

Abstract
Knowing the precise locations of nucleosomes in a genome is key to understanding how genes are regulated. Recent ‘next generation’ ChIP–chip and ChIP–Seq technologies have accelerated our understanding of the basic principles of chromatin organization. Here we discuss what high-resolution genome-wide maps of nucleosome positions have taught us about how nucleosome positioning demarcates promoter regions and transcriptional start sites, and how the composition and structure of promoter nucleosomes facilitate or inhibit transcription. A detailed picture is starting to emerge of how diverse factors, including underlying DNA sequences and chromatin remodelling complexes, influence nucleosome positioning.
The genetic code resides within a negatively charged DNA polymer. The resulting electrostatic repulsion from neighbouring phosphates stiffens the polymer such that it cannot fit within the small confines of a nucleus. A solution to this problem has evolved in the form of highly basic histone proteins that bind to DNA and neutralize the negative charges. The formation of chromatin through the binding of histones to DNA allows the DNA to be folded into chromosomes and compacted by as much as a factor of 10,000. The packaging of DNA creates both a problem and an opportunity: wrapping DNA around histones potentially obstructs access to the genetic code; however, the ubiquity of the histones that are bound at all regions of chromosomal DNA can be exploited so that enzymes that read, replicate and repair DNA can be directed to the appropriate entry sites. In this way, RNA polymerase (Pol) II initiates transcription at the beginning of genes rather than in the middle, DNA polymerase initiates replication at replication origins and DNA repair enzymes are directed to sites of DNA damage.
How does the cell package a helical DNA polymer in a way that is both refractory and accessible? The evolutionary solution to the packaging problem is the nucleosome1,2 (FIG. 1a). The nucleosome is the basic unit of eukaryotic chromatin, consisting of a histone core around which DNA is wrapped. Each histone core is composed of two copies of each of the histone proteins H2A, H2B, H3 and H4 (FIG. 1b). Approximately 147 bp of DNA coils 1.65 times around the histone octamer in a left-handed toroid2. Amino-terminal histone ‘tails’ emanate from the nucleosome core, past the DNA. The polypeptide chains of the histone tails are subject to covalent modifications, including acetylation and methylation. At active genes or at genes that are poised for activation, histones H2A and H3 are replaced by the histone variants H2A.Z and H3.3 (see REFS 3,4 for reviews of histone variants). Beyond the nucleosome core is the linker histone, H1. Nucleosomes are arranged as a linear array along the DNA polymer as ‘beads on a string’. This structure can be further compacted by H1 into higher-order transcriptionally inactive 30 nm fibres.
Figure 1. Nucleosome structure.

a | Structure of a nucleosome core particle (front and side view)2,131. Histones are shown in light grey, and the DNA helix is shown in dark grey with a pink backbone. Basic amino acids (lysine and arginine) within 7 Å of the DNA are shown in blue to emphasize the electrostatic contacts between the DNA phosphates and the histones. b | A schematic of DNA wrapped around a nucleosome. Examples of histone tail modifications (Ac, acetylation; Me, methylation) and histone variants (H2A.Z and H3.3) are shown. Arrows indicate the replacement of canonical histones with histone variants. Part a courtesy of S. Tan, Pennsylvania State University, USA.
The combination of nucleosome positions and their chemical and compositional modifications are key to genome regulation. In this Review, we focus specifically on nucleosome positioning rather than on histone modifications and variants. Here we interrelate past and recent developments in our understanding of the basic organization of nucleosomes on chromosomes, and show how DNA sequences and chromatin remodelling complexes selectively position and organize nucleosomes so that they can regulate genomic function. Importantly, massively parallel DNA sequencing and microarray hybridization technologies have allowed the location of every nucleosome across a genome to be determined with unprecedented accuracy (BOX 1). We discuss how these maps reveal a common organizational theme at nearly every gene, including a nucleosome-free region (NFR) at the beginning and end of genes. We also discuss how the underlying DNA sequence and the action of chromatin remodelling complexes influence where nucleosomes are positioned. There is emerging evidence that nucleosomes regulate transcriptional initiation, and therefore understanding how nucleosomes are positioned has implications for how cells respond to external stimuli or how misregulation of nucleosome positioning leads to developmental defects and cancer.
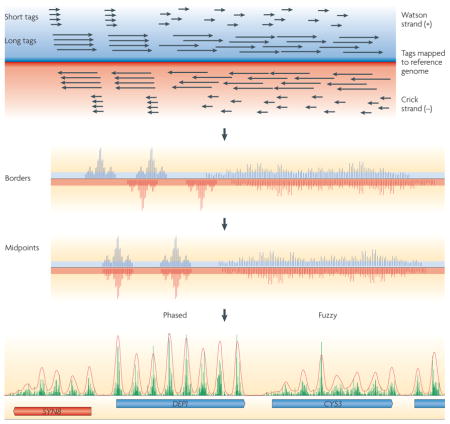
Box 1. ChIP–Seq nucleosome mapping technology.
A stringent procedure for high-resolution mapping of nucleosomes by ChIP–Seq involves an initial step to cross-link histones to nucleosomal DNA by formaldehyde treatment of living cells. In principle, cross-linking traps nucleosomes at their in vivo locations. Next, linker DNA is removed from isolated chromatin by digestion with high levels of micrococcal nuclease (MNase). Subsets of nucleosome particles are isolated by immunoprecipitation using antibodies directed against histones, histone variants or histone modifications. Fragments of mononucleosomal DNA that are ~150 bp long are size-selected by agarose gel electrophoresis. The 5′ ends of millions of individual DNA molecules in this library are then sequenced in parallel. Short-read technology sequences 25–35 bp fragments (called tags), whereas long-read technology produces read lengths of 100 bp or more. Sequence tags are then mapped to the reference genome using alignment algorithms (see the figure; black arrows represent the reads derived from long-and short-read technology) on either the Watson (blue) or Crick (red) strand. The 5′ ends of each tag, which correspond to nucleosome borders, are then plotted as a bar graph at each coordinate in the genome (as displayed in some publications16,22). Next, the tag location is adjusted to represent the nucleosome midpoint (typically +73 bp on the ‘W’ or ‘+’ strand and −73 bp on the ‘C’ or ‘−’ strand is added to the genomic coordinate of the 5′ end of each tag)17,19,20. Clusters of tags show a consensus nucleosome position. Two clusters are shown. The tighter the cluster, the more phased the corresponding nucleosome is. Randomly distributed tags reflect random (‘fuzzy’) positioning. A sample of tag distribution in a small section of the yeast genome is shown at the bottom of the figure, in which the red and blue tags are collapsed into a single bar graph. Each peak equates to a single consensus nucleosome position.
Three DNA sequencing technologies have been used to map nucleosomes16,17,19,23,24. Pyrosequencing using the Roche 454 GS20/FLX sequences nucleosomal DNA end-to-end, allowing both nucleosome borders to be linked in a single sequence. This method provides the greatest mapping accuracy, particularly in genomic regions of low complexity. By contrast, other platforms, such as those provided by the Illumina–Solexa Genome Analyzer and Applied Biosystems SOLiD, generate only 25–35 bp sequence tags, requiring both nucleosome borders to be inferred. Nonetheless, these short-read technologies produce >100 times the number of sequence tags at a similar cost as the long-read technologies, and so the short-read technology is currently the only practical technology for mapping nucleosomes in large genomes. The higher tag count of the short-read technology enhances mapping accuracy and thus provides a practical way of mapping nucleosomes.
Genomic organization of nucleosomes
Until recently, it was unclear whether deposition of histones on DNA during DNA replication occurs at random positions. Random deposition implies that nucleosomes lack positional cues and that histones are simply DNA packaging proteins that are removed and redeposited as DNA and RNA polymerases pass through them. Alternatively, individually positioned nucleosomes could take on specific physiological functions depending on where they reside in the genome. In this section, we will discuss how cells use both random deposition and specific positioning of histones to organize nucleosomes. This understanding has arisen through the development of technologies that have allowed genome-wide mapping of nucleosome positioning; we start by describing this progress then we discuss the genomic properties of nucleosomes.
A brief history of nucleosome cartography
In 2004, the exact genomic location of only a few hundred nucleosomes was known because techniques were limited to the individual interrogation of specific genomic loci. The early development of microarrays, which consisted of ~500–2,000 bp DNA probes that spanned each genic and intergenic region, provided a comprehensive view of the nucleosome landscape across the simple genome of the budding yeast Saccharomyces cerevisiae. The long (~1 kb) probe lengths of these early microarrays precluded the assessment of individual nucleosome states, which occur at <200 bp intervals. Nevertheless, for the first time, these pioneering studies showed a general depletion of nucleosomes in the intergenic regions where promoters are found5–7. It was initially unclear how the presence or absence of nucleosomes related to transcription, because the upstream intergenic regions of active and quiescent yeast genes were depleted of nucleosomes compared with other regions of the genome. However, higher-resolution nucleosome maps clarified that gene activation resulted in additional nucleosome depletion5,7–11. Furthermore, antibodies that were specific for individual histone post-translational modifications showed that the promoter regions of highly transcribed genes were particularly enriched with nucleosomes that contained acetylated and methylated histones12–14. The function of these modified histones remains an area of active research.
By 2005, microarrays had been developed that had shorter DNA probes and shorter probe–probe genomic distances, which provided higher resolution views of nucleosomes. However, printing technology limited the search space to small sections of small genomes. Nonetheless, these arrays showed that the nucleosomes at most genes are organized around the beginning of genes in basically the same way15: a NFR flanked by two well-positioned nucleosomes (the −1 and +1 nucleosomes), which is followed by a nucleosomal array that packages the gene (FIG. 2). This basic pattern has also held true for metazoans16,17.
Figure 2. Nucleosomal landscape of yeast genes.

The consensus distribution of nucleosomes (grey ovals) around all yeast genes is shown, aligned by the beginning and end of every gene. The resulting two plots were fused in the genic region. The peaks and valleys represent similar positioning relative to the transcription start site (TSS). The arrow under the green circle near the 5′ nucleosome-free region (NFR) represents the TSS. The green –blue shading in the plot represents the transitions observed in nucleosome composition and phasing (green represents high H2A.Z levels, acetylation, H3K4 methylation and phasing, whereas blue represents low levels of these modifications). The red circle indicates transcriptional termination within the 3′ NFR. Figure is reproduced, with permission, from REF. 20 © (2008) Cold Spring Harbor Laboratory Press.
A complete and comprehensive map of nucleosome locations in a eukaryotic genome (S. cerevisiae) was completed in 2007 owing to two impressive technological advances. First, the densities of commercially printed probes on microarrays increased dramatically, allowing millions of genomic loci to be interrogated by ChIP–chip analysis in a single experiment. The genomic distances between probes were reduced to 5 bp in S. cerevisiae and 36 bp in Drosophila melanogaster17,18. Second, massively parallel shotgun sequencing allowed individual nucleosomal DNA molecules to be sequenced, initially at the level of hundreds of thousands of nucleosomes, and now at the level of tens of millions of nucleosomes (BOX 1). The first such ultra-high-resolution genome-wide ChIP–Seq nucleosome map was achieved for nucleosomes containing H2A.Z in S. cerevisiae19. The map showed the precise nucleosomal contexts in which gene regulatory elements function on a genomic scale. Nucleosome maps of a similar resolution in yeast, worms, flies and humans have now been published16–18,20–24 and are likely to be produced for other model organisms soon. Future nucleosome mapping endeavours will probably focus on how nucleosome positions and histone modifications depend on cellular factors, and how they change in response to environmental signals, tissue differentiation and cellular disease states.
Lessons learned from global nucleosome maps
Genome-wide nucleosome maps allow us to explore the genomic properties of chromatin. At any given genomic locus, the preferential positioning of nucleosomes — called phasing — can be described (FIG. 3a). At most loci, there is an approximately Gaussian (normal) distribution of nucleosome positions around particular genomic coordinates, ranging from ~30 bp for highly phased nucleosomes to a random continuous distribution throughout an array. How much of this variation is due to genuine positional heterogeneity and how much is an artefact that is caused by overtrimming or undertrimming of the DNA at nucleosome borders by micrococcal nuclease during sample preparation remains to be determined.
Figure 3. Phasing information and rotational setting.

a | In a population, individual nucleosomes are either positioned within a small range of a genomic locus (phased) or with a continuous distribution throughout an array (fuzzy). b | The bar graph is an idealized distribution of nucleosomal sequence tags, which form a large cluster and several subclusters, in which the subclusters are spaced about 10 bp apart and represent multiple translational settings with a single predominant rotational setting (see also BOX 1). Also shown is a schematic of alternative rotational settings of DNA and its effect on site accessibility (indicated by the black ‘rungs’ on the DNA helix).
Within each Gaussian distribution, nucleosomes have preferred positions; these positions tend to be about 10 bp apart19 (FIG. 3b). This means that, owing to the helical nature of DNA, a DNA sequence will tend towards the same rotational setting (facing inwards or outwards) on the histone surface when a nucleosome is in alternative preferred positions (translational settings). This is important because the orientation of a DNA sequence on the histone surface determines the accessibility of its sequence and thus its activity (FIG. 3b).
Positioned nucleosomes tend to be spaced at a fixed distance from each other, with short stretches of linker DNA between them. The most common distance between adjacent nucleosome midpoints is approximately 165 bp (~18 bp linker) in S. cerevisiae18,20,23, 175 bp (~28 bp linker) in D. melanogaster 17 and Caenorhabditis elegans24, and 185 bp (~38 bp linker) in humans16,22. Chromatin remodelling or spacing complexes of the imitation switch (ISWI) class, such as ATP-dependent chromatin assembly and remodelling factor (ACF) and chromatin accessibility complex (CHRAC), establish nucleosome spacing25–28. These complexes bind nucleosomes and a finite amount of adjacent linker DNA, then use energy from ATP hydrolysis to move nucleosomes in the direction of the linker DNA29–31. As a result, the linker shortens until it can no longer bind the ISWI complex. Linker length is likely to be further constrained by the linker-binding histone H1 (REFS 32–34), which might reduce the amount of linker DNA that is available to the ISWI complexes. The different linker lengths in evolutionarily diverged eukaryotes might reflect the presence of evolutionarily divergent ISWI subunits or H1 proteins that have species-specific DNA length requirements for binding in these eukaryotes. Shorter linkers might result in a reduced availability of sequences for protein binding and thus these linkers might have regulatory functions. Very long linkers, or NFRs (~140 bp in length17,20,22), are present in the genome where a nucleosome seems to be missing or where the DNA is depleted of nucleosomes relative to the rest of the genome. As we will discuss in a later section, these NFRs are key to unlocking the mystery of how nucleosome organization and gene regulation are linked.
The organization of nucleosomes on genes
The genome-wide maps of nucleosome location have also provided insights into the organization of nucleosomes around protein-coding genes. The S. cerevisiae genome provides the clearest example of a consensus pattern of organization (FIG. 2). The first predominant nucleosome located upstream of the transcription start site (TSS) (designated −1, see BOX 2) covers a region from −300 to −150 relative to the TSS, and can regulate the accessibility of promoter regulatory elements in that region. During a transcription cycle, the −1 nucleosome will experience many changes that affect its stability, including histone replacement, acetylation and methylation, as well as translational repositioning, and ultimately eviction after pre-initiation complex (PIC) formation. Whether the −1 nucleosome remains evicted during multiple rounds of transcription, or returns between each transcription cycle, remains an important unanswered question. The answer to this question would help elucidate whether reinitiation of transcription is mechanistically distinct from the initial activation event.
Box 2. Nucleosome numbering.
In yeast and flies, the first nucleosome upstream of the 5′ nucleosome-free region (NFR) is considered the −1 nucleosome, whereas the first nucleosome downstream of the NFR is considered the +1 nucleosome17–21,23 (FIG. 2). In humans, the rare nucleosome that appears in the consensus NFR regions has been defined as −1, which leaves the more predominant first upstream nucleosome to be called −2 (REF. 22). As this nomenclature inconsistency between organisms could be confusing, some standardization of nucleosome numbering might be necessary, particularly as different nucleosome positions have been shown to have specific functions.
Downstream of the −1 nucleosome is a NFR (the 5′ NFR), then the TSS (discussed in a later section), which is followed by the +1 nucleosome. Of all the nucleosomes found in and around genes, the +1 nucleosome displays the tightest positioning (or phasing)20. The +1 nucleosome often contains histone variants (H2A.Z and H3.3)35 and histone tail modifications (methylation and acetylation)36–38, all of which might facilitate nucleosome eviction and PIC assembly. During transcription, the +1 nucleosome is likely to be evicted, but it seems to rapidly return to its original place after Pol II has passed, as it is only modestly depleted at highly transcribed genes19. The +2 nucleosome is found immediately downstream of the +1 nucleosome. It shares some properties with the +1 nucleosome but contains less H2A.Z, and displays less methylation, acetylation and phasing38,39. The +3 nucleosome and the more downstream nucleosomes each have less of these properties than the previous upstream nucleosome. The reduction in these properties might reflect a limitation in the functional distance of histone remodelling or modifying enzymes that are tethered to the 5′ end of genes.
Beyond ~1 kb from the TSS, consensus spacing from the TSS dissipates. Although phased nucleosomes are found, there is an increasing tendency for random nucleosome positions15,20. This might represent a loss in the functional constraints that are imposed on nucleosomes at the beginning of genes.
The array of nucleosomes that covers a gene terminates with a NFR at the 3′ end of the gene (the 3′ NFR). The 3′ NFR is the region at which Pol II terminates transcription, which is precipitated by the cleavage of the nascent RNA transcript near the 3′ end of the gene. Whether the nucleosome located at the end of the 3′ NFR contributes to termination is not known. Overall, these high-resolution genomic maps show that genes are packaged into a regular array of nucleosomes that starts at a fixed position from the TSS and are bracketed by nucleosome-free or nucleosome-depleted zones. In the next section, we discuss how this pattern might be set up.
Origins of nucleosome positions
So far, we have learned that nucleosomes adopt canonical positions around promoter regions and more random positions in the interior of genes. But how is this organization established? We describe one view using an analogy of a roulette wheel (an analogy of a parking lot is described elsewhere40). In a roulette wheel, the ball is allowed to land only in the designated slots (FIG. 4a). Regardless of how many balls are used, the possible positions of the balls are predetermined. Every positioned nucleosome could have an underlying DNA sequence structure (a ‘slot’) that favours positioning in that location. Randomly positioned nucleosomes would not be associated with any positioning sequence. This model implies that the positions of adjacent nucleosomes are independently controlled. An alternative possibility, called statistical positioning41–44, arises from the close packing of nucleosomes into an array. The positioning of one nucleosome in the array (FIG. 4b, left side) forces the positioning of all other nucleosomes, because the tight packing restricts their lateral movement (this is termed probabilistic positioning, as indicated by the distribution trace in FIG. 4b). Thus a single genomic barrier can potentially position many nucleosomes without the need for individual positioning sequences. Below, we describe how a combination of both models might exist (FIG. 4c).
Figure 4. Sequence-based packing versus statistical packing.

a | Individual slots represent nucleosome positioning sequences that define where a nucleosome (grey circle) will reside on a length of DNA. b | In its purest form, statistical positioning relies on a single positional barrier (left side), against which nucleosomes are ordered. A probabilistic density trace of where nucleosomes would reside in a population is shown. c | The true cellular state is likely to be a combination of both independent and statistical positioning.
DNA sequence patterns
The +1 nucleosome could provide the barrier for statistical positioning. So, are there DNA sequence patterns that are associated with well-positioned nucleosomes? The idea behind pattern searching is to align the 147 bp DNA sequence of thousands of well-positioned nucleosomes and determine whether particular base pair combinations are statistically enriched at particular positions along the DNA molecule. Such pattern searching began in the 1980s with a few hundred nucleosomal sequences, and showed that AA, TT and TA dinucleotides occurred at 10 bp intervals42,45–47. There were also 10 bp periodicities of GC dinucleotides, but their periodicity was offset by 5 bp compared with the AA, TT and TA patterns. Current alignments of thousands of nucleosomal DNAs show essentially the same pattern, including changes in nucleotide composition in linker regions17,19,20,24,45,48–53. Other nucleotide and DNA structural elements also exist, but they might be less universal and might be tailored for specific positioning purposes that remain to be elucidated18.
What do the periodic AA, TT and GC patterns tell us? The 10 bp periodical presence of certain dinucleotides probably provides a rotational setting of the DNA on the histone surface because AA or TT dinucleotides tend to expand the major groove of DNA, whereas GC dinucleotides tend to contract the major groove. These alterations of the major groove might facilitate DNA wrapping around the histone core when the dinucleotides are placed in phase with the helical twist of DNA. In addition, other sequence combinations could create subtle bends in the DNA or alter the flexibility of DNA to contribute to the rotational setting of nucleosomal DNA18,48. Owing to rotational phasing, translational repositioning of a resident nucleosome into an adjacent linker region or NFR could obscure a DNA regulatory element in the linker without affecting the accessibility of another regulatory site that is already rotationally exposed on the surface of the nucleosome (FIG. 3b).
A key observation which showed that rotational phasing does not necessarily establish translational phasing was the inability of a 10 bp repeating pattern of AA and TT dinucleotides to predict the genomic locations of nucleosomes20. Instead, the nucleosome positions were more accurately predicted when the search pattern was enriched with AA dinucleotides towards the 5′ end and TT dinucleotides towards the 3′ end. Thus, partitioning of AA and TT dinucleotides towards the 5′ and 3′ ends, respectively, helps define translational positioning, whereas periodic AA and TT dinucleotides help define rotation positioning.
Despite the statistical enrichment of AA, TT and GC patterns associated with nucleosomes, the presence of these dinucleotide patterns in individual nucleosomes only occurs modestly above a random distribution and is largely limited to the −1 and +1 nucleosomes20,46. Thus, sequence-directed positioning might be subtle or diffuse, meaning that a small number of sequence determinants could be spread throughout the 147 bp nucleosomal DNA. Positioning is also likely to involve a combination of these favourable positioning sequences plus linker-enriched unfavourable sequences. It might be advantageous to have a mixture of favourable and unfavourable sequences, which results in only marginally stable nucleosome positions. An optimum mixture might strike an important balance between a state that can be disrupted to allow transcription and replication and a stable state that prevents inappropriate access to DNA. Indeed, the entire genome can be thought of as a continuous thermodynamic landscape of nucleosome occupancy, in which NFRs represent the thermodynamically least favourable regions and the +1 or −1 nucleosome positions represent the thermodynamically most favourable regions.
Predicting nucleosome positions
Many studies have attempted to computationally predict in vivo nucleosome locations de novo in yeast, flies and humans based on properties of the underlying DNA sequence17,42,44,46,48,49,53,54, and more sophisticated strategies are now emerging. Such predictions have been successful from a statistical perspective (that is, better than random guessing), but are limited compared with the experimental determination of nucleosome positions. Two studies used a support vector machine classifier that incorporated an experimental data set of nucleosome positions to identify characteristics that could discriminate between nucleosome-forming and nucleosome-avoiding DNA sequences (AT versus GC sequences)44,49. Another study used a combination of favourable short distance dinucleotide periodicities and short unfavourable sequence patterns to provide a probalistic model of nucleosome positions53. A third, and possibly the most accurate method, involved the use of wavelet transformation sequence periodicities that were spread throughout a training set of nucleosomal DNA sequences, which were combined with nucleosomal and linker sequence differences to create discriminatory signatures that were then used to make de novo predictions of nucleosome positions by hidden Markov modelling54.
It seems unlikely that a simple sequence-based algorithm will ever accurately predict all nucleosome locations. Factors other than the surrounding DNA sequence might contribute to nucleosome positioning in vivo. For example, nucleosome remodelling complexes, such as Isw2 in S. cerevisiae, override the sequence preferences of nucleosomes, causing nucleosomes to encroach into the 5′ and 3′ NFRs, thereby suppressing cryptic transcription that arises from the NFRs55,56. In addition, as a mechanism of gene repression, Isw2 uses the energy from ATP hydrolysis to position nucleosomes onto promoter regions that are intrinsically designed to repel nucleosomes56. Such nucleosomes are said to be ‘spring-loaded’, because removal of Isw2 would quickly result in intrinsic nucleosome eviction or repositioning of the nucleosome away from the unfavourable sequence.
Structure and function of NFRs
Both DNA sequence and protein factors are important for establishing NFRs. It is striking that regions of the genome that possess the strongest nucleosome positioning sequences (at the +1 nucleosome) are adjacent to regions that have the strongest anti-positioning sequences (5′ NFRs). An important factor in the establishment of a 5′ NFR might be the presence of poly(dA:dT) tracts15,20,53,57–59. Nucleosomes tend to be excluded from these tracts owing to the rigidity imparted to the DNA by the bifurcating hydrogen bonds present between adenosine bases on one strand (at position n) and thymines located on the other strand at positions n and n + 1 (REFS 59–61). In addition, specific DNA-binding proteins, such as the Myb-related protein Reb1 in yeast, might be important in positioning nucleosomes to create NFR boundaries62.
NFRs and transcription
The discovery of NFRs changed the way we think about how the transcription machinery assembles at promoters. We expected that promoter regions would be occluded by nucleosomes except when they were activated. This is still largely true for many genes that are repressed in specific tissues. However, the discovery of NFRs demonstrated that open promoter states are stable and common, even at genes that are transcribed so infrequently (<1% of the maximum level) that they are essentially turned off 17–19,23,63. Thus, although a NFR is permissive for transcription, it is not sufficient to activate genes. NFRs might allow low basal levels of leaky transcription, which could be interpreted as meaningless biological noise, particularly if the transcripts are rapidly degraded64–66. However, low levels of genic transcription might have a general housekeeping function whereby gene products are constitutively produced at low levels.
5′ NFRs are likely to be sites for the assembly of the transcription machinery, whereas 3′ NFRs are likely to be sites for the disassembly of the transcription machinery, although in compact genomes (for example, yeast, flies and worms), the 3′ NFR of one gene could be the 5′ NFR of the next downstream gene. It is currently unclear whether the open architecture of the 5′ NFR is necessary for the initial ‘pioneering’ polymerase or whether transcription itself establishes the NFR from the closed state, although at heat-shock genes in D. melanogaster, some domain-wide chromatin reorganization occurs after heat-shock treatment but before Pol II traverses the gene67. Given the reasonable expectation that Pol II and nucleosomal histones cannot simultaneously occupy the same DNA sequence, any nucleosome that is present in the promoter region is likely to be evicted or substantially remodelled before Pol II binding occurs68. When transcription is initiated and Pol II has cleared the promoter, the resident chromatin might not return to its original closed state (at least, not immediately) but might maintain an open state in which the composition of the nucleosomes is better suited for eviction during multiple rounds of transcription. This scenario is still hypothetical and remains fertile ground for experimentation.
Transcription start site selection by nucleosomes?
Because many PIC components, such as the SAGA complex and TFIID, have nucleosome-binding subunits, positioned nucleosomes might define the location of the TSS by positioning the PIC. The conventional view is that most genes contain a predominant TSS, the location of which is defined by core promoter elements69. In PIC assembly, general transcription factors, such as TATA-binding protein (TBP) or TFIID, bind to core promoter elements and position other initiation factors, such as TFIIB and TFIIF, which then direct Pol II to initiate transcription at the initiator element (INR element) (the consensus sequence is TCAKTY in flies and YYANWYY in humans)70,71. The problem with this view is that, despite extensive bioinformatic searches, most promoters seem to lack core promoter elements, including a TATA box, the TFIIB recognition element (BRE), INR, downstream promoter element (DPE) or motif ten element (MTE). In the absence of core promoter elements, how does the transcription machinery establish the location of the TSS? The answer is not known. Below, we speculate that positioned nucleosomes might determine the location of the TSS.
For about 80% of the 5,700 genes in S. cerevisiae, there is one TSS72. Remarkably, this specificity is achieved without a well-defined initiator element. The minimal consensus TSS in S. cerevisiae is YR (a C or T followed by an initiating A or G, although other nearby sequences might influence start site selection)73,74. YR is predicted to occur once every 4 bp in the genome, and thus lacks selectivity. However, TSSs are tightly distributed ~10–15 bp inside of the upstream border of the +1 nucleosome19 (FIG. 2). Given this tight linkage, it is difficult to envision how positioning of the TSS and the +1 nucleosome could have arisen independently at thousands of genes in yeast, yet maintained a fixed distance from each other.
How might the TSS be tightly linked to the position of the +1 nucleosome? First, during transcriptional activation, which is promoted by sequence-specific transcriptional activators, the −1 and +1 nucleosomes are acetylated and methylated (FIG. 5). The acetylation marks are recognized by bromodomain modules, which are found in many chromatin regulatory complexes, including the SAGA histone acetyltransferase complex75 (step 1 in FIG. 5) and TFIID (which is contained in the TFIID-interacting protein bromodomain-containing factor 1 (Bdf1) in yeast)76,77. In mammals, TFIID also binds H3K4me3 (histone H3 methylated at lysine 4), which is a mark of active transcription78. SAGA and TFIID then deliver TBP to promoters79,80 (step 2 in FIG. 5). Therefore, in principle, TBP positioning at promoters could be directed in part by SAGA and/or TFIID bound to nucleosomes, without the need for a positioning element.
Figure 5. Mechanistic differences between transcription initiation in budding yeast and metazoans.

A current model of how nucleosomes might direct start site selection in yeast, compared with metazoans is shown. Each step is also described in the main text. In step 1, the acetylation marks are recognized by bromodomain modules, which are found in many chromatin regulatory complexes, including the SAGA histone acetyltransferase complex and TFIID. In step 2, SAGA and TFIID then deliver TATA binding protein (TBP) to promoters. In step 3, TBP binds TFIIB and places it immediately downstream towards the transcription start site (TSS). In step 4, TFIIB positions RNA polymerase II (Pol II) at the promoter. The diagram for metazoans is a simplified version of that shown for yeast, in which the relationship between Pol II and the initiator (INR) is emphasized. The dashed arrows in both panels indicate sliding of Pol II before transcription initiation. Acetylation marks are indicated by green stars. The green colouring represents H2A.Z enrichment in the nucleosome array.
TBP binds TFIIB and places it immediately downstream towards the TSS81 (step 3 in FIG. 5) and TFIIB positions Pol II at the promoter82,83 (step 4 in FIG. 5). There is experimental evidence that TFIIB controls TSS selection; for example, the TSS location can be shifted by mutations in TFIIB84 or by replacement of TFIIB and Pol II with the same proteins from an evolutionarily diverged eukaryote that normally has a shifted TSS74,85,86. None of these steps invokes a need for core promoter elements. A similar scenario occurs during transcription of eukaryotic tRNA genes by Pol III. The protein complex TFIIIC binds to specific DNA sequences that are internal to the tRNA genes and positions the TBP-containing TFIIIB complex at a precise distance from the TSS without an underlying positioning element87, and TFIIIB then positions Pol III at the TSS.
Core promoter elements might have been adopted later in evolution. In metazoans, such elements might focus the TSS69. At least in vertebrates, genes that lack core promoter elements tend to have many TSSs dispersed over a distance of 50–100 bp69. It will be interesting to learn whether such promoters also have a dispersed (fuzzy) nucleosome architecture, which might be expected if nucleosome positions define at least some of the TSSs.
Evolutionary shifts in the TSS location
Compared with S. cerevisiae, metazoans have a genome-wide shift in the location of the TSS with respect to the position of the +1 nucleosome (FIG. 5). The predominant metazoan TSS resides in the NFR, ~60 bp upstream of the +1 nucleosome border16,20,24, whereas S. cerevisiae initiates transcription just inside the +1 nucleosome border. Part of this downstream shift in the TSS location in S. cerevisiae might be due to the initiation of metazoan transcription 30 bp downstream of the site at which TBP is bound70, whereas S. cerevisiae initiates transcription ~60 bp downstream of TBP. Therefore TBP might reside at the same distance from the +1 nucleosome in both eukaryotic branches. Although this hypothesis remains to be tested, if it is true, this would suggest that the distance between the +1 nucleosome and the PIC is a fundamental constant in eukaryotes and that the species-specific differences in TSS location are due to species-specific differences in TFIIB and Pol II74,85,86.
One model for how TFIIB and Pol II select a TSS is that after they are recruited to the site where TBP binds, TFIIB directs Pol II to scan downstream in a manner that does not require transcription74,88 (step 4, dashed arrow in FIG. 5). Any mechanism that causes Pol II to dwell at a particular site might increase the probability of initiation at that site. Such a mechanism might be based in part on the nature of the TFIIB–Pol II interactions in combination with core promoter sequences and/or nucleosome positions, all of which could affect Pol II scanning efficiency. In metazoans, increased dwelling might be caused by core promoter elements (INR, DPE and MTE), whereas in S. cerevisiae, the +1 nucleosome might provide the predominant impediment because core promoter elements beyond TATA boxes might not exist. Such a mechanism remains highly speculative until tested.
What are the implications of species-specific shifts in the TSS? The location of the TSS relative to the +1 nucleosome in S. cerevisiae compared with its location in metazoans indicates that yeast have to displace or remodel the +1 nucleosome before initiation of transcription, whereas metazoans only need to contend with the +1 nucleosome after initiation (FIG. 5). Indeed, a large fraction of metazoan genes have an initiated, but paused, Pol II immediately upstream of, and in contact with, the +1 nucleosome17,89–91.
Control of DNA access
We have discussed how genome-wide patterns of nucleosome positioning might influence transcription. In this final section, we turn briefly to the general question of how nucleosome disruption or displacement might allow nucleic acid polymerases to translocate along the underlying DNA or to allow transcription factor binding. There are a number of mechanisms by which the effect of a positioned nucleosome can be modulated to regulate DNA accessibility and therefore gene expression.
DNA accessibility without catalysis
Widom and colleagues have proposed a ‘site exposure’ model whereby thermal fluctuation of DNA on the nucleosome surface transiently exposes DNA-binding sites for transcriptional regulators92,93. Site exposure through thermal fluctuation posits that DNA unwrapping originates from the DNA entry and exit points of the nucleosome, and becomes energetically less favourable towards the midpoint of the nucleosome59,94. Consistent with this model, DNA regulatory sites tend to reside near the entry and exit sites of nucleosomes19,55. Binding of one factor might stabilize a partially disassembled state, allowing other transcription factors to access cognate sites that were previously buried95. Alternatively, certain regulatory proteins might bind to the rotationally exposed major groove of DNA that rests on the nucleosome surface (FIGS 3b, 6a)19 or bind to sites located in the NFR, where they might be constitutively accessible15.
Figure 6. Mechanisms that allow DNA accessibility.

a | A stable nucleosome. b | A remodelled nucleosome. c | An evicted nucleosome. Three transcription factor binding sites are shown in red, green and blue, respectively. The red and blue sites become accessible only during remodelling, either by nucleosome sliding, as indicated by the arrows in a, or by chromatin remodelling complexes (for example, ISW2, SWR1 and SWI/SNF) that ‘extract’ DNA from the nucleosome surface, as shown in b. Owing to rotational phasing, the green site is always accessible in the various states. Nucleosome eviction (c) might be necessary to assemble a pre-initiation complex and to transcribe the underlying DNA. Anti-silencing function 1 (Asf1) and H2A.Z-specific chaperone (Chz1) are examples of histone chaperones. Ac, acetylation.
DNA accessibility and remodelling complexes
Access to DNA sites that are internal to a nucleosome might require catalysed remodelling (FIG. 6b). Regulated nucleosome dynamics are driven by ATP-dependent chromatin remodelling complexes (for example, SWI/SNF43,96–99) in many ways: DNA ‘breathing’ on the nucleosome surface, in which SWI/SNF transiently exposes DNA regulatory sites by creating DNA loops on the nucleosome surface; translational repositioning (nucleosome sliding), in which complexes containing Isw2 move nucleosomes laterally to expose or cover DNA regulatory sites; nucleosome removal and deposition by the RSC complex and histone chaperones, for example, FACT (facilitates chromatin transcription), Asf1 (anti-silencing function 1) and Chz1 (H2A.Z-specific chaperone 1); and replacement of histone subunits, such as the replacement of H2A with H2A.Z by the SWR1 remodelling complex100–102 and replacement of H3 with H3.3 by the CHD1 (chromodomain-helicase-DNA-binding 1) remodelling complex103. Nucleosome dynamics are important because they regulate DNA accessibility, which is key to proper gene regulation and transcription fidelity.
Nucleosome sliding might be an important way of regulating access to DNA sites that are near nucleosome borders. For example, the TATA box of the yeast PHO5 (repressible acid phosphatase 5) gene resides near the −1 nucleosome border, and movement of this nucleosome by as little as a few base pairs alters the accessibility of the TATA box, which ultimately alters the composition of the assembled transcription machinery104. In mammalian cells, induction of the interferon-β promoter by viruses involves nucleosome sliding, which is promoted by the combined action of SWI/SNF on nucleosomes and TBP binding to TATA105. Thus, nucleosome sliding, remodelling and/or eviction seem to be important mechanisms for promoting TSS access and gene activation.
Because the binding of sequence-specific transcription factors to their cognate sites might be largely controlled by the −1 nucleosome in S. cerevisiae, remodelling complexes that promote site access might be specifically targeted to such nucleosomes68 (FIG. 6b). Consistent with this suggestion, SWI/SNF promotes the binding of the Gal4 activator to nucleosomal DNA in vitro106,107 and can modulate Gal4 binding to sites near the −1 nucleosome to promote transcription in vivo108.
The activity of SWI/SNF (and related complexes) can be enhanced by histone acetylation109,110. For example, acetylation might reduce histone–DNA electrostatic interactions by neutralizing positively charged lysines111, which might disrupt higher-order, repressive chromatin structures112 and also provide acetyl-lysine binding sites for SWI/SNF and other bromodomain-containing complexes75. A detailed discussion of this subject is beyond the scope of this Review, but it is clear from a wide range of studies that histone acetylation is an important contributor to gene activation12,111,113–115.
Nucleosome eviction
In addition to shifting the contacts between DNA and histones, eviction of a nucleosome from a particular genomic location allows DNA-binding factors to access the DNA (FIG. 6c) and can therefore affect gene expression. Remodelling complexes remove nucleosomes, and this process is likely to be influenced by histone variants116. Nucleosome loss can occur as a specific response to environmental stresses or signals, leading to transcriptional reprogramming. For example, yeast genes that are activated in response to heat shock or changes in the cell cycle often lose nucleosomes in the promoter region23,117. Genes in which expression is turned down gain nucleosomes5,7,10,11,23,117,118.
Nucleosome ejection during gene activation has been studied for some time at model target genes in Saccharomyces spp.118–125 and other organisms126. For example, during erythropoiesis in humans, induction of the β-globin gene results in histone loss over DNA sequences in the locus control region (LCR), which contains enhancer elements that direct expression of the globin genes. Indeed, loss of nucleosomes allows the haematopoietic, cell-specific transcriptional activator NF-E2 (nuclear factor, erythroid-derived 2) to bind to the LCR127. In other cell types, nucleosomes create a closed chromatin state over the LCR, which precludes globin expression.
Conclusions and future directions
Our understanding of how nucleosome positions and dynamics regulate gene expression has increased dramatically in recent years. This is in large part due to the discovery and characterization of proteins that write, read and erase the many kinds of histone modifications, and the convergence of this knowledge on the involvement of chromatin remodelling factors, histone variants and histone chaperones in gene regulation. The ability to determine where in the genome proteins are bound and how much is bound using ChIP–chip and ChIP–Seq technologies has opened our eyes to the general mechanism or specificity of nucleosomal regulation. Because misregulation of nucleosomes can lead to developmental defects and cancer128–130, such as mixed-lineage leukaemia, when the ability to methylate histone H3 is disrupted, understanding the extent to which nucleosome organization and modification of this organization are altered throughout a genome in normal and disease states will be an important step towards chromatin-based therapy.
Some of the next steps towards increasing our understanding of global chromatin structure will be to identify the cellular components and mechanisms that determine the canonical positioning of nucleosomes. This might be best achieved through molecular and/or genetic techniques that remove candidate nucleosome organizing factors and determine whether canonical positions are altered. The observation that many proteins contain conserved domains that interact with histone modifications raises the question of whether such proteins bind to specific nucleosomes in the genome. Addressing this question might require the development of methods to cross-link proteins to nucleosomes in vivo and map such interactions across the genome using ChIP–Seq technology. The development of high-resolution genome-wide mapping technologies will allow us to answer many new questions regarding the function of genomic chromatin organization and its interplay with the transcription machinery.
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
UniProtKB:www.uniprot.org
FURTHER INFORMATION
B.F. Pugh’s laboratory homepage: http://www.bmb.psu.edu/faculty/pugh/pugh.html
Penn State Genome Cartography project: http://atlas.bx.psu.edu
UCSC Genome Bioinformatics http://genome.ucsc.edu
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Acknowledgments
We thank S. Tan for providing the image for FIG. 1a. Support from National Institutes of Health grant HG004160 is gratefully acknowledged.
Glossary
- Chromatin remodelling complex
An ATP-dependent enzyme that is catalysed by different types of ATPase to alter nucleosome structure. The net effect of all chromatin remodelling enzymes is to modify nucleosome position or to increase accessibility of nucleosomal DNA.
- Nucleosome-free region (NFR)
An ~140 bp region lacking nucleosomes that is found at the beginning and end of genes. Many regions might not be completely nucleosome free, but are depleted of nucleosomes compared with the surrounding region. Certain environmental conditions can cause nucleosomes to occupy an NFR; for example, when genes are repressed.
- ChIP–chip
A method for detecting the location of proteins throughout a genome using chromatin-immunoprecipitation followed by microarray analysis.
- ChIP–Seq
A method for detecting the location of proteins throughout a genome using chromatin-immunoprecipitation followed by high-throughput DNA sequencing.
- Phasing
The distribution of nucleosomes around a particular coordinate in a population of cells.
- Rotational setting
The local orientation of the DNA helix on the histone surface.
- Translational setting
The nucleosomal DNA midpoint position relative to a chromosomal locus.
- Linker DNA
A short length of DNA located between nucleosomes. Long linker DNA can be considered to be a nucleosome-free region (NFR) — the DNA length cut-off for the two classes is arbitrary. However, NFRs tend to be sites of RNA and DNA polymerase loading and unloading.
- Pre-initiation complex (PIC)
This assembly is found at the promoter and before the complex has initiated transcription. It includes the general transcription factors (TFIIA, TFIIB, TFIID, TFIIE, TFIIF and TFIIH), the mediator, the RNA polymerase II complex, and activator or co-activator proteins (including SAGA).
- Support vector machine classifier
A widely used method of classifying training data (for example, nucleosomal compared with non-nucleosomal genomic DNA), which can then be used to make predictions de novo.
- Hidden Markov modelling
A method of identifying unknown or hidden states (for example, nucleosome positions) from observable states (for example, measured nucleosome positions).
- Cryptic transcription
A low level of presumably unregulated transcription that originates from nucleosome-free regions. The transcripts are usually rapidly degraded.
- SAGA complex
A multisubunit multifunctional complex that delivers TATA-binding protein (TBP) to promoters (by Spt3 and Spt8 subunits), acetylates nucleosomes (by the Gcn5 subunit) and is associated with activities that remodel (by Chd1) and deubiquitylate (by Ubp8) nucleosomes.
- TFIID
A multisubunit general transcription factor composed of TATA-binding protein (TBP) and ~15 other subunits (TBP-associated factors).
- Core promoter element
A widely used DNA sequence element that helps position the transcription initiation complex, and is typically located within 60 bp of the transcription start site.
- General transcription factor
A protein that is widely considered to be required to set up a transcription initiation complex at all promoters (examples include TFIIA, TFIIB, TFIID, TFIIE, TFIIF and TFIIH).
- TATA-binding protein (TBP)
This protein is important for assembling the transcription initiation complex.
- Initiator element (INR element)
A DNA sequence that specifies the transcription start site (consensus abbreviations include: K = G or T; Y = C or T; W = A or T; N = G, A, T or C).
- Histone chaperone
A member of a class of proteins that help to deposit histones onto DNA, but are not components of nucleosomes.
References
- 1.Kornberg RD, Klug A. The nucleosome. Sci Am. 1981;244:52–64. doi: 10.1038/scientificamerican0281-52. [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 3.Kamakaka RT, Biggins S. Histone variants: deviants? Genes Dev. 2005;19:295–310. doi: 10.1101/gad.1272805. [DOI] [PubMed] [Google Scholar]
- 4.Sarma K, Reinberg D. Histone variants meet their match. Nature Rev Mol Cell Biol. 2005;6:139–149. doi: 10.1038/nrm1567. [DOI] [PubMed] [Google Scholar]
- 5.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nature Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 6.Sekinger EA, Moqtaderi Z, Struhl K. Intrinsic histone–DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell. 2005;18:735–748. doi: 10.1016/j.molcel.2005.05.003. An early study that showed, using in vitro reconstituted nucleosomes at specific loci, that NFRs and nucleosomes positioned nearby might be dictated largely by intrinsic DNA sequence preference rather than by trans-acting factors. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Global nucleosome occupancy in yeast. Genome Biol. 2004;5:R62. doi: 10.1186/gb-2004-5-9-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guillemette B, et al. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol. 2005;3:e384. doi: 10.1371/journal.pbio.0030384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwabish MA, Struhl K. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2004;24:10111–10117. doi: 10.1128/MCB.24.23.10111-10117.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zanton SJ, Pugh BF. Full and partial genome-wide assembly and disassembly of the yeast transcription machinery in response to heat shock. Genes Dev. 2006;20:2250–2265. doi: 10.1101/gad.1437506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, Roberts DN, Cairns BR. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell. 2005;123:219–231. doi: 10.1016/j.cell.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733. doi: 10.1016/j.cell.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Vogelauer M, Wu J, Suka N, Grunstein M. Global histone acetylation and deacetylation in yeast. Nature. 2000;408:495–498. doi: 10.1038/35044127. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein BE, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci USA. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan GC, et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. The first high-resolution genome-wide study to reveal a NFR and a canonical arrangement of nucleosomes, including the DNA sequences that contribute to this arrangement. [DOI] [PubMed] [Google Scholar]
- 16.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. One of the most extensive catalogues of the positions of post-translationally modified nucleosomes throughout the human genome. The study used ChIP–Seq and reports on patterns associated with each nucleosome modification. [DOI] [PubMed] [Google Scholar]
- 17.Mavrich TN, et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee W, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nature Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 19.Albert I, et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. This paper provides the first report of the use of ChIP–Seq to develop high-resolution maps of nucleosome positions, which allowed the rotational and translational context of DNA regulatory elements to be determined. [DOI] [PubMed] [Google Scholar]
- 20.Mavrich TN, et al. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18:1073–1083. doi: 10.1101/gr.078261.108. This paper provides evidence that sequence-based nucleosome positioning is largely restricted to promoter regions, and that adjacent positions are dictated largely by packing principles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mito Y, Henikoff JG, Henikoff S. Histone replacement marks the boundaries of cis-regulatory domains. Science. 2007;315:1408–1411. doi: 10.1126/science.1134004. [DOI] [PubMed] [Google Scholar]
- 22.Schones DE, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shivaswamy S, et al. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008;6:e65. doi: 10.1371/journal.pbio.0060065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valouev A, et al. A high-resolution, nucleosome position map of C elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–1063. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;90:145–155. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 26.Varga-Weisz PD, et al. Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature. 1997;388:598–602. doi: 10.1038/41587. [DOI] [PubMed] [Google Scholar]
- 27.Saha A, Wittmeyer J, Cairns BR. Mechanisms for nucleosome movement by ATP-dependent chromatin remodeling complexes. Results Probl Cell Differ. 2006;41:127–148. doi: 10.1007/400_005. [DOI] [PubMed] [Google Scholar]
- 28.Gangaraju VK, Bartholomew B. Mechanisms of ATP-dependent chromatin remodeling. Mutat Res. 2007;618:3–17. doi: 10.1016/j.mrfmmm.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kagalwala MN, Glaus BJ, Dang W, Zofall M, Bartholomew B. Topography of the ISW2–nucleosome complex: insights into nucleosome spacing and chromatin remodeling. EMBO J. 2004;23:2092–2104. doi: 10.1038/sj.emboj.7600220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira H, Owen-Hughes T. Lighting up nucleosome spacing. Nature Struct Mol Biol. 2006;13:1047–1049. doi: 10.1038/nsmb1206-1047. [DOI] [PubMed] [Google Scholar]
- 31.Rippe K, et al. DNA sequence- and conformation-directed positioning of nucleosomes by chromatin-remodeling complexes. Proc Natl Acad Sci USA. 2007;104:15635–15640. doi: 10.1073/pnas.0702430104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blank TA, Becker PB. Electrostatic mechanism of nucleosome spacing. J Mol Biol. 1995;252:305–313. doi: 10.1006/jmbi.1995.0498. [DOI] [PubMed] [Google Scholar]
- 33.Fan Y, et al. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol Cell Biol. 2003;23:4559–4572. doi: 10.1128/MCB.23.13.4559-4572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan Y, et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123:1199–1212. doi: 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 35.Malik HS, Henikoff S. Phylogenomics of the nucleosome. Nature Struct Biol. 2003;10:882–891. doi: 10.1038/nsb996. [DOI] [PubMed] [Google Scholar]
- 36.Cosgrove MS, Wolberger C. How does the histone code work? Biochem Cell Biol. 2005;83:468–476. doi: 10.1139/o05-137. [DOI] [PubMed] [Google Scholar]
- 37.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 39.Lieb JD, Clarke ND. Control of transcription through intragenic patterns of nucleosome composition. Cell. 2005;123:1187–1190. doi: 10.1016/j.cell.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 40.Kiyama R, Trifonov EN. What positions nucleosomes? A model. FEBS Lett. 2002;523:7–11. doi: 10.1016/s0014-5793(02)02937-x. [DOI] [PubMed] [Google Scholar]
- 41.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184:868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 42.Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nucleosome positions predicted through comparative genomics. Nature Genet. 2006;38:1210–1215. doi: 10.1038/ng1878. [DOI] [PubMed] [Google Scholar]
- 43.Rando OJ, Ahmad K. Rules and regulation in the primary structure of chromatin. Curr Opin Cell Biol. 2007;19:250–256. doi: 10.1016/j.ceb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Gupta S, et al. Predicting human nucleosome occupancy from primary sequence. PLoS Comput Biol. 2008;4:e1000134. doi: 10.1371/journal.pcbi.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satchwell SC, Drew HR, Travers AA. Sequence periodicities in chicken nucleosome core DNA. J Mol Biol. 1986;191:659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- 46.Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. Together with Reference 42, this study provides evidence that at least some genomic sequences favour nucleosome assembly, which can be used to approximately predict nucleosome positions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JP, Widom J. Improved alignment of nucleosome DNA sequences using a mixture model. Nucleic Acids Res. 2005;33:6743–6755. doi: 10.1093/nar/gki977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miele V, Vaillant C, d’Aubenton-Carafa Y, Thermes C, Grange T. DNA physical properties determine nucleosome occupancy from yeast to fly. Nucleic Acids Res. 2008;36:3746–3756. doi: 10.1093/nar/gkn262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peckham HE, et al. Nucleosome positioning signals in genomic DNA. Genome Res. 2007;17:1170–1177. doi: 10.1101/gr.6101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trifonov EN. Sequence-dependent deformational anisotropy of chromatin DNA. Nucleic Acids Res. 1980;8:4041–4053. doi: 10.1093/nar/8.17.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Widom J. Role of DNA sequence in nucleosome stability and dynamics. Q Rev Biophys. 2001;34:269–324. doi: 10.1017/s0033583501003699. [DOI] [PubMed] [Google Scholar]
- 52.Wang JP, et al. Preferentially quantized linker DNA lengths in Saccharomyces cerevisiae. PLoS Comput Biol. 2008;4:e1000175. doi: 10.1371/journal.pcbi.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Field Y, et al. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuan GC, Liu JS. Genomic sequence is highly predictive of local nucleosome depletion. PLoS Comput Biol. 2008;4:e13. doi: 10.1371/journal.pcbi.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450:1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 56.Whitehouse I, Tsukiyama T. Antagonistic forces that position nucleosomes in vivo. Nature Struct Mol Biol. 2006;13:633–640. doi: 10.1038/nsmb1111. [DOI] [PubMed] [Google Scholar]
- 57.Radwan A, Younis A, Luykx P, Khuri S. Prediction and analysis of nucleosome exclusion regions in the human genome. BMC Genomics. 2008;9:186. doi: 10.1186/1471-2164-9-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iyer V, Struhl K. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995;14:2570–2579. doi: 10.1002/j.1460-2075.1995.tb07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson JD, Widom J. Poly(dA–dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol Cell Biol. 2001;21:3830–3839. doi: 10.1128/MCB.21.11.3830-3839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nelson HC, Finch JT, Luisi BF, Klug A. The structure of an oligo(dA). oligo(dT) tract and its biological implications. Nature. 1987;330:221–226. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- 61.Struhl K. Naturally occurring poly(dA–dT) sequences are upstream promoter elements for constitutive transcription in yeast. Proc Natl Acad Sci USA. 1985;82:8419–8423. doi: 10.1073/pnas.82.24.8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raisner RM, et al. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005;123:233–248. doi: 10.1016/j.cell.2005.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nature Genet. 2005;37:1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 64.Arigo JT, Eyler DE, Carroll KL, Corden JL. Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol Cell. 2006;23:841–851. doi: 10.1016/j.molcel.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 65.Thiebaut M, Kisseleva-Romanova E, Rouge-maille M, Boulay J, Libri D. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the Nrd1–Nab3 pathway in genome surveillance. Mol Cell. 2006;23:853–864. doi: 10.1016/j.molcel.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 66.Thompson DM, Parker R. Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27:92–101. doi: 10.1128/MCB.01023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petesch SJ, Lis JT. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell. 2008;134:74–84. doi: 10.1016/j.cell.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venters BJ, Pugh BF. A canonical promoter organization of the transcription machinery and its regulators in the Saccharomyces genome. Genome Res. 2009 Jan 5; doi: 10.1101/gr.084970.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Juven-Gershon T, Hsu JY, Theisen JW, Kadonaga JT. The RNA polymerase II core promoter — the gateway to transcription. Curr Opin Cell Biol. 2008;20:253–259. doi: 10.1016/j.ceb.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 71.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 72.David L, et al. A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci USA. 2006;103:5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Z, Dietrich FS. Mapping of transcription start sites in Saccharomyces cerevisiae using 5′ SAGE. Nucleic Acids Res. 2005;33:2838–2851. doi: 10.1093/nar/gki583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuehner JN, Brow DA. Quantitative analysis of in vivo initiator selection by yeast RNA polymerase II supports a scanning model. J Biol Chem. 2006;281:14119–14128. doi: 10.1074/jbc.M601937200. [DOI] [PubMed] [Google Scholar]
- 75.Hassan AH, et al. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- 76.Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAF(II)250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- 77.Matangkasombut O, Buratowski RM, Swilling NW, Buratowski S. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev. 2000;14:951–962. [PMC free article] [PubMed] [Google Scholar]
- 78.Vermeulen M, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 79.Pugh BF, Tjian R. Mechanism of transcriptional activation by Sp1: evidence for coactivators. Cell. 1990;61:1187–1197. doi: 10.1016/0092-8674(90)90683-6. [DOI] [PubMed] [Google Scholar]
- 80.Sermwittayawong D, Tan S. SAGA binds TBP via its Spt8 subunit in competition with DNA: implications for TBP recruitment. EMBO J. 2006;25:3791–3800. doi: 10.1038/sj.emboj.7601265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nikolov DB, et al. Crystal structure of a TFIIB–TBP–TATA-element ternary complex. Nature. 1995;377:119–128. doi: 10.1038/377119a0. [DOI] [PubMed] [Google Scholar]
- 82.Hausner W, Wettach J, Hethke C, Thomm M. Two transcription factors related with the eucaryal transcription factors TATA-binding protein and transcription factor IIB direct promoter recognition by an archaeal RNA polymerase. J Biol Chem. 1996;271:30144–30148. doi: 10.1074/jbc.271.47.30144. [DOI] [PubMed] [Google Scholar]
- 83.Bushnell DA, Westover KD, Davis RE, Kornberg RD. Structural basis of transcription: an RNA polymerase II–TFIIB cocrystal at 4.5 Angstroms. Science. 2004;303:983–988. doi: 10.1126/science.1090838. [DOI] [PubMed] [Google Scholar]
- 84.Pardee TS, Bangur CS, Ponticelli AS. The N-terminal region of yeast TFIIB contains two adjacent functional domains involved in stable RNA polymerase II binding and transcription start site selection. J Biol Chem. 1998;273:17859–17864. doi: 10.1074/jbc.273.28.17859. [DOI] [PubMed] [Google Scholar]
- 85.Ghazy MA, Brodie SA, Ammerman ML, Ziegler LM, Ponticelli AS. Amino acid substitutions in yeast TFIIF confer upstream shifts in transcription initiation and altered interaction with RNA polymerase II. Mol Cell Biol. 2004;24:10975–10985. doi: 10.1128/MCB.24.24.10975-10985.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y, Flanagan PM, Tschochner H, Kornberg RD. RNA polymerase II initiation factor interactions and transcription start site selection. Science. 1994;263:805–807. doi: 10.1126/science.8303296. [DOI] [PubMed] [Google Scholar]
- 87.Geiduschek EP, Kassavetis GA. The RNA polymerase III transcription apparatus. J Mol Biol. 2001;310:1–26. doi: 10.1006/jmbi.2001.4732. [DOI] [PubMed] [Google Scholar]
- 88.Giardina C, Lis JT. DNA melting on yeast RNA polymerase II promoters. Science. 1993;261:759–762. doi: 10.1126/science.8342041. [DOI] [PubMed] [Google Scholar]
- 89.Muse GW, et al. RNA polymerase is poised for activation across the genome. Nature Genet. 2007;39:1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeitlinger J, et al. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nature Genet. 2007;39:1512–1516. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. This study showed that most genes in human embryonic stem cells seem to have a stalled RNA polymerase II at their 5′ ends (although such sites might actually have low occupancy levels) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 93.Polach KJ, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 94.Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 95.Adams CC, Workman JL. Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol Cell Biol. 1995;15:1405–1421. doi: 10.1128/mcb.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith CL, Peterson CL. ATP-dependent chromatin remodeling. Curr Top Dev Biol. 2005;65:115–148. doi: 10.1016/S0070-2153(04)65004-6. [DOI] [PubMed] [Google Scholar]
- 97.Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995;23:2715–2723. doi: 10.1093/nar/23.14.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cairns BR. Chromatin remodeling complexes: strength in diversity, precision through specialization. Curr Opin Genet Dev. 2005;15:185–190. doi: 10.1016/j.gde.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 99.Gutierrez JL, Chandy M, Carrozza MJ, Workman JL. Activation domains drive nucleosome eviction by SWI/SNF. EMBO J. 2007;26:730–740. doi: 10.1038/sj.emboj.7601524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kobor MS, et al. A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol. 2004;2:e131. doi: 10.1371/journal.pbio.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mizuguchi G, et al. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004;303:343–348. doi: 10.1126/science.1090701. [DOI] [PubMed] [Google Scholar]
- 102.Krogan NJ, et al. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell. 2003;12:1565–1576. doi: 10.1016/s1097-2765(03)00497-0. [DOI] [PubMed] [Google Scholar]
- 103.Konev AY, et al. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science. 2007;317:1087–1090. doi: 10.1126/science.1145339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinez-Campa C, et al. Precise nucleosome positioning and the TATA box dictate requirements for the histone H4 tail and the bromodomain factor Bdf1. Mol Cell. 2004;15:69–81. doi: 10.1016/j.molcel.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 105.Lomvardas S, Thanos D. Nucleosome sliding via TBP DNA binding in vivo. Cell. 2001;106:685–696. doi: 10.1016/s0092-8674(01)00490-1. [DOI] [PubMed] [Google Scholar]
- 106.Kwon H, Imbalzano AN, Khavari PA, Kingston RE, Green MR. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature. 1994;370:477–481. doi: 10.1038/370477a0. [DOI] [PubMed] [Google Scholar]
- 107.Cote J, Peterson CL, Workman JL. Perturbation of nucleosome core structure by the SWI/SNF complex persists after its detachment, enhancing subsequent transcription factor binding. Proc Natl Acad Sci USA. 1998;95:4947–4952. doi: 10.1073/pnas.95.9.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burns LG, Peterson CL. The yeast SWI–SNF complex facilitates binding of a transcriptional activator to nucleosomal sites in vivo. Mol Cell Biol. 1997;17:4811–4819. doi: 10.1128/mcb.17.8.4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suganuma T, et al. ATAC is a double histone acetyltransferase complex that stimulates nucleosome sliding. Nature Struct Mol Biol. 2008;15:364–372. doi: 10.1038/nsmb.1397. [DOI] [PubMed] [Google Scholar]
- 110.Hassan AH, Neely KE, Workman JL. Histone acetyltransferase complexes stabilize SWI/SNF binding to promoter nucleosomes. Cell. 2001;104:817–827. doi: 10.1016/s0092-8674(01)00279-3. [DOI] [PubMed] [Google Scholar]
- 111.Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci USA. 2005;102:5501–5506. doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang X, Hayes JJ. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol Cell Biol. 2008;28:227–236. doi: 10.1128/MCB.01245-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu CL, et al. Single nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328. doi: 10.1371/journal.pbio.0030328. This study showed that acetylation of histones at specific residues does not elicit a specific transcriptional response, indicating that acetylation might have cumulative effects rather than being encoded. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pokholok DK, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 115.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 116.Santisteban MS, Kalashnikova T, Smith MM. Histone H2A.Z regulates transcription and is partially redundant with nucleosome remodeling complexes. Cell. 2000;103:411–422. doi: 10.1016/s0092-8674(00)00133-1. [DOI] [PubMed] [Google Scholar]
- 117.Hogan GJ, Lee CK, Lieb JD. Cell cycle-specified fluctuation of nucleosome occupancy at gene promoters. PLoS Genet. 2006;2:e158. doi: 10.1371/journal.pgen.0020158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol Cell. 2004;14:667–673. doi: 10.1016/j.molcel.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 119.Korber P, Luckenbach T, Blaschke D, Horz W. Evidence for histone eviction in trans upon induction of the yeast PHO5 promoter. Mol Cell Biol. 2004;24:10965–10974. doi: 10.1128/MCB.24.24.10965-10974.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reinke H, Horz W. Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol Cell. 2003;11:1599–1607. doi: 10.1016/s1097-2765(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 121.Adkins MW, Howar SR, Tyler JK. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol Cell. 2004;14:657–666. doi: 10.1016/j.molcel.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 122.Moreira JM, Holmberg S. Transcriptional repression of the yeast CHA1 gene requires the chromatin-remodeling complex RSC. EMBO J. 1999;18:2836–2844. doi: 10.1093/emboj/18.10.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhao J, Herrera-Diaz J, Gross DS. Domain-wide displacement of histones by activated heat shock factor occurs independently of Swi/Snf and is not correlated with RNA polymerase II density. Mol Cell Biol. 2005;25:8985–8999. doi: 10.1128/MCB.25.20.8985-8999.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang H, Reese JC. Exposing the core promoter is sufficient to activate transcription and alter coactivator requirement at RNR3. Proc Natl Acad Sci USA. 2007;104:8833–8838. doi: 10.1073/pnas.0701666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Almer A, Rudolph H, Hinnen A, Horz W. Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J. 1986;5:2689–2696. doi: 10.1002/j.1460-2075.1986.tb04552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cartwright IL, Elgin SC. Nucleosomal instability and induction of new upstream protein–DNA associations accompany activation of four small heat shock protein genes in Drosophila melanogaster. Mol Cell Biol. 1986;6:779–791. doi: 10.1128/mcb.6.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Armstrong JA, Emerson BM. NF-E2 disrupts chromatin structure at human β-globin locus control region hypersensitive site 2 in vitro. Mol Cell Biol. 1996;16:5634–5644. doi: 10.1128/mcb.16.10.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bu P, Evrard YA, Lozano G, Dent SY. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol Cell Biol. 2007;27:3405–3416. doi: 10.1128/MCB.00066-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 130.Whittle CM, et al. The genomic distribution and function of histone variant HTZ-1 during C elegans embryogenesis. PLoS Genet. 2008;4:e1000187. doi: 10.1371/journal.pgen.1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]