. A fit of the data to a quadruple equation (equation 4) revealed a Kd (m1G37-tRNA) of 0.2 ± 0.1 µM.
| (equation 4) |
where y represents the methylated tRNA product after one turnover; Eo is the active enzyme concentration; S is the tRNA concentration; Kd is the dissociation constant for tRNA (Dupasquier et al., 2008; Zhang et al., 2006). The Kd (m1G37-tRNA) is lower than the Kd (G37-tRNA) of the enzyme-substrate complex that was determined from single turnover kinetic assays (Christian et al., 2010b).
The burst amplitude can be further exploited to determine the parameters for AdoMet, such as Kd (AdoMet) and Kd (AdoHcy). For example, we determined Kd (AdoMet) by pre-forming a series of Trm5-AdoMet complexes. Analysis of the series of complexes for methyl transfer in pre-steady-state assays showed progressive increases of the burst amplitude with increasing concentration of the methyl donor. Fitting the amplitude data as a function of AdoMet concentration to a hyperbolic equation (equation 5) revealed a Kd (AdoMet) of 0.44 ± 0.09 µM. Conversely, we determined Kd (AdoHcy) by pre-forming a series of Trm5-AdoHcy complexes and showed that increasing concentration of AdoHcy progressively decreased the burst amplitude in pre-steady-state assays. Fitting the amplitude data as a function of AdoHcy concentration to a hyperbolic equation (equation 6) revealed a Kd (AdoHcy) of 20 ± 5 nM, which is nearly 20-fold lower than the Kd (AdoMet). Interestingly, between the two products of the methyl transfer reaction, we noticed that the Kd (AdoHcy) is 20-fold lower than the Kd (m1G37-tRNA), indicating that it is AdoHcy that has the highest affinity for Trm5 and that it is the release of AdoHcy that controls the overall catalytic turnover for Trm5.
| (equation 5) |
where A is maximum amplitude in burst kinetics, S is the AdoMet concentration (Dupasquier et al., 2008; Zhang et al., 2006).
| (equation 6) |
where yo is the initial burst amplitude in the absence of AdoHcy; A is the scaling constant for amplitude change upon addition of AdoHcy; I is the concentration of AdoHcy (Dupasquier et al., 2008; Zhang et al., 2006).
5. Single turnover assays
In single turnover assays, the enzyme is in excess of the tRNA substrate and the AdoMet concentration is saturating, so that the reaction proceeds only once. This single turnover condition offers two advantages. First, because the observed rate constant (kobs or kapp) reports the kinetics of just one methyl transfer, the plot of kobs vs. concentration leads to the determination of kinetic parameters intrinsically associated with the methyl transfer. Second, because the enzyme and AdoMet are both saturating relative to the tRNA substrate, the enzyme-substrate affinity is measured under the condition of rapid equilibrium binding, so that the determined Kd reflects the true thermodynamic binding affinity.
5.1. Methyl transfer in single turnover conditions
- Prepare the tRNA solution for syringe #1 of the RQF-3 instrument:
Syringe 1 Volume 2× Concentration 1× Concentration G37-tRNA transcript (100 µM) 1.5 µL 0.5 µM 0.25 µM ddH2O 209.5 µL 5× buffer 60.0 µL 1× Working stock of 3H-AdoMet 29.0 µL 30.0 µM 15.0 µM Total 300.0 µL In single turnover assays, the reaction rate is driven by the enzyme concentration, not by the tRNA concentration (Dupasquier et al., 2008; Liu et al., 2011; Liu et al., 2007; Zhang et al., 2006). Therefore, a series of reactions are designed with increasing concentration of the enzyme, so that the plot of kobs vs. concentration gives the Kd of the enzyme for the tRNA substrate (Kd (tRNA)). The 6 reactions designed below are to provide an initial evaluation of the Kd (tRNA). Once an initial Kd (tRNA) is obtained, the experiments should be repeated to vary the enzyme concentration in the range of 1/5 and 5× of the Kd.
- Using EcTrmD as an example, prepare a series of the enzyme solutions (ranging from 2 to 32 µM as the 2×) for syringe #2 of the RQF-3 instrument. For each concentration, prepare a 300 µL solution. A total of 6 concentrations are prepared.
Reaction 1 2 3 4 5 6 2× EcTrmD 2 µM 4 µM 8 µM 16 µM 24 µM 32 µM 1× EcTrmD 1 µM 2 µM 4 µM 8 µM 12 µM 16 µM EcTrmD stock (400 µM) 1.5 µL 3.0 µL 6 µL 12 µL 18 µL 24 µL 5× buffer 60 µL 60 µL 60 µL 60 µL 60 µL 60 µL ddH2O 238.5 µL 237 µL 234 µL 228 µL 222 µL 216 µL Total 300 µL 300 µL 300 µL 300 µL 300 µL 300 µL On the RQF-3 instrument, fill the 1× buffer solution, 5% TCA solution, the tRNA solution, and one enzyme solution to the appropriate syringes as described in step 4 of Section 4.1. After the specified time lapse, the reaction is quenched with 54 µL of 5% TCA. Put the quenched solution in an Eppendorf tube and collect samples up to 17 time points (e.g., 0, 1, 3, 5, 7, 9, 12, 15, 20, 25, 30, 40, 50, 60, 80, 100, and 120 sec).
Spot 20 µL of each aliquot onto a 1 cm2 Whatman 3 MM filter pad. Wash all filter pads with 5% TCA twice, with 95% ethanol twice, and with ether once. Dry filter pads in a hood and measure radioactivity of each filter pad in a scintillation counter. Correct the counts for the quenching factor, calculate the synthesis of m1G37-tRNA based on the specific activity of 3H-AdoMet, convert the data into fractional conversion from G37 to m1G37-tRNA, and plot the data vs. time. These procedures are as described in steps 6–12 of section 3.2.
- Data points for each time course are fit to the single exponential equation:
where yo is the y intercept, A is the scaling constant, kapp is the apparent rate constant, and t is the time in seconds to determine kobs (or kapp) (Dupasquier et al., 2008; Zhang et al., 2006). The data of kobs vs. enzyme concentration for single turnover analysis of m1G37-tRNA synthesis are fit to the hyperbolic equation:(equation 7)
where kchem is the rate constant for the steps associated with the methyl transfer chemistry, Kd is the enzyme affinity for the tRNA substrate (Kd (tRNA)), and Eo is the enzyme concentration (Dupasquier et al., 2008; Zhang et al., 2006). Note that in single turnover assays using chemical quench, kchem can be the chemistry of methyl transfer or the pre-chemistry rearrangement of the enzyme active site. The latter possibility can be addressed by dynamics experiments using fluorescent tRNA (Liu et al., 2009) in stopped-flow methodologies.(equation 8)
5.2. Experimental considerations
In single turnover assays, control experiments should be performed in which enzyme, tRNA, and AdoMet are mixed in different orders and in different syringes. For example, syringe #1 may contain just the tRNA solution, while syringe #2 may contain AdoMet and the enzyme solution. If all of the mixing control experiments return the same kobs value, then this is evidence that the rate of methyl transfer is independent of the mixing order, and that the reaction is proceeding under a rapid equilibrium binding condition.
For TrmD, which lacks burst kinetics, the determination of Kd (AdoMet) cannot take advantage of the burst amplitude as in the case of Trm5. Instead, the value of Kd (AdoMet) for TrmD can be determined using single turnover assays, in which the enzyme concentration is in excess of the AdoMet substrate, while the tRNA concentration is saturating. For example, prepare the tRNA solution for syringe #1 of RQF-3 at the 2× concentration of 20 µM of G37-tRNA and 1 µM of the working stock of 3H-AdoMet in 1× buffer. The specific activity of 3H-AdoMet is 3,400 dpm/pmole, higher than the specific activity (2,650 dpm/pmole) for determination of Kd (tRNA), to compensate for the lower amount of AdoMet used in the reaction. Prepare a range of TrmD concentration for syringe #2 as in step 3 of Section 5.1. After rapid mixing of the tRNA solution with an enzyme solution, monitor the synthesis of m1G37-tRNA over time, and determine the kobs from the single exponential equation (equation 7) for the specific enzyme concentration. Fitting the data of kobs as a function of enzyme concentration to the hyperbola equation (equation 8) will then determine the Kd (AdoMet) for the enzyme. This assay has also been successfully applied to HsTrm5 without relying on the burst kinetics of the enzyme (Christian et al., 2013).
6. Conclusions
All tRNA molecules contain multiple post-transcriptional modifications by AdoMet-dependent methyl transfer reactions. These methylation reactions are subject to regulation, due to the variability of AdoMet levels depending on the energy state of a cell. However, because different methyl transferases bind AdoMet differently, their reactions are likely to be subject to different regulation. To understand the regulation of each methyl transferase and how the regulation contributes to the biology of a tRNA, kinetic analysis of the methyl transfer reaction is necessary. This chapter describes three types of kinetic assays that collectively form the foundation to address the regulation. The steady state assays are important for establishing a global view of the methyl transfer reaction and the data provide the basis for comparing data of other assays. The pre-steady-state assays are performed under conditions closely similar to those in a cell and as such the data are relevant to biology, particularly with respect to the rate-limiting step in catalytic turnovers. The single turnover assays are performed upon rapid equilibrium binding of a methyl transferase to its substrates, so the data are most appropriate for correlating with the structural information of the enzyme. Although the three assays are described for the TrmD and Trm5 reactions, they are easily adaptable to other AdoMet-dependent methyl transferases.
Acknowledgments
This work was supported by NIH grant R01GM81601 to YMH. The authors thank Megumi Shigematsu for preparation of Figure 1.
References
- Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. Embo J. 2003;22:2593–2603. doi: 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez-Paez A, Villarroya M, Douthwaite S, Gabaldon T, Armengod ME. YibK is the 2'-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNA(Leu) isoacceptors. RNA. 2010;16:2131–2143. doi: 10.1261/rna.2245910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP. A primordial tRNA modification required for the evolution of life? Embo J. 2001;20:231–239. doi: 10.1093/emboj/20.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrom AS, Bjork GR. Chromosomal location and cloning of the gene (trmD) responsible for the synthesis of tRNA (m1G) methyltransferase in Escherichia coli K-12. Mol Gen Genet. 1982a;188:440–446. doi: 10.1007/BF00330046. [DOI] [PubMed] [Google Scholar]
- Bystrom AS, Bjork GR. The structural gene (trmD) for the tRNA(m1G)methyltransferase is part of a four polypeptide operon in Escherichia coli K-12. Mol Gen Genet. 1982b;188:447–454. doi: 10.1007/BF00330047. [DOI] [PubMed] [Google Scholar]
- Cantoni GL. Biological methylation: selected aspects. Annu Rev Biochem. 1975;44:435–451. doi: 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- Christian T, Evilia C, Williams S, Hou YM. Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol. 2004;339:707–719. doi: 10.1016/j.jmb.2004.04.025. [DOI] [PubMed] [Google Scholar]
- Christian T, Gamper H, Hou YM. Conservation of structure and mechanism by Trm5 enzymes. RNA. 2013;19:1192–1199. doi: 10.1261/rna.039503.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Hou YM. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J Mol Biol. 2007;373:623–632. doi: 10.1016/j.jmb.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hoffmann K, Perona JJ, Hou YM. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA. 2010a;16:2484–2492. doi: 10.1261/rna.2376210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hou YM. Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J Mol Biol. 2010b;400:204–217. doi: 10.1016/j.jmb.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bie LG, Roovers M, Oudjama Y, Wattiez R, Tricot C, Stalon V, Droogmans L, Bujnicki JM. The yggH gene of Escherichia coli encodes a tRNA (m7G46) methyltransferase. J Bacteriol. 2003;185:3238–3243. doi: 10.1128/JB.185.10.3238-3243.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupasquier M, Kim S, Halkidis K, Gamper H, Hou YM. tRNA integrity is a prerequisite for rapid CCA addition: implication for quality control. J Mol Biol. 2008;379:579–588. doi: 10.1016/j.jmb.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins PA, Watts JM, Zalacain M, van Thiel A, Vitazka PR, Redlak M, Andraos-Selim C, Rastinejad F, Holmes WM. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J Mol Biol. 2003;333:931–949. doi: 10.1016/j.jmb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Fechter P, Rudinger J, Giege R, Theobald-Dietrich A. Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: production and activity. FEBS Lett. 1998;436:99–103. doi: 10.1016/s0014-5793(98)01096-5. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Golovina AY, Sergiev PV, Golovin AV, Serebryakova MV, Demina I, Govorun VM, Dontsova OA. The yfiC gene of E. coli encodes an adenine-N6 methyltransferase that specifically modifies A37 of tRNA1Val(cmo5UAC) Rna. 2009;15:1134–1141. doi: 10.1261/rna.1494409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, Yokoyama S. Crystal structure of archaeal tRNA(m(1)G37)methyltransferase aTrm5. Proteins. 2008;72:1274–1289. doi: 10.1002/prot.22019. [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Kuratani M, Bessho Y, Yokoyama S. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat Struct Mol Biol. 2009;16:1109–1115. doi: 10.1038/nsmb.1653. [DOI] [PubMed] [Google Scholar]
- Hou YM. High-Purity enzymatic synthesis of site-specifically modified tRNA. In: Conn GL, editor. Preparation of RNA for biochemical and biophysical analysis: Methods and protocols. Humana Press; 2012. [DOI] [PubMed] [Google Scholar]
- Hou YM, Li Z, Gamper H. Isolation of a site-specifically modified RNA from an unmodified transcript. Nucleic Acids Res. 2006;34:e21. doi: 10.1093/nar/gnj018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YM, Perona JJ. Stereochemical mechanisms of tRNA methyltransferases. FEBS Lett. 2010;584:278–286. doi: 10.1016/j.febslet.2009.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Montange RK, Malik HS, Phizicky EM. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. Rna. 2003;9:574–585. doi: 10.1261/rna.5070303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C, Zheng M, Rudisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. Rna. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Miyauchi K, Ikeuchi Y, Thiaville PC, Crecy-Lagard V, Suzuki T. Discovery of the beta-barrel-type RNA methyltransferase responsible for N6-methylation of N6-threonylcarbamoyladenosine in tRNAs. Nucleic Acids Res. 2014;42:9350–9365. doi: 10.1093/nar/gku618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahoud G, Goto-Ito S, Yoshida K, Ito T, Yokoyama S, Hou YM. Differentiating analogous tRNA methyltransferases by fragments of the methyl donor. RNA. 2011;17:1236–1246. doi: 10.1261/rna.2706011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Betteridge T, Hou YM. Fluorophore labeling to monitor tRNA dynamics. Methods in Enzymology. 2009;469:69–93. doi: 10.1016/S0076-6879(09)69004-2. [DOI] [PubMed] [Google Scholar]
- Liu C, Gamper H, Liu H, Cooperman BS, Hou YM. Potential for interdependent development of tRNA determinants for aminoacylation and ribosome decoding. Nat Commun. 2011;2:329. doi: 10.1038/ncomms1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Gamper H, Shtivelband S, Hauenstein S, Perona JJ, Hou YM. Kinetic Quality Control of Anticodon Recognition by a Eukaryotic Aminoacyl-tRNA Synthetase. J Mol Biol. 2007;367:1063–1078. doi: 10.1016/j.jmb.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sanders JM, Pascal JM, Hou YM. Adaptation to tRNA acceptor stem structure by flexible adjustment in the catalytic domain of class I tRNA synthetases. RNA. 2012;18:213–221. doi: 10.1261/rna.029983.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham GD, Hafner EW, Tabor CW, Tabor H. S-Adenosylmethionine synthetase from Escherichia coli. J Biol Chem. 1980;255:9082–9092. [PubMed] [Google Scholar]
- Masuda I, Sakaguchi R, Liu C, Gamper H, Hou YM. The Temperature Sensitivity of a Mutation in the Essential tRNA Modification Enzyme tRNA Methyltransferase D (TrmD) J Biol Chem. 2013;288:28987–28996. doi: 10.1074/jbc.M113.485797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookhtiar KA, Peluso PS, Muller DK, Dunn JJ, Coleman JE. Processivity of T7 RNA polymerase requires the C-terminal Phe882-Ala883-COO- or "foot". Biochemistry. 1991;30:6305–6313. doi: 10.1021/bi00239a032. [DOI] [PubMed] [Google Scholar]
- Nureki O, Shirouzu M, Hashimoto K, Ishitani R, Terada T, Tamakoshi M, Oshima T, Chijimatsu M, Takio K, Vassylyev DG, et al. An enzyme with a deep trefoil knot for the active-site architecture. Acta Crystallogr D Biol Crystallogr. 2002;58:1129–1137. doi: 10.1107/s0907444902006601. [DOI] [PubMed] [Google Scholar]
- Nureki O, Watanabe K, Fukai S, Ishii R, Endo Y, Hori H, Yokoyama S. Deep knot structure for construction of active site and cofactor binding site of tRNA modification enzyme. Structure (Camb) 2004;12:593–602. doi: 10.1016/j.str.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Ny T, Bjork GR. Cloning and restriction mapping of the trmA gene coding for transfer ribonucleic acid (5-methyluridine)-methyltransferase in Escherichia coli K-12. J Bacteriol. 1980;142:371–379. doi: 10.1128/jb.142.2.371-379.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson BC, Jager G, Gustafsson C. The spoU gene of Escherichia coli, the fourth gene of the spoT operon, is essential for tRNA (Gm18) 2'-O-methyltransferase activity. Nucleic Acids Res. 1997;25:4093–4097. doi: 10.1093/nar/25.20.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham JS, Sakaguchi R, Yeoh LM, De Silva NS, McFadden GI, Hou YM, Ralph SA. A dual-targeted aminoacyl-tRNA synthetase in Plasmodium falciparum charges cytosolic and apicoplast tRNACys. Biochem J. 2014;458:513–523. doi: 10.1042/BJ20131451. [DOI] [PubMed] [Google Scholar]
- Purta E, van Vliet F, Tkaczuk KL, Dunin-Horkawicz S, Mori H, Droogmans L, Bujnicki JM. The yfhQ gene of Escherichia coli encodes a tRNA:Cm32/Um32 methyltransferase. BMC molecular biology. 2006;7:23. doi: 10.1186/1471-2199-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers M, Hale C, Tricot C, Terns MP, Terns RM, Grosjean H, Droogmans L. Formation of the conserved pseudouridine at position 55 in archaeal tRNA. Nucleic Acids Res. 2006;34:4293–4301. doi: 10.1093/nar/gkl530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi R, Giessing A, Dai Q, Lahoud G, Liutkeviciute Z, Klimasauskas S, Piccirilli J, Kirpekar F, Hou YM. Recognition of guanosine by dissimilar tRNA methyltransferases. RNA. 2012;18:1687–1701. doi: 10.1261/rna.032029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi R, Lahoud G, Christian T, Gamper H, Hou YM. A divalent metal ion-dependent N(1)-methyl transfer to G37-tRNA. Chem Biol. 2014;21:1351–1360. doi: 10.1016/j.chembiol.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinehart WE, Henderson JC, Jackman JE. Unexpected expansion of tRNA substrate recognition by the yeast m1G9 methyltransferase Trm10. RNA. 2013;19:1137–1146. doi: 10.1261/rna.039651.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor S, Huber HE, Richardson CC. Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J Biol Chem. 1987;262:16212–16223. [PubMed] [Google Scholar]
- Taya Y, Nishimura S. Biosynthesis of 5-methylaminomethyl-2-thiouridylate. I. Isolation of a new tRNA-methylase specific for 5-methylaminomethyl-2-thiouridylate. Biochem Biophys Res Commun. 1973;51:1062–1068. doi: 10.1016/0006-291x(73)90035-1. [DOI] [PubMed] [Google Scholar]
- Yi C, Pan T. Cellular dynamics of RNA modification. Acc Chem Res. 2011;44:1380–1388. doi: 10.1021/ar200057m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CM, Liu C, Slater S, Hou YM. Aminoacylation of tRNA with phosphoserine for synthesis of cysteinyl-tRNA(Cys) Nat Struct Mol Biol. 2008;15:507–514. doi: 10.1038/nsmb.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CM, Perona JJ, Ryu K, Francklyn C, Hou YM. Distinct kinetic mechanisms of the two classes of Aminoacyl-tRNA synthetases. J Mol Biol. 2006;361:300–311. doi: 10.1016/j.jmb.2006.06.015. [DOI] [PubMed] [Google Scholar]