

Introduction
While the phenomenon of electron transfer (ET) between small molecules in solution is reasonably well described,1 the factors that influence protein ET reactions are less understood.2,3 Biological systems are far from ideal experimental systems for the study of a fundamental physical process such as ET. A complete description of biological ET reactions requires not only knowledge of the physics that describes ET, but also an understanding of how the biological milieu, most commonly a protein, influences this fundamental process. The studies described in this Account are patterned after the approaches that biochemists have long used to elucidate protein structure-function relationships in enzymes. First one determines the rate-limiting step for the overall reaction and then the activation parameters (i. e., ΔG°, ΔH°, ΔS°) for that reaction step. In conjunction with structural studies and often site-directed mutagenesis, it is then possible to describe the roles of specific amino acid residues and features of protein structure in the catalytic process. For protein ET reactions the same approach may be used, but there are complications. In contrast to chemical reactions, no bond making or breaking is associated with ET reactions. The redox centers which serve as electron donor and acceptor are not in direct contact, and the reactants and products may be structurally indistinguishable. As such, the reaction coordinate and transition state are poorly defined and analysis of true ET reactions requires a modified form of transition state theory with different activation parameters (i.e., λ, HAB). Furthermore, when ET occurs between and through proteins, non-ET reaction steps in the overall process, including protein-protein interactions, may complicate the assignment of the rate-limiting step.
Many biologically relevant interprotein ET reactions, as in the respiratory and photosynthetic ET chains, occur within and between integral membrane proteins. These are inherently difficult to study because the proteins are difficult to purify and cannot readily be studied under aqueous conditions. Soluble redox proteins are more amenable to mechanistic studies. Several soluble ET proteins have been characterized; however, relatively few complexes of soluble ET protein partners have been described and defined at the structural level.4–10 This Account will focus on results obtained from studies of the methylamine dehydrogenase (MADH)-amicyanin-cytochrome c-551i complex from Paracoccus denitrificans (Figure 1) which is perhaps the best characterized physiological protein ET complex. X-ray crystal structures are available for the complex of MADH and amicyanin9 and for the ternary protein complex.10 It was demonstrated by single-crystal polarized absorption microspectroscopy 11 and EPR spectroscopy12 that in the crystalline state MADH is catalytically competent and transfers electrons from its tryptophan tryptophylquinone (TTQ) cofactor to the cytochrome heme via the type 1 copper center of amicyanin. The steady-state kinetic parameters for methylamine-dependent cytochrome c-551i reduction by the MADH-amicyanin complex in solution have been characterized,13 and rates of the individual ET reactions which occur within the complex have been determined by monitoring characteristic changes in the absorption spectra of the proteins which occur during the redox reactions.14–16 Site-directed mutagenesis studies of MADH and amicyanin have identified specific amino acid residues that stabilize specific protein-protein interactions,17,18 modulate the Em value of the copper,19 and influence ET parameters for the reactions which occur within the complex.20–23 It was also possible to generate mutations of amicyanin that alter the kinetic mechanisms of ET reactions within the complex by converting true ET reactions to ones which are gated or coupled.24,25 These studies showed that ET rates may be significantly altered by subtle changes in protein structure by a variety of mechanisms.
Figure 1.

The MADH-amicyanin-cytochrome c-551i complex. One half of the symmetrical complex of the crystal structure (PDB, 2MTA)10 is shown.
Electron Transfer Theory
In the classical model, ET occurs at the intersection of the potential energy surfaces for the reactant and product states. For simplicity, these multi-dimensional energy surfaces are typically presented as parabolas described by the free energy (ordinate) and reaction coordinate (abscissa) (Figure 2). The reorganization energy (λ) is the energy difference between the reactant and product states at the equilibrium nuclear configuration of the reactant (i.e., the minimum). When ΔG° is equal to zero the activation energy is λ/4. HAB is the electronic coupling matrix element which represents the extent to which the wave functions of the reactant and product states overlap. The splitting at the intersection point is equal to 2HAB. This describes the degree of nonadiabicity (i.e., probability of the reaction occurring when the activation energy has been achieved). When HAB is zero (a diabatic system) there is no chance of the reaction occurring regardless of the energy. At the other extreme, the adiabatic system, the value of HAB is so large that the probability of crossover when the activation energy is achieved is unity. Such adiabatic reactions are described by transition-state theory. In a true ET reaction, the gap represented as 2HAB is relatively small and this system is said to be nonadiabatic. Because of the weak coupling, the activation energy may have to be achieved several times before the crossover from reactant to product states occurs. For nonadiabatic ET, kET is described by a modified form of transition-state theory (eq 1).1 The activation energy for the reaction is equal to (ΔG°+λ)2/4λ where ΔG° is determined from the redox potential difference for the ET reaction. The other parameters are Planck’s constant (h), the gas constant (R) and temperature (T). In simple systems HAB and, consequently kET will decrease exponentially with distance. This is reflected in eq 2, where ko is the characteristic frequency of the nuclei (1013 s−1), which is the limit for kET when donor and acceptor are in van der Waals’ contact and λ = −ΔG°. The donor to acceptor distance is r, and ro is the close contact distance (3 Å). The parameter β is used to quantitate the nature of the intervening medium with respect to its efficiency to mediate ET.26,27 The challenge for those wishing to understand biological ET reactions is to make the transition from the realm of parabolas to the world of proteins and to describe protein structure-function relationships that reveal how the protein influences these ET parameters. Examples of the use of site-directed mutagenesis to selectively alter the values of ET parameters for true ET reactions in the MADH-amicyanin-cytochrome c-551i complex (Table 1) will be discussed.
Figure 2.

Potential energy diagrams for electron transfer reactions. In each set, the parabolas on the left and right represent the reactant and product states, respectively. In these examples ΔG°=0.
Table 1.
Electron transfer reactions within native and mutant complexes of methylamine dehydrogenase, amicyanin and cytochrome c-551i.
| ET donor | ET acceptor | k, 30 °C (s−1) | ΔG° (kJ/mol) | λ (kJ/mol)a | λ (eV) | HAB (cm−1)b | Reaction Type | Ref |
|---|---|---|---|---|---|---|---|---|
| O-quinol MADH | amicyanin | 9.8 | −3.2 | 222 | 2.3 | 12±7 | True | 31 |
| N-quinol MADH | amicyanin | 275 | −3.2c | 341 | 3.5 | 23,000 | Gated | 41 |
| O-quinol αF55A MADH | amicyanin | 45 | −3.2 | 174 | 1.8 | 3±1 | True | 22 |
| O-quinol MADH | M98Q amicyanin | 0.2 | −3.3 | 261 | 2.7 | 12±4 | True | 23 |
| O-quinol MADH | M98A amicyanin | 9.6 | −3.2 | 203 | 2.1 | 6±2 | True | 23 |
| O-quinol MADH | F97E amicyanin | 0.2 | −3.2 | 222 | 2.3 | 3±0.1 | True | 21 |
| O-quinol MADH | P94F amicyanin | 53 | −21.7 | 222 | 2.3 | 5±1 | True | 39 |
| O-quinol MADH | P94A amicyanin | 82 | −18.9 | 212 | 2.2 | 3.8±1.2 | True | 25 |
| O-quinol MADH | P52G amicyanin | 3.0 | −4.82 | 270 | 2.8 | 78±30 | Gated | 24 |
| O-quinol MADH | M51A amicyanin | 1.3 | −2.8 | 299 | 3.1 | 142±20 | Gated | 48 |
| amicyanin | cytochrome c-551i | 87 | +3.2 | 116 | 1.2 | 0.3±0.1 | True | 32 |
| P94F amicyanin | cytochrome c-551i | 0.6 | +21.7 | 125 | 1.3 | 0.3±0.1 | True | 39 |
| P94A amicyanin | cytochrome c-551i | 0.4 | +18.9 | 222 | 2.3 | 8.3±5.5 | Coupled | 25 |
λ is sometimes expressed in units of kJ/mol and sometimes as eV so both values are given.
Errors are listed only for HAB which is the parameter that yields the most uncertainty. For the other parameters errors are typically much less than 10%.
Values of ΔG° for gated reactions refer to the ET reaction step and not the non-ET reaction which gates ET.
| (1) |
| (2) |
Kinetic Complexity of Protein ET
For long-range protein ET reactions it is often difficult to ascertain whether or not the observed rate of the redox reaction (kobs) is a true ET rate constant (kET) (Scheme 1). In some reactions, a non-ET event (where Kx is the equilibrium constant for this reaction) may be required to optimize or activate the system for ET.2,3,28,29 This applies to both interprotein and intraprotein ET reactions. Kinetic models have been developed that define such kinetically complex ET reactions as true, gated or coupled. ET reactions from TTQ in MADH are true or gated depending on the whether the reduced cofactor is in the O-quinol or N-quinol state, respectively (Figure 3).30,31 ET from copper to heme is a true ET reaction.32 Using site-directed mutagenesis it has been possible to convert these true ET reactions to gated and coupled reactions (Table 1). This has provided insight into how the protein may dictate the kinetic mechanism of the ET process, as well as control true ET parameters.
Scheme 1.
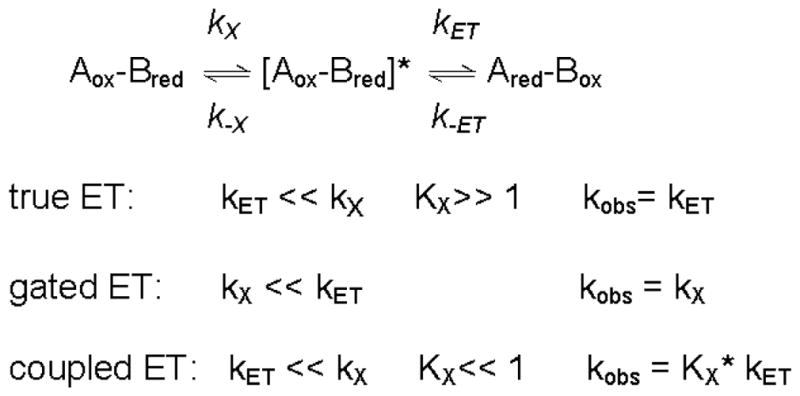
Models for kinetically complex ET
Figure 3.

Different forms of reduced TTQ in MADH. Reduction of MADH by dithionite yields O-quinol TTQ whereas reduction of MADH by the amine substrate yields N-quinol TTQ in which the substrate-derived nitrogen has displaced a quinone oxygen.
Alteration of λ by site-directed mutagenesis
It is presumed that the ET event occurs only when the system is completely optimized for ET to occur in the absence of nuclear motions. The energy required to bring the reactant and product states to this common intermediate state is λ. It comprises the inner sphere λin which reflects changes in ligand bond lengths and bond angles that accompany the redox reaction, and the outer sphere λout that reflects reorientation of solvent molecules that accompanies the redox reaction. For reactions in solution between small molecules, these distinctions are reasonably well-defined, but not so for protein ET reactions. For protein-bound redox cofactors such as quinones or flavins one does not have a simple set of metal ligands but a large number of bonds and an asymmetric electron distribution in a complex organic molecule. Even with metalloproteins, amino acid residues provide metal ligands and reorientation of the protein matrix may accompany the redox reaction. For these reasons the distinction between λin and λout becomes blurred and the influence of the protein environment on each difficult to ascertain. Mutations of MADH and amicyanin are described that appear to selectively alter λin and λout for the true ET reaction from O-quinol MADH to amicyanin.
Alteration of λout
Phe55 of the α subunit of MADH is present in a substrate channel that connects the protein surface with the active site and is a key determinant of amine substrate specificity.33 An αF55A MADH mutation (Figure 4) significantly increased kET from O-quinol MADH to amicyanin.22 Thermodynamic analysis of the ET reaction revealed that kET increased because λ for this reaction decreased by 0.5 eV as a consequence of the mutation. The crystal structure of αF55A MADH was determined in complex with amicyanin and cytochrome c-551i and compared with that of the native complex. Very little difference in the overall structure was seen, but there was a change in the solvent content of the active-site and substrate channel (Figure 4). Two waters are in the native MADH active site in close proximity to TTQ and shielded from bulk solvent by αPhe55. While water fills the void left by removal of the phenyl ring in the channel, only one of the active-site waters is present in αF55A MADH. The observed decrease in λ is consistent with TTQ being less solvated in αF55A MADH than in native MADH. The dramatic influence of water on λ may explain why proteins which evolved to function solely as ET mediators usually contain redox centers that are buried within the protein matrix, thus minimizing λ. Redox enzymes with bi-functional cofactors such as TTQ, which participate in catalysis as well as ET, must be at least partially exposed to solvent at the active site, which may explain why their ET reactions tend to exhibit relatively large values of λ.
Figure 4.
Active-site structures of MADH (blue) and αF55A MADH (red). The structures of the ternary protein complexes with native (PDB, 2MTA) and αF55A MADH (PDB, 1GM2)22 are superimposed with the electron density for αF55A MADH included. The single water present in the native active-site and absent in the mutant is indicated by the arrow.
Alteration of λin
Spectroscopic and crystallographic analysis of M98Q amicyanin revealed that this substitution of the axial ligand of copper caused a significant rhombic distortion of the type 1 site34 (Figure 5). The EPR spectra of native and M98Q amicyanins exhibited All values of 53 G and 23 G, respectively. Comparison of isomorphous crystals of native and M98Q amicyanins at atomic resolution revealed no significant change in the distances and orientations of the three equatorial copper ligands but indicated that the mutation increased the distance of the copper from the equatorial plane that is formed by the other three copper ligands from 0.20 to 0.42 Å. The mutation had little effect on the Em value but kET for the reaction from O-quinol MADH to amicyanin was reduced 45-fold in M98Q amicyanin. Thermodynamic analysis of these ET reactions showed that the decrease in kET was due to an increase of 0.4 eV in λ. No change in the experimentally-determined HAB or ET distance was observed confirming that the mutation had not altered the rate determining step for ET and that this was still a true ET reaction. The basis for the increased λ for the reaction with M98Q amicyanin is not solely the nature of the atom which provides the axial ligand (Gln98 OE2). M98A amicyanin also uses an oxygen, from water, for the axial ligand and no such change in λ was observed (Table 1).19,23 These results correlate well with results of quantum chemical calculations of λ of model compounds of the type 1 copper site with Gln and Met axial ligands. The calculated λin for a Cu(Im)2(SCH3)(CH3CONH2) model was approximately 0.3 eV greater than for a Cu(Im)2(SCH3)(SCH3)2 model.35 This relationship between the extent of rhombicity and λ, most likely λin, highlights the importance of the geometry of the type 1 copper site in controlling λ, consistent with the concept of “rack-induced” folding of type 1 copper proteins facilitating rapid ET by reducing λ.36
Figure 5.

A. Position of copper relative to the plane formed by equatorial ligands in native (green sphere) and M98Q (purple sphere) amicyanins. The superimposed structures are native (PDB, 2OV0, 0.75 Å) and M98Q (PDB, 2IDT, 1.0 Å).34 B. EPR spectra of native and M98Q amicyanins.23
Alteration of HAB by Site-Directed Mutagenesis
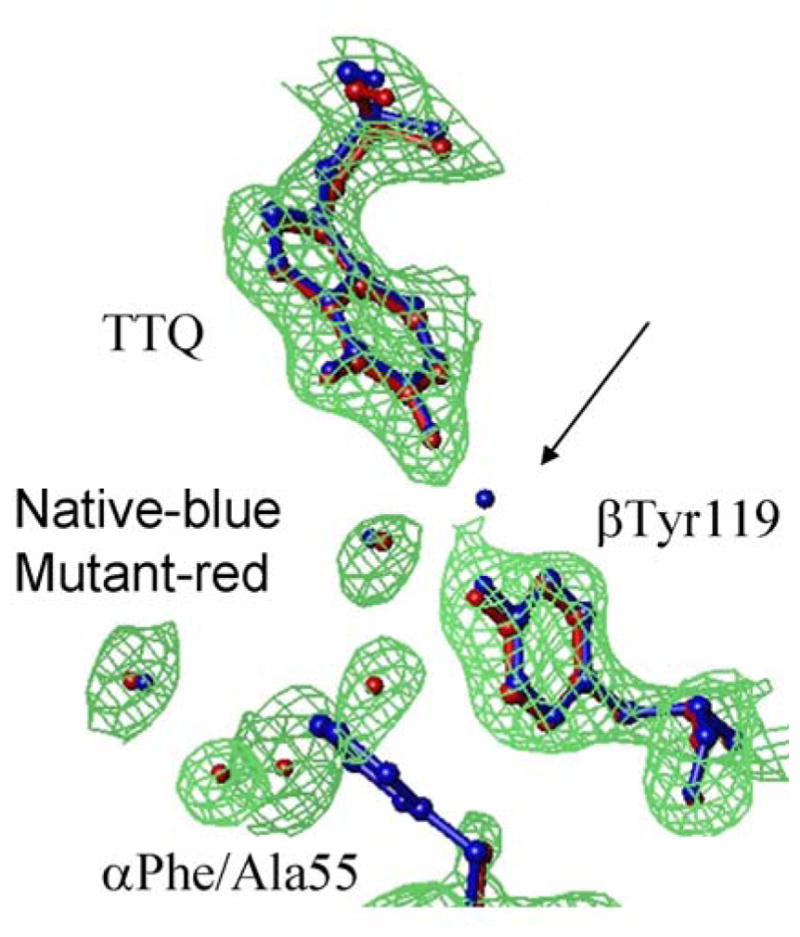
HAB depends on the ET distance and the nature of the intervening medium (HAB ~ e−βr). Two approaches have been used to predict relative HAB values for protein ET reactions from known structures. In one, the overall HAB is proportional to the product of the HAB for each individual through-bond or through-space step along the ET pathway.26 In the other, the overall HAB is proportional to the direct distance using a single average β that is related to the packing density of the intervening protein.27 Because the β value for ET through a vacuum (2.8 Å−1) is much larger than for ET through a covalent bond (0.7 Å−1),37 it follows that efficient ET through proteins would occur primarily through bonds. An F97E mutation of amicyanin decreased kET for the reaction from O-quinol MADH to amicyanin.21 The ΔG° and λ associated with the ET reaction were unaffected by the mutation and the decrease in kET was due solely to a decrease in HAB (Table 1). Phe97 is located at the MADH-amicyanin interface (Figure 6). Inspection of the structure of the protein complex reveals that an interprotein though-space jump of at least 2.6 Å is required for ET from TTQ to copper (Figure 6B). On the basis of the native structure and analysis of the ET reactions of native and F97E amicyanin it was concluded that the F97E mutation causes an increase in this critical interprotein distance of 0.9 Å which accounts for the observed decreases in HAB and consequently kET. This demonstrates that small changes in the length of through-space segments of ET pathways, particularly interprotein gaps, can significantly alter HAB.
Figure 6.
The amicyanin-MADH interface. A. Residues of amicyanin (green) and MADH (brown) near the site of interprotein ET are shown as ball and stick with their van der Waals radii colored as oxygen (red), nitrogen (blue), carbon (grey) and sulfur (yellow). B. Predicted point of interprotein ET from the backbone oxygen of Pro54 of amicyanin to TTQ.
Alteration of ΔG° by site-directed mutagenesis
The ΔG° for true ET reactions depends on the ΔEm for the donor and acceptor redox centers. Factors that influence the Em value include the identity of ligands for metal cofactors, protein-imposed constraints on organic cofactor conformation or metal ligation geometry, H-bonding pattern around the cofactor, presence of water, hydrophobicity, and electrostatic effects. An example of the use of site-mutagenesis to alter kET by altering the Em value of amicyanin is described to illustrate the predictable effect of altering ΔG° on kET for a true ET reaction.
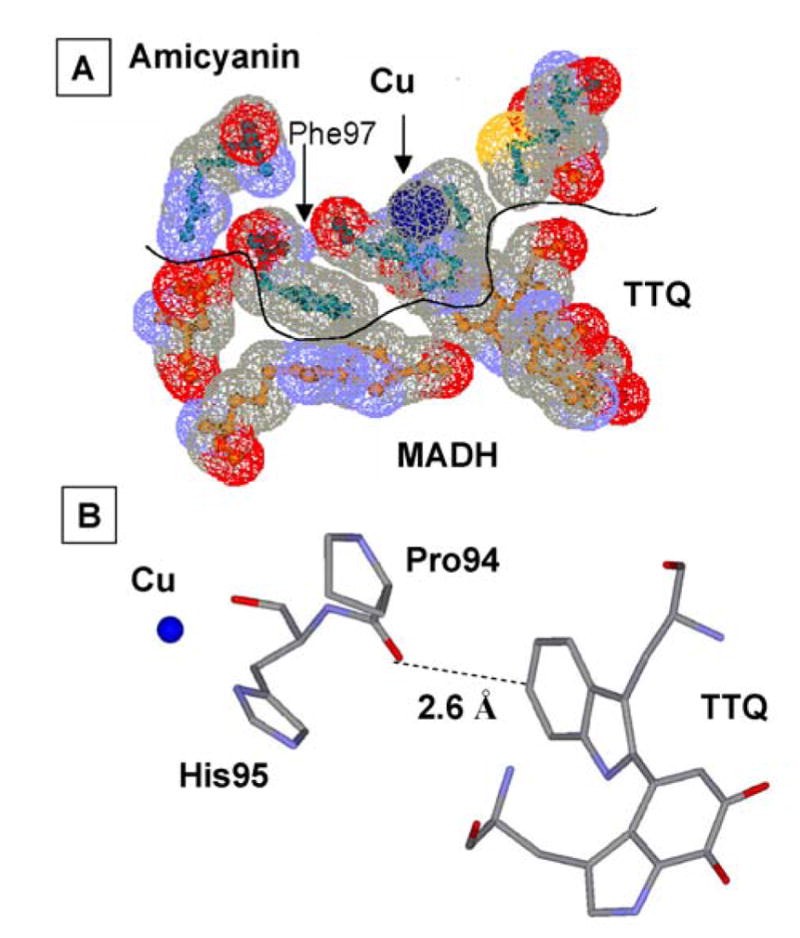
Pro94 resides in the “ligand loop” of amicyanin, a sequence of amino acids that contains three of the four copper ligands (Figure 7). P94F and P94A mutations of amicyanin increased its Em value by 150 and 115 mV, respectively.38 Atomic resolution structures of P94F and P94A amicyanins34 revealed that the bond lengths and angles of the copper ligands were unchanged as a consequence of mutation of Pro94, but in each mutant, a hydrogen-bond to the copper-coordinating thiolate sulfur of Cys92 is introduced by movement of the amide nitrogens of Phe94 and Ala94 closer to the thiolate sulfur than the nitrogen of Pro94. This is the likely explanation for the increased Em values which result in a more negative ΔG° for ET from MADH and more positive ΔG° for ET to cytochrome c-551i. As expected for true ET reactions, this causes an increase in kET from TTQ to copper and decrease in kET from copper to heme39 (Table 1). Analysis of the temperature-dependence of these reactions indicated that the λ values for these ET reactions were unchanged by mutations, despite the large change in Em value. Steady-state kinetic studies of methylamine-dependent reduction of heme by the three-protein complex indicated that the P94F mutation decreases kcat because the now unfavorable uphill ET reaction from copper to heme becomes the rate-limiting step in the overall reaction.39 This has important implications for understanding the roles of individual redox centers in regulating the rate of flux through biological ET chains.
Figure 7.
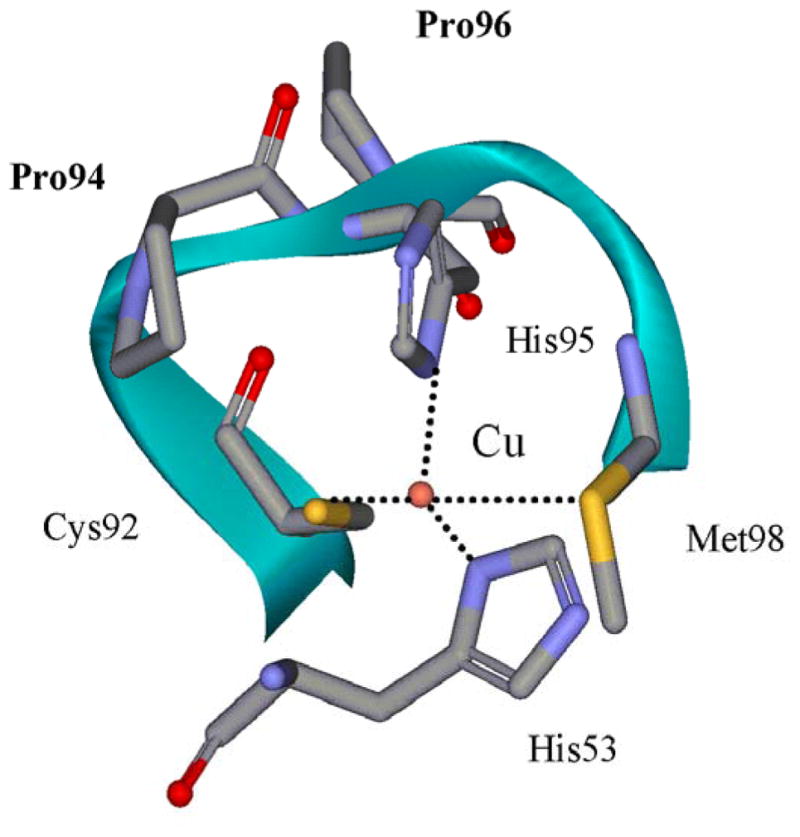
The copper center of amicyanin. The “loop” indicated as a ribbon from Cys92 to Met98 contains three of the amino acids providing copper ligands as well a two proline residues which have been targets for site-directed mutagenesis.
Protein control of the kinetic mechanism of ET
To investigate the kinetic control of protein ET reactions it is desirable to obtain systems where it is possible to compare a true and a gated ET reaction between the same redox centers within the same protein matrix. A primary criterion for initially classifying an ET reaction as gated is an experimentally-determined HAB which exceeds the non-adiabatic limit.40 Supporting evidence includes demonstration that the reaction rate is influenced by factors which would not be expected to affect a true ET reaction, such as ionic strength or viscosity. ET reactions from TTQ in MADH will be true or gated depending on the whether the reduced cofactor is in the O-quinol or N-quinol state, respectively (Figure 2). ET from amicyanin to cytochrome c-551i in the complex is another true ET reaction.32 By using site-directed mutagenesis it was possible to convert the true ET reaction from O-quinol into a conformationally-gated ET reaction,24 and the true ET reaction from amicyanin to the cytochrome to a kinetically-coupled ET reaction.25
Chemically-gated ET from N-quinol MADH to amicyanin
The phenomenon of “chemically-gated” ET 29 is one in which a prerequisite chemical reaction activates the system for ET that is so rapid that the reaction becomes rate-limited by the chemical step, yet the chemical step is much faster than the corresponding true ET reaction in the absence of activation. ET from N-quinol MADH to amicyanin is chemically-gated by the transfer of a solvent exchangeable proton as indicated by the large observed solvent kinetic isotope effect on the apparent kET for the reaction.30,41 Analysis of the temperature dependence of the reaction rate yielded unreasonable values of HAB of 23,000 cm−1 and an ET distance of −4.9 Å, which provided strong evidence that the rate did not describe a non-adiabatic ET reaction and is more appropriately described by standard transition-state theory. The rate was also dependent on pH and the concentration of monovalent cations. The pH-dependence was attributed to an ionizable group that is involved in binding the cation which stabilizes a negatively-charged transient reaction intermediate that is formed by the rate-limiting deprotonation of the N-quinol to generate the activated ET complex. This model provided a detailed description of how a chemical reaction that occurs at an enzyme active-site can gate an ET reaction which occurs at the protein surface.30 Chemically-gated ET was also described for another TTQ-dependent enzyme, aromatic amine dehydrogenase [AADH], which catalyzes the oxidative deamination of phenylethylamines42 and donates substrate-derived electrons to the copper protein azurin.43 As with MADH and amicyanin, ET from the O-quinol AADH to azurin was a true ET reaction, but ET from the substrate-reduced N-quinol AADH to azurin was gated by deprotonation of the N-quinol TTQ.44 Other protein ET reactions described in the literature that may be considered examples of chemically-gated ET controlled by chemical reactions other than deprotonation include the ET reaction between the iron protein and molybdenum-iron protein of the nitrogenase complex45,46 and the ET reaction from thiamine pyrophosphate to an iron-sulfur cluster in pyruvate-ferredoxin oxidoreductase in the presence of CoA.47
Conformationally-gated ET from MADH to P52G and M51A amicyanins
For interprotein ET, if the ideal orientations of the proteins for binding and ET are different, then some rearrangement of proteins within the complex must occur to maximize kET. This phenomenon has been elucidated by site-directed mutagenesis of amicyanin residues Met51 and Pro52 which are present at the MADH-amicyanin interface (Figure 8).24,48 A portion of Pro52 is in close proximity to βVal127 of MADH, and the side-chain of Met51 makes close contacts with βVal56 of MADH. A P52G mutation both increased the Kd for complex formation and decreased kET for the true ET reaction from O-quinol MADH.24 Thermodynamic analysis of kET yielded an increase in HAB from 12 cm−1 to 78 cm−1 with a corresponding decrease in ET distance of 3 Å, which is physically impossible given the known structures. A significant increase in the experimentally-determined λ further suggested that the ET reaction was now gated by a slower non-ET process. Analysis of the crystal structure of P52G amicyanin revealed that in addition to the loss of three carbons of Pro52 the position of the side chain of Met51 was altered such that contacts with the side-chain of βVal56 of MADH were lost. When Met51 was mutated to alanine to mimic the loss of the side-chain, a decrease in kET and increased values of λ and HAB were observed, similar to what was seen with P52G amicyanin (Table 1). In contrast to P52G amicyanin, the Kd for complex formation between M51A amicyanin and MADH was the same as for native amicyanin.48 Therefore, the loss of the interactions involving Pro52 were primarily responsible for the change in Kd for P52G amicyanin while the interactions involving Met51 are entirely responsible for the change in ET parameters seen with both the P52G and M51A amicyanins. Because the Kd is unaffected by the M51A mutation, the relative orientations of the proteins immediately upon binding are likely the same. Therefore, this mutation probably slows the rate of an existing but previously unrecognized conformational rearrangement that normally occurs rapidly in the native amicyanin-MADH complex subsequent to binding and prior to ET, thus causing ET to become gated. This demonstrates that subtle perturbations of protein-protein interactions may have significant effects on the rates of interprotein ET by altering the kinetic mechanism for the overall reaction. Thus, surface residues of redox proteins not only dictate specificity for their redox protein partners, but also may be critical to optimize the orientations of the redox centers and intervening media within the protein complex for the ET event.
Figure 8.

Interactions involving amicyanin residues Met51 and Pro52 at the MADH-amicyanin interface. Protein backbones of the MADH β subunit (green), native amicyanin (orange), M51A amicyanin (purple) and P52G amicyanin (cyan) are shown as solid ribbons with amicyanin residues 51 and 52, and residues on MADH with which they interact are shown as sticks. Structures (A) M51A amicyanin (PDB, 2QDV)48 and (B) P52G amicyanin (PDB, 2GB2)24 are shown in relation to the MADH structure after alignment with the amicyanin portion of the native complex structure (PDB, 2GC4).
Kinetically-coupled ET from P94A amicyanin to cytochrome c-551i
As stated earlier, mutation of Pro94, which resides in the “ligand loop” of amicyanin (Figure 7), to alanine alters ΔG° for the reaction with O-quinol MADH, but it remains a true ET reaction with values of HAB and λ similar to those for the reaction with native amicyanin. In contrast, the parameters for the ET reaction from reduced P94A amicyanin to cytochrome c-551i were significantly altered as a consequence of the mutation. These values of HAB and λ are 8.3 cm−1 and 2.3 eV, respectively, compared to values of 0.3 cm−1 and 1.2 eV for the reaction of native reduced amicyanin.25 The crystal structure of reduced P94A amicyanin exhibits two alternate conformations (Figure 9) with the positions of the copper 1.4 Å apart.19 In one of these conformations a water has replaced Met98 as a copper ligand and the ET distance to the heme of the cytochrome is increased by 1.4 Å. The kET from the copper in this conformation to heme is expected to be significantly diminished for three reasons. The overall ET distance is greater, the predicted ET pathway from Cu1+ via Met98 is disrupted, and the presence of water is likely to increase the λ associated with the ET reaction. To explain these data a kinetic mechanism was proposed in which after the reduction of Cu2+ by MADH, ET from the favored conformation is coupled to an unfavorable equilibrium with the unfavorable conformation.
Figure 9.

Model for coupled ET from alternate conformations of reduced P94A amicyanin to cytochrome c-551i.
It is also possible for an interprotein ET reaction to be conformationally coupled. An example of this is the ET reaction from pyrroloquinoline quinone in methanol dehydrogenase to heme in cytochrome c-551i. It was shown that the optimal orientation for ET was different from that for binding such that kET was reduced by the Keq for the rapid but unfavorable conformational rearrangement.49 Thus, the most stable conformation of the redox protein or protein complex is not necessarily the optimum conformation for ET. This can lead to coupled ET.
Conclusion
The application of nonadiabatic ET theory to protein ET reactions is a challenging task. Redox enzymes are structurally, chemically and kinetically complex. Kinetic and thermodynamic studies, coupled with structural information and biochemical data, are necessary to fully describe protein ET reactions. Site-directed mutagenesis may then be used to elucidate structure-function relationships. When mutations selectively alter ΔG°, HAB or λ, then one can determine how specific amino acid residues and features of protein structure influence kET by influencing the magnitude of these ET parameters. When mutations alter the kinetic mechanism for ET, one can determine the mechanisms by which adiabatic non-ET processes control the rates of ET reactions, and how specific amino acid residues and features of protein structure influence these non-ET reactions.
Acknowledgments
Work performed in this laboratory was supported by NIH grant GM-41574. I gratefully acknowledge the contributions of my co-workers whose names are included in our joint publications that are cited here.
Biography
Victor Davidson received his B.S. in Biochemistry from the University of Illinois (1973) and Ph.D. in Chemistry from Texas Tech University (1987). After postdoctoral training at Purdue University (1982–1984) and a research position at UCSF (1984–1988) he joined the faculty at the University of Mississippi Medical Center where he is currently Professor of Biochemistry.
References
- 1.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 2.Davidson VL. What controls the rates of interprotein electron-transfer reactions. Acc Chem Res. 2000;33:87–93. doi: 10.1021/ar9900616. [DOI] [PubMed] [Google Scholar]
- 3.Davidson VL. Unraveling the kinetic complexity of interprotein electron transfer reactions. Biochemistry. 1996;35:14035–14039. doi: 10.1021/bi961577p. [DOI] [PubMed] [Google Scholar]
- 4.Leys D, Basran J, Talfournier F, Sutcliffe MJ, Scrutton NS. Extensive conformational sampling in a ternary electron transfer complex. Nat Struct Biol. 2003;10:219–225. doi: 10.1038/nsb894. [DOI] [PubMed] [Google Scholar]
- 5.Pelletier H, Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 6.Toogood HS, van Thiel A, Basran J, Sutcliffe MJ, Scrutton NS, Leys D. Extensive domain motion and electron transfer in the human electron transferring flavoprotein.medium chain Acyl-CoA dehydrogenase complex. J Biol Chem. 2004;279:32904–32912. doi: 10.1074/jbc.M404884200. [DOI] [PubMed] [Google Scholar]
- 7.Kurisu G, Kusunoki M, Katoh E, Yamazaki T, Teshima K, Onda Y, Kimata-Ariga Y, Hase T. Structure of the electron transfer complex between ferredoxin and ferredoxin-NADP(+) reductase. Nat Struct Biol. 2001;8:117–121. doi: 10.1038/84097. [DOI] [PubMed] [Google Scholar]
- 8.Sukumar N, Chen ZW, Ferrari D, Merli A, Rossi GL, Bellamy HD, Chistoserdov A, Davidson VL, Mathews FS. Crystal structure of an electron transfer complex between aromatic amine dehydrogenase and azurin from Alcaligenes faecalis. Biochemistry. 2006;45:13500–13510. doi: 10.1021/bi0612972. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Durley R, Poliks BJ, Hamada K, Chen Z, Mathews FS, Davidson VL, Satow Y, Huizinga E, Vellieux FM. Crystal structure of an electron-transfer complex between methylamine dehydrogenase and amicyanin. Biochemistry. 1992;31:4959–4964. doi: 10.1021/bi00136a006. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Durley RC, Mathews FS, Davidson VL. Structure of an electron transfer complex: methylamine dehydrogenase, amicyanin, and cytochrome c551i. Science. 1994;264:86–90. doi: 10.1126/science.8140419. [DOI] [PubMed] [Google Scholar]
- 11.Merli A, Brodersen DE, Morini B, Chen Z, Durley RC, Mathews FS, Davidson VL, Rossi GL. Enzymatic and electron transfer activities in crystalline protein complexes. J Biol Chem. 1996;271:9177–9180. doi: 10.1074/jbc.271.16.9177. [DOI] [PubMed] [Google Scholar]
- 12.Ferrari D, Di Valentin M, Carbonera D, Merli A, Chen ZW, Mathews FS, Davidson VL, Rossi GL. Electron transfer in crystals of the binary and ternary complexes of methylamine dehydrogenase with amicyanin and cytochrome c551i as detected by EPR spectroscopy. J Biol Inorg Chem. 2004;9:231–237. doi: 10.1007/s00775-003-0513-0. [DOI] [PubMed] [Google Scholar]
- 13.Davidson VL, Jones LH. Intermolecular electron transfer from quinoproteins and its relevance to biosensor technology. Anal Chim Acta. 1991;249:235–240. [Google Scholar]
- 14.Davidson VL, Brooks HB, Graichen ME, Jones LH, Hyun YL. Detection of intermediates in tryptophan tryptophylquinone enzymes. Methods Enzymol. 1995;258:176–190. doi: 10.1016/0076-6879(95)58046-8. [DOI] [PubMed] [Google Scholar]
- 15.Husain M, Davidson VL, Smith AJ. Properties of Paracoccus denitrificans amicyanin. Biochemistry. 1986;25:2431–2436. doi: 10.1021/bi00357a020. [DOI] [PubMed] [Google Scholar]
- 16.Husain M, Davidson VL. Characterization of two inducible periplasmic c-type cytochromes from Paracoccus denitrificans. J Biol Chem. 1986;261:8577–8580. [PubMed] [Google Scholar]
- 17.Davidson VL, Jones LH, Graichen ME, Mathews FS, Hosler JP. Factors which stabilize the methylamine dehydrogenase-amicyanin electron transfer protein complex revealed by site-directed mutagenesis. Biochemistry. 1997;36:12733–12738. doi: 10.1021/bi971353m. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Z, Jones LH, Graichen ME, Davidson VL. Molecular basis for complex formation between methylamine dehydrogenase and amicyanin revealed by inverse mutagenesis of an interprotein salt bridge. Biochemistry. 2000;39:8830–8836. doi: 10.1021/bi000502p. [DOI] [PubMed] [Google Scholar]
- 19.Carrell CJ, Sun D, Jiang S, Davidson VL, Mathews FS. Structural studies of two mutants of amicyanin from Paracoccus denitrificans that stabilize the reduced state of the copper. Biochemistry. 2004;43:9372–9380. doi: 10.1021/bi049634z. [DOI] [PubMed] [Google Scholar]
- 20.Davidson VL, Jones LH, Graichen ME, Zhu Z. Tyr(30) of amicyanin is not critical for electron transfer to cytochrome c-551i: implications for predicting electron transfer pathways. Biochim Biophys Acta. 2000;1457:27–35. doi: 10.1016/s0005-2728(00)00052-9. [DOI] [PubMed] [Google Scholar]
- 21.Davidson VL, Jones LH, Zhu Z. Site-directed mutagenesis of Phe 97 to Glu in amicyanin alters the electronic coupling for interprotein electron transfer from quinol methylamine dehydrogenase. Biochemistry. 1998;37:7371–7377. doi: 10.1021/bi973020v. [DOI] [PubMed] [Google Scholar]
- 22.Sun D, Chen ZW, Mathews FS, Davidson VL. Mutation of αPhe55 of methylamine dehydrogenase alters the reorganization energy and electronic coupling for its electron transfer reaction with amicyanin. Biochemistry. 2002;41:13926–13933. doi: 10.1021/bi026654x. [DOI] [PubMed] [Google Scholar]
- 23.Ma JK, Mathews FS, Davidson VL. Correlation of rhombic distortion of the type 1 copper site of M98Q amicyanin with increased electron transfer reorganization energy. Bochemistry. 2007;46:8561–8568. doi: 10.1021/bi700303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma JK, Carrell CJ, Mathews FS, Davidson VL. Site-directed mutagenesis of proline 52 to glycine in amicyanin converts a true electron transfer reaction into one that is conformationally gated. Biochemistry. 2006;45:8284–8293. doi: 10.1021/bi0605134. [DOI] [PubMed] [Google Scholar]
- 25.Sun D, Li X, Mathews FS, Davidson VL. Site-directed mutagenesis of proline 94 to alanine in amicyanin converts a true electron transfer reaction into one that is kinetically coupled. Biochemistry. 2005;44:7200–7206. doi: 10.1021/bi050288a. [DOI] [PubMed] [Google Scholar]
- 26.Onuchic JN, Beratan DN, Winkler JR, Gray HB. Pathway analysis of protein electron-transfer reactions. Ann Rev Biophys Biomol Struct. 1992;21:349–377. doi: 10.1146/annurev.bb.21.060192.002025. [DOI] [PubMed] [Google Scholar]
- 27.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 28.Davidson VL. Effects of kinetic coupling on experimentally determined electron transfer parameters. Biochemistry. 2000;39:4924–4928. doi: 10.1021/bi992671j. [DOI] [PubMed] [Google Scholar]
- 29.Davidson VL. Chemically gated electron transfer. A means of accelerating and regulating rates of biological electron transfer. Biochemistry. 2002;41:14633–14636. doi: 10.1021/bi026812k. [DOI] [PubMed] [Google Scholar]
- 30.Bishop GR, Davidson VL. Catalytic role of monovalent cations in the mechanism of proton transfer which gates an interprotein electron transfer reaction. Biochemistry. 1997;36:13586–13592. doi: 10.1021/bi970586a. [DOI] [PubMed] [Google Scholar]
- 31.Brooks HB, Davidson VL. Free energy dependence of the electron transfer reaction between methylamine dehydrogenase and amicyanin. J Am Chem Soc. 1994;116:11201–11202. [Google Scholar]
- 32.Davidson VL, Jones LH. Electron transfer from copper to heme within the methylamine dehydrogenase-amicyanin-cytochrome c-551i complex. Biochemistry. 1996;35:8120–8125. doi: 10.1021/bi952854f. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Z, Sun D, Davidson VL. Conversion of methylamine dehydrogenase to a long-chain amine dehydrogenase by mutagenesis of a single residue. Biochemistry. 2000;39:11184–11186. doi: 10.1021/bi001568n. [DOI] [PubMed] [Google Scholar]
- 34.Carrell CJ, Ma JK, Antholine WE, Hosler JP, Mathews FS, Davidson VL. Generation of novel copper sites by mutation of the axial ligand of amicyanin. Atomic resolution structures and spectroscopic properties. Biochemistry. 2007;46:1900–1912. doi: 10.1021/bi0619674. [DOI] [PubMed] [Google Scholar]
- 35.Olsson MH, Ryde U, Roos BO. Quantum chemical calculations of the reorganization energy of blue-copper proteins. Protein Sci. 1998;7:2659–2668. doi: 10.1002/pro.5560071220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malmstrom BG. Rack-induced bonding in blue-copper proteins. Eur J Biochem. 1994;223:711–718. doi: 10.1111/j.1432-1033.1994.tb19044.x. [DOI] [PubMed] [Google Scholar]
- 37.Regan JJ, Risser SM, Beratan DN, Onuchic JN. Protein electron transport: Single versus multiple pathways. J Phys Chem. 1993;97:13083–13088. [Google Scholar]
- 38.Machczynski MC, Gray HB, Richards JH. An outer-sphere hydrogen-bond network constrains copper coordination in blue proteins. J Inorg Biochem. 2002;88:375–380. doi: 10.1016/s0162-0134(02)00364-1. [DOI] [PubMed] [Google Scholar]
- 39.Sun D, Davidson VL. Effects of engineering uphill electron transfer into the methylamine dehydrogenase-amicyanin-cytochrome c-551i complex. Biochemistry. 2003;42:1772–1776. doi: 10.1021/bi0271594. [DOI] [PubMed] [Google Scholar]
- 40.Winkler JR, Gray HB. Electron transfer in ruthenium-modified proteins. Chem Rev. 1992;92:369–379. doi: 10.1007/BF02110099. [DOI] [PubMed] [Google Scholar]
- 41.Bishop GR, Davidson VL. Intermolecular electron transfer from substrate-reduced methylamine dehydrogenase to amicyanin is linked to proton transfer. Biochemistry. 1995;34:12082–12086. doi: 10.1021/bi00037a052. [DOI] [PubMed] [Google Scholar]
- 42.Hyun YL, Davidson VL. Mechanistic studies of aromatic amine dehydrogenase, a tryptophan tryptophylquinone enzyme. Biochemistry. 1995;34:816–823. doi: 10.1021/bi00003a015. [DOI] [PubMed] [Google Scholar]
- 43.Hyun YL, Davidson VL. Electron transfer reactions between aromatic amine dehydrogenase and azurin. Biochemistry. 1995;34:12249–12254. doi: 10.1021/bi00038a020. [DOI] [PubMed] [Google Scholar]
- 44.Hyun YL, Zhu Z, Davidson VL. Gated and ungated electron transfer reactions from aromatic amine dehydrogenase to azurin. J Biol Chem. 1999;274:29081–29086. doi: 10.1074/jbc.274.41.29081. [DOI] [PubMed] [Google Scholar]
- 45.Chiu H, Peters JW, Lanzilotta WN, Ryle MJ, Seefeldt LC, Howard JB, Rees DC. MgATP-Bound and nucleotide-free structures of a nitrogenase protein complex between the Leu 127 Delta-Fe-protein and the MoFe-protein. Biochemistry. 2001;40:641–650. doi: 10.1021/bi001645e. [DOI] [PubMed] [Google Scholar]
- 46.Lanzilotta WN, Parker VD, Seefeldt LC. Electron transfer in nitrogenase analyzed by Marcus theory: evidence for gating by MgATP. Biochemistry. 1998;37:399–407. doi: 10.1021/bi971681m. [DOI] [PubMed] [Google Scholar]
- 47.Furdui C, Ragsdale SW. The roles of coenzyme A in the pyruvate:ferredoxin oxidoreductase reaction mechanism: rate enhancement of electron transfer from a radical intermediate to an iron-sulfur cluster. Biochemistry. 2002;41:9921–9937. doi: 10.1021/bi0257641. [DOI] [PubMed] [Google Scholar]
- 48.Ma JK, Wang Y, Carrell CJ, Mathews FS, Davidson VL. A single methionine residue dictates the kinetic mechanism of interprotein electron transfer from methylamine dehydrogenase to amicyanin. Biochemistry. 2007;46:11137–11146. doi: 10.1021/bi7012307. [DOI] [PubMed] [Google Scholar]
- 49.Harris TK, Davidson VL, Chen L, Mathews FS, Xia ZX. Ionic strength dependence of the reaction between methanol dehydrogenase and cytochrome c-551i: evidence of conformationally coupled electron transfer. Biochemistry. 1994;33:12600–12608. doi: 10.1021/bi00208a010. [DOI] [PubMed] [Google Scholar]