

Abstract
Background
Baculoviruses are insect-associated viruses carrying large, circular double-stranded-DNA genomes with significant biotechnological applications such as biological pest control, recombinant protein production, gene delivery in mammals and as a model of DNA genome evolution. These pathogens infect insects from the orders Lepidoptera, Hymenoptera and Diptera, and have high species diversity which is expressed in their diverse biological properties including morphology, virulence or pathogenicity. Spodoptera frugiperda (Lepidoptera: Noctuidae), the fall armyworm, represents a significant pest for agriculture in America; it is a host for baculoviruses such as the Spodoptera frugiperda multiple nucleopolyhedrovirus (SfMNPV) (Colombia strain, genotype A) having been classified as a Group II alphabaculovirus making it a very attractive target for bioinsecticidal use.
Results
Genome analysis by pyrosequencing revealed that SfMNPV ColA has 145 ORFs, 2 of which were not present in the other sequenced genotypes of the virus (SfMNPV-NicB, SfMNPV-NicG, SfMNPV-19 and SfMNPV-3AP2). An in-depth bioinformatics study showed that ORF023 and ORF024 were acquired by a recent homologous recombination process between Spodoptera frugiperda and Spodoptera litura (the Oriental leafworm moth) nucleopolyhedroviruses. Auxiliary genes are numerous in the affected locus which has a homologous region (hr3), a repetitive sequence associated with genome replication which became lost in SfColA along with 1 ORF. Besides, the mRNAs associated with two acquired genes appeared in the virus’ life-cycle during the larval stage. Predictive studies concerning the theoretical proteins identified that ORF023 protein would be a phosphatase involved in DNA repair and that the ORF024 protein would be a membrane polypeptide associated with cell transport.
Conclusions
The SfColA genome was thus revealed to be a natural recombinant virus showing evidence of recent horizontal gene transfer between different baculovirus species occurring in nature. This feature could be the cause of its high insecticidal power and therefore SfColA becomes a great candidate for bioinsecticide formulations.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2218-5) contains supplementary material, which is available to authorized users.
Keywords: Baculovirus, Spodoptera frugiperda, Horizontal gene transfer, Homologous recombination
Background
Baculoviruses are double-stranded DNA viruses which infect insects from the orders Lepidoptera, Diptera and Hymenoptera. Significant baculovirus characteristics include the presence of two phenotypes during the cell cycle: budded viruses (BVs), and occlusion derived viruses (ODVs) which are embedded into protein crystals called occlusion bodies (OBs); they display very high diversity expressed in hundreds of species spread worldwide having different host ranges and/or virulence. Four genera have been recognized: Alphabaculovirus (lepidopteran-nucleopolyhedroviruses), Betabaculovirus (lepidopteran-granuloviruses), Gammabaculovirus (hymenopteran-nucleopolyhedroviruses), and Deltabaculovirus (dipteran-nucleopolyhedroviruses) [1–3]. The Spodoptera frugiperda multiple nucleopolyhedrovirus (SfMNPV) has been classified into the Baculoviridae family within the Alphabaculovirus [1, 4] and has been extensively studied for its potential regarding the biological control of fall armyworm, an important pest causing economic losses regarding several American crops, mainly corn fields [5]. Since the first reports about SfMNPV [6, 7], work has been focused on studying its genomic constitution and the inter- and intra-population diversity by comparing different isolates. Restriction profiles initially revealed genetic heterogeneity in field isolates, in addition to providing information for determining the first physical maps of the genome [8–10]. Sequencing of single genes and genomic variable regions [11–15] and subsequent analysis showed that SfMNPV phylogenetically clustered with other members of the Group II Alphabaculovirus clade [1].
Four complete SfMNPV genomes have been reported recently, one from a field isolate (SfMNPV-19) [16] and another three from genotypic variants recovered using in vitro plaque assay techniques in insects cells (SfMNPV-3AP2, SfMNPV-NicB, SfMNPV-NicG) [17–19]. The SfMNPV-NicB isolate was the predominant genotype having the largest genome and both SfMNPV-3AP2 and SfMNPV-NicG had deletions regarding the former. Phylogenetic analysis revealed that they were very closely related and also closely related to Spodoptera exigua MNPV, Agrotis spp. NPVs and Mamestra spp. NPVs.
SfMNPV inter-population diversity evaluated in Colombia by analyzing 38 isolates from three different geographical regions revealed that one isolate (SfMNPV-Col or SfCol) had the highest prevalence (92 %). SfCol had minimal genetic differences compared to the SfMNPV isolate from Nicaragua (SfMNPV-NicB or SfNicB) based on restriction profiles; however, it had large differences regarding virulence against S. frugiperda larvae from Colombia, SfCol being more potent than SfNicB for the local insect population [20]. Subsequent intra-population diversity studies have revealed 10 different genotypic variants within SfCol (SfColA to SfColJ), SfColA being the most prevalent (72 %) and having the largest genome, while the other variants had different sized deletions. SfColA was 4.4 times more potent than and as virulent as SfCol for local insect pests [21]. Such biological differences should correlate with genome organization; structural mutations (replacements, inversions, insertions or deletions) would presumably be how baculoviruses evolve in nature and improve their fitness, not forgetting the importance of single nucleotide mutations. According to previous genome evidence, natural recombination events are probably one of the most important processes involved in baculovirus genome plasticity [22]. DNA crossover may occur between two loci from one genome, between genotype variants of the same species, or between genomes from different virus species co-infecting the same host [23]. In any case, the resulting recombinant genomes may be affected by their prior gene content. Baculovirus diversity has been associated with the ubiquitous presence of transposons, which may collaborate in horizontal gene transfer and insertion/deletion (indel) mutations. Different kinds of transposable elements have been detected in baculovirus genomes from many species, sometimes affecting gene functions [24, 25]. Baculoviruses should thus be efficient vectors between animals and such ability would have an important impact on gene content and genome organization because they can provide the sequence homology required for crossover events [26].
Baculovirus genome variability has an undeniable effect on the virus’ life-cycle in the host and affects different parameters such as pathogenicity, virulence and OB’s production (yield) [27, 28]. Some genome regions are more prone to sequence variation than others; these would include loci containing homologous regions (hr) and Bro genes, both being the kind of sequences recognized as target sites for intragenomic recombination because they are usually found in more than one copy [28]. Moreover, most variability is concentrated in regions having auxiliary genes (encoding non-essential proteins) as they are more tolerant to mutations because sequence changes do not affect the production of essential factors needed to complete the viral cycle.
The SfColA isolate was molecularly characterized in the present work to provide extra evidence to explain biological activities and to further understand how baculoviruses evolve in nature, losing ancient sequences or gaining new regions and thereby altering virus fitness.
Methods
Virus isolate
The virus used here [SfMNPV ColA (SfColA)] had been previously isolated by plaque purification in the Sf9 cell line exposed to a natural SfMNPV isolated in Colombia (SfMNPV-Col or SFCol) [21]. SfColA was propagated in S. frugiperda fourth instar larvae reared in laboratory conditions (25 ± 1 °C, 75 ± 5 % relative humidity, 16 h light: 8 h dark photoperiod and a wheat germ-based semisynthetic diet) and maintained as OB suspension in sterile distilled water.
Sequencing, assembly and ORFeome determination
SfColA DNA was purified from OBs by alkaline lysis and cesium chloride gradient [29] and used for sequencing with the 454 Genome Sequencer (GS) FLX™ Standard (Roche) (Centro Nacional de Secuenciación Genómica, CNSG; Universidad de Antioquia, Medellín, Colombia). De novo assembly was performed using NewBler assembler (GS FLX Data Analysis Software) to define whole genome sequence. The reads were independently assembled five times without using a reference genome; in all the runs the resulting sequence was essentially the same. This assembly was then compared to the genomes from baculoviruses which infect Spodoptera spp. (SfMNPV-3AP2, SfMNPV-NicB, SfMNPV-NicG, SeMNPV and SpltNPV-II) and the genome organisation was conserved, thereby validating the previous result. The SfMNPV-ColA assembly correlated with experimental physical map data and the region containing differential genes was confirmed by Sanger sequencing. Open reading frames (ORFs) were identified using ARTEMIS [30]. ATG initiated ORFs having at least 150 nt (50 aa) showing minimal overlap with other putative encoding sequences were selected for further analysis. BlastN, BlastP, tBlastN, tBlastX and PSI-Blast were used for homology searches [31], initially against other SfMNPV genomes and then against other baculovirus species. Homologous genes’ identity and similarity values were obtained by global alignment using ClustalX [32, 33] with default parameters. The baculovirus genomic sequences used in the bioinformatics studies are listed in Table 1.
Table 1.
Baculoviruses used in bioinformatics studies including SfMNPV ColA
| Baculovirus | Acc. number | Abbreviation |
|---|---|---|
| Antheraea pernyi MNPV Isolate L2 | EF207986 | AnpeMNPV |
| Antheraea pernyi NPV Isolate Z | NC_008035 | AnpeNPV |
| Anticarsia gemmatalis MNPV | NC_008520 | AgMNPV |
| Autographa californica MNPV Clone C6 | NC_001623 | AcMNPV |
| Bombyx mandarina NPV S1 | NC_012672 | BomaNPV S1 |
| Bombyx mandarina NPV S2 | JQ071499 | BomaNPV S2 |
| Bombyx mori NPV Isolate T3 | NC_001962 | BmNPV |
| Choristoneura fumiferana MNPV | NC_004778 | CfMNPV |
| Choristoneura fumiferana Defective MNPV | NC_005137 | CfDEFMNPV |
| Choristoneura murinana NPV Strain Darmstadt | NC_023177 | ChmuNPV |
| Choristoneura occidentalis NPV Isolate BC1 | NC_021925 | ChocNPV |
| Choristoneura rosaceana NPV Isolate NB1 | NC_021924 | ChroNPV |
| Epiphyas postvittana NPV | NC_003083 | EppoNPV |
| Hyphantria cunea NPV | NC_007767 | HycuNPV |
| Maruca vitrata NPV | NC_008725 | MaviNPV |
| Orgyia pseudotsugata MNPV | NC_001875 | OpMNPV |
| Philosamia cynthia ricini NPV | JX404026 | PhcyNPV |
| Plutella xylostella MNPV Isolate CL3 | NC_008349 | PlxyMNPV |
| Rachiplusia ou MNPV | NC_004323 | RoMNPV |
| Thysanoplusia orichalcea NPV P2 | NC_019945 | ThorNPV P2 |
| Adoxophyes honmai NPV | NC_004690 | AdhoNPV |
| Adoxophyes orana NPV | NC_011423 | AdorNPV |
| Agrotis ipsilon MNPV | NC_011345 | AgipMNPV |
| Agrotis segetum NPV | NC_007921 | AgseNPV |
| Apocheima cinerarium NPV | NC_018504 | ApciNPV |
| Buzura suppressaria NPV Isolate Hubei | NC_023442 | BusuNPV |
| Chrysodeixis chalcites NPV | NC_007151 | ChchNPV |
| Clanis bilineata NPV Isolate DZ1 | NC_008293 | ClbiNPV |
| Ectropis obliqua NPV Strain A1 | NC_008586 | EcobNPV |
| Euproctis pseudoconspersa NPV | NC_012639 | EupsNPV |
| Helicoverpa armigera MNPV | NC_011615 | HearMNPV |
| Helicoverpa armigera NPV Isolate Australia | JN584482 | HearNPV Aus |
| Helicoverpa armigera NPV Strain C1 | NC_003094 | HearNPV C1 |
| Helicoverpa armigera NPV Strain G4 | NC_002654 | HearNPV G4 |
| Helicoverpa armigera SNPV Strain NNg1 | NC_011354 | HearSNPV |
| Helicoverpa zea NPV | NC_003349 | HzSNPV |
| Hemileuca sp. NPV | NC_021923 | HespNPV |
| Leucania separata NPV Strain AH1 | NC_008348 | LeseNPV |
| Lymantria dispar MNPV | NC_001973 | LdMNPV |
| Lymantria xylina MNPV | NC_013953 | LyxyMNPV |
| Mamestra brassicae MNPV Isolate Chb1 | JX138237 | MabrMNPV Chb1 |
| Mamestra brassicae MNPV Isolate K1 | NC_023681 | MabrMNPV K1 |
| Mamestra configurata NPV Strain 90-2 | NC_003529 | MacoNPV 90 2 |
| Mamestra configurata NPV Strain A90-4 | AF539999 | MacoNPV A90 4 |
| Mamestra configurata NPV Strain B | NC_004117 | MacoNPV B |
| Orgyia leucostigma NPV IsolateCFS77 | NC_010276 | OrleNPV |
| Spodoptera exigua MNPV | NC_002169 | SeMNPV |
| Spodoptera frugiperda MNPV Isolate 3AP2 | NC_009011 | SfMNPV 3AP2 |
| Spodoptera frugiperda MNPV Isolate Nicaraguan B | HM595733 | SfMNPV NicB |
| Spodoptera frugiperda MNPV Isolate Nicaraguan DefG | JF899325 | SfMNPV NicG |
| Spodoptera frugiperda MNPV Strain 19 | EU258200 | SfMNPV 19 |
| Spodoptera frugiperda MNPV ColA | KF891883 | SfMNPV ColA |
| Spodoptera littoralis NPV Isolate AN1956 | JX454574 | SpliMNPV AN1956 |
| Spodoptera litura II MNPV | NC_011616 | SpltNPV-II |
| Spodoptera litura MNPV Strain G2 | NC_003102 | SpltMNPV G2 |
| Trichoplusia ni SNPV | NC_007383 | TnSNPV |
| Adoxophyes orana GV | NC_005038 | AdorGV |
| Agrotis segetum GV | NC_005839 | AgseGV |
| Choristoneura occidentalis GV | NC_008168 | ChocGV |
| Clostera anastomosis GV Strain Henan | NC_022646 | CaLGV |
| Cryptophlebia leucotreta GV | NC_005068 | CrleGV |
| Cydia pomonella GV | NC_002816 | CpGV |
| Epinotia aporema GV | NC_018875 | EpapGV |
| Helicoverpa armigera GV | NC_010240 | HearGV |
| Phthorimaea operculella GV | NC_004062 | PhopGV |
| Pseudaletia unipuncta GV Strain Hawaiin | NC_013772 | PsunGV |
| Pieris rapae GV | NC_013797 | PiraGV |
| Plutella xylostella GV | NC_002593 | PlxyGV |
| Spodoptera frugiperda GV | KM371112 | SpfrGV |
| Spodoptera litura GV Strain K1 | NC_009503 | SpliGV |
| Xestia c-nigrum GV | NC_002331 | XecnGV |
| Neodiprion abietis NPV | DQ317692 | NeabNPV |
| Neodiprion lecontei NPV | NC_005906 | NeleNPV |
| Neodiprion sertifer NPV | NC_005905 | NeseNPV |
| Culex nigripalpus NPV | NC_003084 | CuniNPV |
For detecting homologous regions (hrs) in the SfColA genome, the SfMNPV NicB hr-1 sequence was used as computational probe. All individual palindromes (44 residue lengths) were then recovered from SfColA, SfMNPV Nic, SfMNPV 3AP2 and SfMNPV 19 genomes and multiple alignments were performed using the ClustalX algorithm with default parameters. Sequence logos were constructed using the WebLogo server (http://weblogo.berkeley.edu/) [34]. The secondary DNA structure prediction was obtained using the Mfold server of Michael Zuker website [35] and using RNADraw program [36]. A/T-content was profiled using a sliding windows strategy (window = 500 nucleotides, displacement = 50 nucleotides) [37]. Relationships between each point and the A/T-content average were obtained and peaks of 1.12 or above were considered as A/T-rich regions.
Colinearity genome studies and phylogenetic analysis
Nucleotide synteny blocks were searched using BlastN routine with the following parameters: expected value = 0.1 (−e 0.1), penalty for a nucleotide mismatch = −2 (−q −2), reward for a nucleotide match = 1 (−r 1) and filter query sequence = false (−F F). Output files for each genome comparison were drawn using the GenomeComp v1.2 software [38]. A color code was used for showing different ranges of nucleotide identity. Baculoviridae phylogeny was inferred using the 37 core genes in silico translated from 75 baculovirus genomes (Table 1) which were independently aligned using ClustalX program with the following parameters: Pairwise alignment (Gap Open Penalty = 10, Gap Extension Penalty = 0.1, protein weight matrix: Blosum 30); Multiple alignment (Gap Open Penalty = 10, Gap Extension Penalty = 0.05, protein weight matrix: Blosum series). A concatemer was then generated by adding complete individual alignments and phylogeny was inferred using MEGA 5 software [39] with the following parameters: UPGMA; Bootstrap with 1000 replicates; Gap/Missing data = pairwise deletion; Model = Amino (Dayhoff Matrix); patterns among sites = Same (Homogeneous); rates among sites = Different (Gamma Distributed); gamma parameter = 0.9839. Besides, a phylogeny inference was similarly performed but using only SfMNPV genomes and the most related baculoviruses (SeMNPV, SpltNPV-II, SpltMNPV-G2, SpliNPV AN1956). The concatemer of individual alignments derived from 100 genes translated in silico which were shared among baculoviruses considered for the study (indicated in Additional file 1: Table S1).
Interspecies horizontal gene transfer studies
The partial SfColA genome sequence, from chitinase ATG to the gp37 stop codon genes, was compared to corresponding SfMNPV-B, SfMNPV 3AP2, SfMNPV 19, SeMNPV and SpltNPV-II regions to detect potential recombination events by running alternative methods. In the first one [37], ClustalX (default parameters) was used for aligning sequence pairs, always involving the putative recombinant candidate (SfColA) and one of the other sequences. Relative similarities were calculated using the ClustalX consensus symbol (* and blank space) as the input sequence, in an overlapping windows-based strategy. Arbitrary values of +1 for identical (*) and −1 for non-identical residues (blank spaces) were set for obtaining similarity profiles. The sum of assigned values for each residue in each window (35 nucleotides) was divided by the window width and allotted to the central position to generate the plots. Profiles were drawn and analyzed for detecting crossover points. Different window lengths were scanned to find good relationships between graph complexity and crosspoint detection sensitivity. Bootscan analysis (Simplot program, version 3.5.1) [40, 41] was performed using the following parameters: Window = 500 residues; Step = 50 residues; Gaps strip = on; Replicates = 100; Model = Kimura 2-parameters; Transition and transversion ratio = 2.0; Phylogenetic method = Neighbor Joining). A G/C-content study was made for the same SfColA, SfNicB and SpltNPV-II genome region. G/C-contents were studied using a sliding window strategy (window = 65 nucleotides, displacement = 1 nucleotide). The profiles were adjusted using in-house software. For this a multiple alignment with the three nucleotide sequences was used as template and the adjustment of numerical profiles consisted in the introduction of blank positions interrupting the curve where there were indel events. SigmaPlot v9 was used for all studies where final numerical profiles were represented and the putative crossover breakpoints were estimated. Multiple alignment of homologous proteins was done to estimate Kimura 2-parameter distances [42] using MEGA 5 software with the following parameters: Scope = Pairs of taxa; Estimate Variance, Variance Estimation Method = Bootstrap method; N° of Bootstrap Replications = 1000; Substitution model, Substitution type = Nucleotide, Model/Method = Kimura 2-parameter model, Substitutions to include = d: Transitions + Transversions; Rates and Patterns, Rates among Sites = Gamma parameter, Gamma Parameter = 0.9839, Pattern among lineages = Same (Homogeneous); Data Subset to Use, Gaps/Missing Data Treatment = Pairwise deletion, Select Codon Positions = All + Noncoding Sites.
SfColA ORF023 and ORF024 transcription studies
Reverse transcription-PCR assays were performed to determine SfColA ORF023 and ORF024 transcription activities. Forty five S. frugiperda second-instar larvae were kept fasting for 8 to 12 h at 26 °C and then allowed to drink from an aqueous suspension containing 10 % (wt/vol) sucrose, 0.001 % (wt/vol) Fluorella blue, and 1x107 OBs/mL. Larvae that ingested the suspension within 10 min were then transferred to individual plastic cups with semisynthetic diet. Total RNA was extracted from two infected larvae at 0, 2, 4, 6, 12, 24, 48, 72, 96, 120 and 144 hpi using TRIzol reagent (Invitrogen), as described in the manufacturers’ protocol. RNA samples were then treated with RNase free DNase (Promega) prior to ensuring the absence of contaminant DNA. First strand cDNA synthesis was done using reverse transcriptase (Promega) and oligo-dT primer. The resulting cDNAs were amplified by PCR with specific primers: Sf23.1 (5′ GCTTGTGCGTTGTCGTTGAT 3′) and Sf23.2 (5′ TTGTAGTCGACTCGGTCCCA 3′) for ORF023; Sf24.1 (5′ TCGTCGGCATCATACTGCTC 3′) and Sf24.2 (5′ CACGTTCGCATGGTTTTCGT 3′) for ORF024; Sfpolh.1 (5′ TTGCGACCCTGACTACGTTC 3′) and Sfpolh.2 (5′ ACGAGCGACAGTTCAAGGAG 3′) for the very late transcribed polyhedrin gene (polh); Sfie-0.1 (5′ CATTTGCCAAGAGAGCAGCG 3′) and Sfie-0.2 (5′ TTTAGCGGCAGTGGGAGTTT 3′) for the early transcribed ie-o gene. The amplification products were resolved in 1 % agarose gels and visualized with ethidium bromide and UV exposure.
Characterization of SfColA ORF023 and ORF024 proteins
Different bioinformatics tools were used for determining the nature of SfColA ORF023 and ORF024 proteins. Hydrophobicity profiles were constructed using a sliding windows strategy (window = 21 amino acids; sliding 1 residue each time). Several hydrophobicity scales were assayed [43–47]. The presence of signal peptides was assessed by using SignalP (http://www.cbs.dtu.dk/services/SignalP/; [48]). Putative functions were predicted using the HHpred server (http://toolkit.lmb.uni-muenchen.de/hhpred; [49]). Secondary and tertiary structures were predicted using the LOcal MEta-Threading-Server (LOMETS; http://zhanglab.ccmb.med.umich.edu/LOMETS; [50]) and the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER; [51]). SfColA ORF023 secondary and tertiary structures were also predicted using the QUARK (http://zhanglab.ccmb.med.umich.edu/QUARK; [52]) ab initio prediction server. Post-translational modifications were predicted by the INTERPROSCAN tool (http://www.ebi.ac.uk/interpro/; [53]).
Results and discussion
The SfColA genome and gene content
Five genomes have been sequenced to date from baculoviruses isolated from Spodoptera frugiperda: 4 alphabaculoviruses (SfMNPV 3AP2, SfMNPV NicB, SfMNPV NicG, SfMNPV 19) and 1 betabaculovirus (SpfrGV) (Table 1). The aforementioned polyhedroviruses were sequenced using a molecular cloning strategy followed by an automated Sanger’s method; only the granulovirus involved using next generation-sequencing (NGS) [37]. The Colombian isolate’s (SfCol) SfColA genotypic variant was molecularly characterized in view of its interesting biological properties to complete the study of the baculovirus group naturally infecting the armyworm and to provide extra information about baculovirus evolution. This genome (GenBank: KF891883) consisted of 134,239 bp, and was covered 64 times by 454 sequencing, this being the first Spodoptera frugiperda alphabaculovirus studied by the NGS approach. Its size was revealed to be the largest regarding previously reported genomes where SfMNPV-NicB (132,954 bp) was the previous head of the ranking. Group II alphabaculoviruses have a broad range of A + T content [42.53 % in Lymantria dispar MNPV [54] to 66.64 % in Apocheima cinerarium NPV (unpublished data)]. SfColA had 59.66 %, thereby agreeing with data published for the other Spodoptera frugiperda nucleopolyhedrovirus [SfNicB 59.72 %, SfMNPV-19 (Sf19) 59.74 %, and SfMNPV-3AP2 (Sf3AP2) 59.75 %].
Analysis of the SfColA genome led to detection of 145 putative open reading frames (ORFs) considering sequences encoding polypeptides having at least 50 amino acids, starting in an ATG codon and having minimal overlap with the closest ones. Thus the ORFeome would cover 92.5 % of the whole nucleotide sequence and each ORF was numbered, starting at the polyhedrin gene (ORF001) and continuing the numbering downstream polyhedrin stop codon. Promoter motifs were searched, an early CAKT initiator sequence (INR) [55] was found in 40 ORFs, irrespective of including the TATA-box, while 29 ORFs had a late INR motif [56] and another 59 had both early and late elements. As expected, the SfColA genome contained the 37 core genes present in all baculoviruses and these sequences were thus identified using current denominations. The other putative genes were mentioned using the most accepted names based on their Blast relationships regarding the annotated ORFs from other baculoviruses [3, 57, 58].
Sequence homology analysis revealed that most ORFs were shared among SfMNPVs, giving close to 100 % similarity but also revealing significant differences in one set of loci (Additional file 1: Table S1). SfColA 005/007/023/024/033/112/124/131 ORFs particularly required more in-depth study because they had less similarity than expected for genotype variants from the same species (having values less than 75 % when the identity average is 98.5 % ± 5.5) or absence regarding their putative orthologs. The region including 005/006/007 ORFs might thus be a putative encoding location for expressing 3 small polypeptides annotated on only SfMNPV genomes (Fig. 1a). It is worth noting that ORF007 is overlapped with ORF006 and probably is not a gene considering the absence of typical promoter motifs and the small size of its theoretical encoded polypeptide. The proteins derived from ORFs 005 and 006 had variability when compared to their orthologs from the other SfMNPVs due to mainly single or double nucleotide insertion-deletions causing frame shifts. What was striking about this unique SfMNPV region was the presence of direct and inverted repeats located on the flanks of a sequence shared with other Group II alphabaculoviruses. This region might thus be considered as non-coding (and until there is experimental evidence) and could be associated with other viral functions such as genomic replication where this kind of sequence seems to be relevant [3]. SfColA ORFs 005/006/007 were annotated since the same was done in SfNicB, Sf19 and Sf3AP2.
Fig. 1.

Genome organization of SfMNPV ColA. The illustrations show the SfColA loci where there are differences regarding other genotypes of this baculovirus species [SfMNPV NicB (SfNicB), SfMNPV 19 (Sf19) and SfMNPV 3AP2 (Sf3AP2)]. In all cases, the SNPs (single nucleotide polymorphisms; asterisks), indels (sequence insertion-deletions; filled circles indicating in parenthesis the number of nucleotides added or deleted) and the annotated ORFs (shown as arrows) are highlighted in each locus. a Region containing SfColA ORF005/006/007. White boxes indicate sequences shared by alphabaculoviruses and direct repeats are shown as red triangles. b Region containing SfColA ORF023/024. Sequences involved in gene replacement are shaded and the respective ORFs located in that position are differentially colored (white in SfColA and black in the other ones). In Sf3AP2 the ORF023 ortholog of SfNicB and Sf19 is annotated as ORF022. c Region containing egt gene in SfColA, SfNicB and Sf19. The unknown gene annotated in all genomes downstream to the egt gene is ORF028 in SfColA, ORF027 in SfNicB, ORF026 in Sf19 and ORF027 in Sf3AP2. The deleted region in Sf3AP2 is shaded in grey. d Regions containing SfColA ORF033 and ORF124. The orthologs of the first one in SfNicB and Sf3AP2 are annotated as ORF032. The orthologs of the second sequence in Sf19 and Sf3AP2 are annotated as ORF121 and ORF122, respectively. e Region containing SfColA ORF131 and ORF131a. The orthologs in Sf19 and Sf3AP2 are ORF 127a/128 and ORF 129/130, respectively. Four SNPs and a deletion determined the absence of a coding sequence equivalent to SfColA ORF131 in the genome of SfNicB. The p26 gene in SfNicB has an insertion of 60 nt. f Genome representations of homologous region distribution
A very different situation occurred with SfColA ORFs 023 and 024 (Fig. 1b); both genes were not present in the other SfMNPV genomes, although these sequences have similarity with some Group II alphabaculovirus genes. By contrast, SfNicB, Sf19 and Sf3AP2 had one ORF in that locus (023, 022 and 023, respectively) that is not present in SfColA and had homology with other baculoviruses. This gene is present in the SpfrGV genome (ORF099) where the encoded polypeptide was hypothesized as being a soluble protein containing ring finger motifs [37]. Such genome replacement (2 genes acquired compared to 1 lost) might thus be considered a recombination product as will be shown below.
Another genome location having differences was the region containing SfColA ORFs 027 and 028 because the theoretical polypeptides encoded by these genes had low identity and similarity values regarding the homologous Sf3AP2 ORFs 026 and 027 (Fig. 1c). This was due to deletion in the Sf3AP2 genome affecting the corresponding carboxy terminal of the ecdysteroid UDP–glucosyltransferase (egt) gene (Sf3AP2 ORF026) and the amino terminal of the other one (Sf3AP2 ORF027).
SfColA ORFs 033 and 124 also had lower similarity values than the expected ones when the in silico translated sequences were compared to the corresponding orthologs (Fig. 1d). Both putative genes encoded unknown proteins; the former only had differences with Sf19 because of a two single nucleotide deletion in this gene affected the reading frame annotated in the other SfMNPVs starting in a later ATG. SfColA ORF124 had differences with only SfNicB due to this sequence having a 5 bp microdeletion. The SfColA ORF131 did not present an annotated ortholog in SfNicB (Fig. 1e). Sequence analysis revealed 6 different nucleotides in the same stretch, including 1 nucleotide deletion affecting the reading frame, even though the region is present in SfNicB and other Group II alphabaculoviruses. In fact, in that location was annotated other ORF (SfNicB ORF130) with similarity with AcMNPV ORF29 and SeMNPV ORF128. It is important to note that homologs of SfNicB ORF130 are also present in the other genotypes of SfMNPVs, including SfColA, and were annotated as ORF130 in Sf3AP2 and ORF128 in Sf19. For these reasons, in the genome of SfColA both putative coding regions were included as ORF131 and ORF131a (Fig. 1e).
Regarding non-encoding loci, baculoviruses homologous regions (hr) are sequence repeats which are dispersed throughout their genomes. All previously described SfMNPVs have 8 h interspersed in different locations; they are characterized by tandem repeats consisting of a 44 bp nucleotide stretch which include an imperfect 34 bp palindromic core. These sequences are variable; however, the local secondary structure motifs are very similar, constituting hairpin loops (see Additional file 2: Figure S1). The hr-1 has 7 repeats; hr-2, hr-3 and hr-6 have only 1 repeat; hr-4 and hr-7 have 6 repeats and hr-5 and hr-8 have 4 repeats. It should be noted that SfColA lacked hr-3 since this sequence was located in the region where gene replacement occurred (Fig. 1f). Sf19 lacked hr-5c and hr-5d, and SfNicB lacked hr-8a and hr-8b. Two unique ORFs (039a and 110a) were annotated in SfNicB but the corresponding sequences were also present in Sf19, Sf3AP2 and SfColA showing few single nucleotide polymorphisms. The locus containing SfNicB ORF039a was located close to hr-4 while the SfNicB ORF110a was close to hr-7 and both postulated encoding sequences were probably not real genes. All repeats from SfMNPV hrs can be summarized in a consensus sequence using the IUPAC ambiguity code: 5′ YNAWSTTDRCTTTYVDYNAHRHDYBTBRNBDAAAKYMAASWTBR 3′. Conserved nucleotides (bold) would be A or T and probably essential for their role as replication origins and/or as transcription enhancers.
Previous results were confirmed by a genome colinearity study showing high nucleotide sequence conservation and genome organization among SfMNPV genotypes (Fig. 2). The exceptions included the locus where SfColA lost a ~1470 bp fragment and acquired another one of ~2970 bp (ORFs 023 and 024), being similar to SpltNPV-II regions, and 3 small insertions present in only SfNicB. The first was 309 bp, located downstream to the odv-e66 gene and producing SfNicB ORF057a. The second insertion was 73 bp, located in the intergenic region of SfNicB ORFs 085 and 086, and the third one was 60 bp positioned 437 bp upstream SfNicB ORF131 (p26 gene) (Fig. 1e).
Fig. 2.

Nucleotide genome synteny. Individual genome comparisons between SfMNPV ColA (SfColA) and the other genotypes of this baculovirus species [SfMNPV NicB (SfNicB), SfMNPV 19 (Sf19) and SfMNPV 3AP2 (Sf3AP2)], SeMNPV and SpltNPV-II are shown. Genome sizes are represented as rules and a colored key is used to show similarity percentages. The locus where SfColA ORF023/024 are located is depicted as a black asterisk. The other asterisks indicate the locations where SfNicB has insertions [309 bp (red), 73 bp (green) and 60 bp (blue)]. Blue blocks in the middle of each graph indicate A + T rich regions
Phylogenetic analysis was based on 37 concatenated core proteins derived from 75 baculovirus genomes (Fig. 3a); as expected, the cladogram reproduced the grouping in 4 genera recognized in the current classification of the virus family [2]. SfColA and the other SfMNPVs formed a clade which was included in Group II Alphabaculovirus, the closest species being SeMNPV and SpltNPV-II. Special attention should be paid to other baculovirus isolates recovered from the same insect species, such as Spodoptera litura, Mamestra configurata, Mamestra brassicae, Helicoverpa armigera, Helicoverpa zea, Agrotis ipsilon and Agrotis segetum NPVs. By contrast with SfMNPV, some members of the previously mentioned set of viruses grouped in different clades, thereby reflecting their greater diversity. Another inference was made regarding phylogeny, but only using the most closest related viruses based on 100 concatenated orthologous proteins (Fig. 3b). This study has revalidated the consistency of SfMNPV relationships with SeMNPV and SpltNPV-II (the closest baculovirus species) and has also highlighted the difficulty of finding groupings among the different genotypes of baculoviruses infecting Spodoptera spp. since non-orthologous proteins were not included in these studies.
Fig. 3.

Phylogenetic inference for SfMNPV ColA. a Cladogram based on a concatemer built with the 37 core proteins obtained from 74 baculoviral genomes and SfMNPV ColA. The phylogenetic tree was inferred using the MEGA 5 program. The four Baculoviridae genera are indicated and Alpha- (Group I), Beta- and Gammabaculovirus clades were collapsed to preserve space. b Cladogram based on a concatemer built with 100 homologous proteins obtained from 8 baculoviral genomes including SfMNPV ColA. The phylogenetic tree was inferred using the MEGA 5 program
Interspecies horizontal gene transfer
The most important difference among SfMNPVs was the sequence acquisition which occurred in SfColA genome; this involved acquiring two genes from other baculovirus species and the loss of one gene present in all remaining molecularly characterized SfMNPV. A detailed study aimed at determining orthology with other baculoviruses showed that SfColA ORF023 and ORF024 were closely related to annotated SpltNPV-II ORFs (Fig. 4). These sequences from both genomes had higher than 95 % identity and similarity, similar to the value when comparing homologous proteins between pairs of SfMNPV genotypes (Additional file 1: Table S1). The study revealed that the ORF023 had putative orthologs in Group II alpha- and betabaculoviruses while ORF024 had homologs only in Group II alphabaculoviruses.
Fig. 4.

Protein relationships for SfMNPV ColA ORFs 023 and 024. The relationships among two SfMNPV ColA ORFs and their orthologs contained in other viruses were calculated by BlastP. Related baculovirus species are shown (three letter abbreviations for species and ORF number) in filled circles (yellow for betabaculoviruses and green for Group II alphabaculoviruses). The BlastP e-value between pairs of species is indicated above each arrow. a Protein relationships for SfMNPV ColA ORF 023. b Protein relationships for SfMNPV ColA ORF 024. AIN: AgipNPV; ASN: AgseNPV; HAA: HearNPV Aus; HA1: HearNPV C1; HA4: HearNPV G4; HAG: HearGV; HAN: HearMNPV; HAS: HearSNPV; HZN: HzSNPV; MC2: MacoNPV 90–2; MC4: MacoNPV A90-4; MBC: MabrNPV CHb1; MBK: MabrNPV K1; MCB: MacoNPV B; PUG: PsunGV; SEN: SeMNPV; SFC: SfMNPV ColA (in red letters); SFG: SpfrGV VG008; SL2: SpltMNPV G2; SLN: SpltNPV-II; SLT: SpliNPV AN1956; XCG: XecnGV
Different approaches were used to explore the recombination hypothesis regarding recent SfMNPV and SpltNPV-II ancestors. The first one consisted in a relative similarity analysis between the genome region involved in the structural mutation from SfColA and the other SfMNPVs, SeMNPV and SpltNPV-II. Poor similarity was revealed regarding the sequences in all the other SfMNPVs, although the chitinase (upstream region) and gp37 (downstream region) genes were almost identical (Fig. 5a–c); by contrast, similarity increased when compared with SeMNPV (Fig. 5d) and reached the maximum value with SpltNPV-II (Fig. 5e). It is worth noting that the only SfColA region regarding the other SfMNPVs was very closely related to SpltNPV-II but that the upstream and downstream sequences had lower similarity values. Another approach based on bootscaning analysis validated previous results showing that a recent ancestor of SpltNPV-II was the most probable DNA donor involved in recombination (Fig. 5f).
Fig. 5.
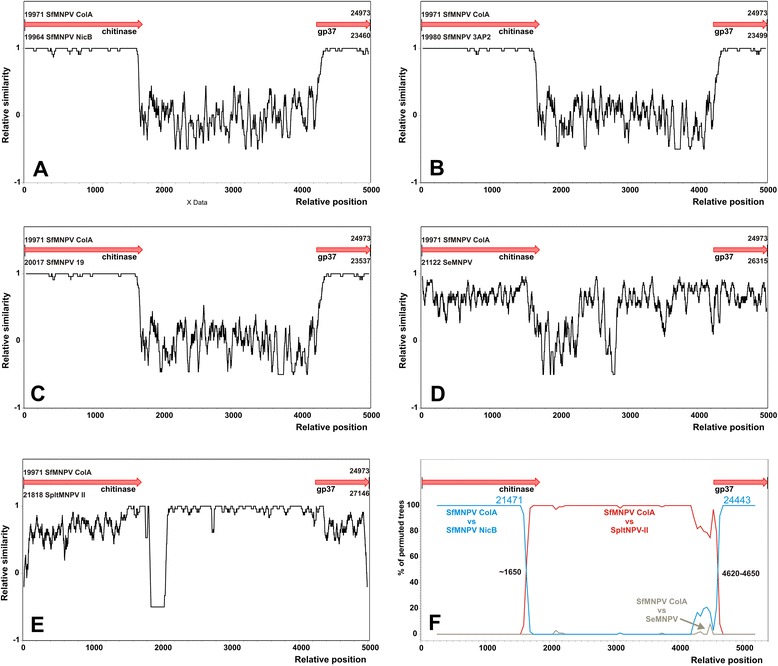
SfMNPV ColA ORFs 023 and 024 origin by horizontal transfer. The recombination process between SfMNPV ColA and SpltNPV-II ancestors was studied by similarity plots and bootscanning analysis. Genome regions analyzed were those containing SfMNPV ColA ORFs 023 and 024, chitinase and gp37 genes. In all cases, colored arrows represent SfMNPV ColA ORFs. Genome positions are indicated at the beginning and the end of the regions analyzed (bp scale). Similarity plots are indicated in black, and different colors (explained in the graphs) are used. a Similarity plot between SfMNPV ColA and SfMNPV NicB. b Similarity plot between SfMNPV ColA and SfMNPV 3AP2. c Similarity plot between SfMNPV ColA and SfMNPV 19. d Similarity plot between SfMNPV ColA and SeMNPV. e Similarity plot between SfMNPV ColA and SpltNPV-II. f Bootscanning using SfMNPV ColA, SfMNPV NicB, SeMNPV and SpltNPV-II
A G/C-content study was performed to demonstrate gene transfer by transposition in SeMNPV [25]. Such approach not based on sequence alignments providing similar results to those aforementioned (Fig. 6). The recombinant region’s G/C profile was more similar to SpltNPV-II than to the other SfMNPVs (43.3 % G/C-content in SfColA and 43.1 % in SpltNPV-II compared to 35.7 % in SfNicB/Sf19/Sf3AP2). By contrast, upstream and downstream regions (chitinase and gp37 genes) had a completely different pattern, having similar values to the G/C-content average (40.3 % in SfMNPVs compared to 45 % in SpltNPV-II). Kimura 2-parameter (K-2-P) distances were estimated to further support the idea of recent recombination (Tables 2 and 3). This approach revealed a very close relationships between SFColA ORF023 and ORF024 regarding SpltNPV-II ORF020 and ORF021, respectively, expressing distances larger than 0.015 but smaller than 0.050. This range of values is currently assumed as an interval in what complementary information is needed to determine whether two sequences are different or genotypes of the same species [1]. The present study thus revealed that the sequences flanked by chitinase and gp37 genes in SfColA and SpltNPV-II genomes would belong to baculovirus isolates from the same species; by contrast, the other genes from both genomes were revealed to be sequences of two different baculovirus species.
Fig. 6.

G + C profile into the locus containing SfMNPV ColA ORFs 023 and 024. The G/C-contents (%) of SfMNPV ColA, SfMNPV NicB and SpltNPV-II were analyzed in the locus of each genome where the recombination process occurred. Profiles are shown with different colors referenced in the graph. A representation of coding regions (indicated as arrows) is featured above the histogram. Grey boxes do not represent sequences and are used to facilitate understanding the graphical positions of chitinase and gp37 genes among analyzed genomes
Table 2.
Kimura 2-parameter distances between ORFs 020/021 of SpltNPV-II and their corresponding orthologs

The K-2-P values are shaded in gray (lower triangles). The other values are the corresponding standard errors (upper triangles)
SLN SpltNPV-II, SFC SfColA, AIN AgipNPV, MCB MacoNPVB, MC2 MacoNPV 90-2, MC4 MacoNPV 90-4, MBK MabrMNPV K1, MBC MabrMNPV Chb1, HAN HearMNPV, SEN SeMNPV, PUG PsunGV, HAG HearGV, XCG XecnGV, SFG SpfrGV, HAS HearsNPV, HAA HearNPV Aus, HA1 HearNPV C1, HA4 HearNPV G4, HZN HzSNPV, ASN AgseNPV, SL2 SpltMNPV G2, SLT SpliNPV AN1956
Table 3.
Kimura 2-parameter distances between ORFs 020/021 of SpltNPV-II and their corresponding orthologs

The K-2-P values are shaded in gray (lower triangles). The other values are the corresponding standard errors (upper triangles)
SLN SpltNPV-II, SFC SfColA, AIN AgipNPV, MCB MacoNPVB, MC2 MacoNPV 90-2, MC4 MacoNPV 90-4, MBK MabrMNPV K1, MBC MabrMNPV Chb1, HAN HearMNPV, SEN SeMNPV, PUG PsunGV, HAG HearGV, XCG XecnGV, SFG SpfrGV, HAS HearsNPV, HAA HearNPV Aus, HA1 HearNPV C1, HA4 HearNPV G4, HZN HzSNPV, ASN AgseNPV, SL2 SpltMNPV G2, SLT SpliNPV AN1956
All the aforementioned analysis suggested that recombination occurred between recent SfMNPV and SpltNPV-II ancestors, involving the end of the chitinase gene and the start of the gp37 gene, causing the replacement of ~1470 bp (including hr-3 and SfNicB/Sf3AP2 ORF023 or Sf19 ORF022) for ~2970 bp carrying 2 complete genes having great similarity to SpltNPV-II 020 and 021 ORFs and a truncated gene similar to SpltNPV-II ORF019. Breakpoints seemed to be inside the reading frames for the chitinase gene (around the position 21,471 in SfColA) and the gp37 gene (around the position 24,443 in SfColA) restoring the continuity of both frames. Regarding the SpltNPV-II ORF019 homolog in SfColA, a sequence analysis revealed 8 different deletions (11 bp, 240 bp, 11 bp, 3 bp, 1 bp, 3 bp, 1 bp and 11 bp, ordered from ATG to stop codon) thereby resulting in a frame shift.
SfColA ORF023 and ORF024
Whole RNAs isolated at different times from S. frugiperda larvae orally infected with SfColA were examined by reverse transcription PCR to determine whether SfColA ORF023 and ORF024 were active transcriptional units (Fig. 7). The very late SfColA ORF001 (polyhedrin) and the inmediate-early SfColA ORF143 (ie-0) were included for reference. Single RT-PCR products were obtained having the expected sizes (214 bp -ORF023-, 166 bp -ORF024-, 255 bp -ORF001- and 163 bp -ORF143-). This experimental approach showed that transcripts from ORF023 appeared at 10 hpi while ORF024 started at 6 hpi, a result in agreement with the presence of early INR promoter motifs (Additional file 1: Table S1).
Fig. 7.

Transcription kinetics of SfMNPV ColA ORFs 023 and 024. Spodoptera frugiperda larvae were exposed to SfMNPV ColA and whole RNAs were isolated from sacrificed animals at different intervals post-infection. Then, cDNA with polyT primer was generated for each sample and PCR assays were done using specific primers which amplify fragments of different ORFs from SfMNPV ColA genome [polyhedrin (polh), immediate-early 0 (ie-0), ORF023 and ORF024]. Figure shows a photo cut-out showing the amplification bands resolved by agarose gel electrophoresis. The SfMNPV ColA genome was used as positive control and water was used as negative control
Predictive studies were then performed for SfColA ORF023 and ORF024 proteins. The SfColA ORF023 theoretical polypeptide thus consisted of 162 residues, 19 being negatively charged (Asp + Glu) and 29 positively charged amino acids (Arg + Lys + His). Based on sequence, the molecular weight is 19,087.1 Da and the theoretical isoelectric point is 9.39. The hydrophobicity profile suggested that this polypeptide was a soluble protein having average hydrophobicity of −0.08. The secondary structure predicted by the LOMETS and I-TASSER servers gave 85.8 % coincidence, revealing the presence of 3 α-helices (28.4 % of residues), 5 β-sheets (17.3 % of residues), and the remaining amino acids constituting loops or turns (Fig. 8a). The QUARK server predicted that ORF023 would be a globular protein having a tertiary structure according to previous results (Fig. 8b). HHpred identified a region (from amino acid 35 to 95) as being a phosphatase domain similar to Schizosaccharomyces pombe Polynucleotide kinase 3 phosphatase (PNK1; [59]) which plays a role in repairing single breaks in DNA induced by several DNA-damaging agents. INTERPROSCAN identified 2 protein kinase C phosphorylation sites (from amino acids 66 to 68 and from amino acid 75 to 77) and 1 tyrosine-kinase phosphorylation site (from amino acid 97 to 104). These post-translational modifications could be part of activation/inactivation processes, but require experimental confirmation.
Fig. 8.

Characterization of theoretical proteins derived from SfMNPV ColA ORFs 023 and 024. The theoretical proteins encoded by SfMNPV ColA ORFs 023 (panels a and c) and 024 (panels b and d) were analyzed and the 3D structures were predicted. Hydrophobicity profiles and predicted secondary structures are shown. Alpha helices are represented as red cylinders and beta sheets as green arrows. The putative signal peptide (SP) and transmembrane domains (TM) are also shown
The SfColA ORF024 theoretical protein consisted of 462 residues, 33 being negatively charged (Asp + Glu) and 41 positively charged amino acids (Arg + Lys + His). Based on sequence, the molecular weight was 52,210.0 Da and the theoretical isoelectric point was 7.60. The hydrophobicity profile suggested that this polypeptide would be a membrane protein having +0.08 average hydrophobicity and having at least 6 transmembrane regions containing 12 α-helices and a signal peptide detected by SignalP (Fig. 8c). The secondary structure predicted by LOMETS and I-TASSER servers gave 85.8 % coincidence, revealing the presence of 19 α-helices (47.8 % of residues), 11 β-sheets (12.5 % of residues), and the remaining amino acids constituting loops or turns. Coincidentally, the LOMETS server predicted a tertiary structure and a secondary motif distribution consistent with a transmembrane motif (Fig. 8d). HHpred identified one region (from amino acid 51 to 451) as a member of the Major Facilitator Superfamily (MFS); these proteins are permeases which act as secondary carriers in cell transport [60]. INTERPROSCAN found several putative post-translational modifications, including 2 N-glycosylation sites (from amino acid 36 to 39 and from amino acid 317 to 320), 1 cAMP- and cGMP-dependent protein kinase phosphorylation site (from amino acid 3 to 6), 2 Protein kinase C phosphorylation sites (from amino acid 452 to 454 and from amino acid 455 to 455) and 1 Casein kinase II phosphorylation site (from amino acid 50 to 53). These post-translational modifications could form part of protein function but experimental confirmation is required. It should be mentioned that the homologous protein encoded by the Helicoverpa armigera nucleopolyhedrovirus (G4 strain) was not detected as a structural protein, suggesting that its role occurs within the infected cells [61, 62].
It is importante to note that all the data sets supporting the results of this article are included within the article and its additional files.
Conclusions
Baculoviruses and other viruses having large dsDNA genomes mainly evolve due to the accumulation of structural mutations (insertions, deletions, replacements, inversions, translocations) affecting gene content, where recombination or transposition appear to be the most relevant examples of mechanisms occurring in nature affecting DNA integrity. Analysis of complete baculovirus genomes has revealed a “core genome” represented by 37 genes encoding essential factors accumulating sequence variability since the last virus ancestor [58]. Such pathogens carry sequences acquired from other entities defining a “plastic genome” which contains sequences included in all members of each genus and other regions present in only some species or variants. Core genes usually produce key factors needed to complete a virus cycle, by contrast many encoding sequences in the plastic genome produce auxiliary proteins collaborating in virus processes even though not being essential for producing infectious progeny increasing fitness for them to perpetuate in nature. New technologies available for acquiring whole genome information have facilitated associating phenotype characteristics with gene content. The SfMNPV ColA genome (from a particular Colombian isolate having better biological properties than others) [21] was thus sequenced having high coverage and compared to other genotypes isolated from other geographical regions.
The most relevant differences occurred in a locus where SfColA underwent recent sequence replacement, losing 1 gene and gaining 2 new encoding sequences. The genome location where recombination occurred has been described as hypervariable since SfMNPV variants have different deletions [17, 18, 21]. These regions include auxiliary genes such as ecdysteroid UDP–glucosyltransferase (egt), protein-tyrosine-phosphatase (ptp), chitinase and cathepsin whose products have activity affecting insect host physiology, development, behavior and integrity [20]. Interestingly this location also contains hr-3, a kind of sequences recognized as being a recombination facilitators [27, 28]. By contrast, most of the other hrs were closer to core genes, thereby decreasing the fitness of natural recombinant viruses due to possible loss of essential functions. It is worth stressing that hr-1 was close to odv-56 (pif5) and f genes, hr-2 was near lef-1, hr-4 next to alk-exo, hr-6 was close to lef-9 and hr-7 was near lef-8 and u-box/ring. The locus containing hr-3 thus seemed to be a hot genome region prone to undergoing structural mutations.
Recombination is an important evolutionary mechanism which might be used as a viral strategy to gain advantage for maintaining adaptability to changing environments [27]. Recombination could facilitate resistance to host range expansion [63–65]. These kinds of interactions between genomes occur if two types of DNA coexist in the same cell, have sequence similarities and they are replicating. Artificial coinfections with AcMNPV and BmNPV in larvae and in cell culture have shown that homologous recombination can occur between viruses belonging to two different species [66]. It has been reported that some SpfrGV genes were acquired by horizontal gene transfer from other baculovirus species including SpltNPV-II [37], such genome having been identified as DNA donor for SfColA ORFs 023 and 024. SpfrGV contains an orthologous gene for SfNicB ORF023, Sf19 ORF022 and Sf3AP2 ORF023 (SpfrGV ORF099), the encoding sequence lost in SfColA. This would suggest that recombination occurred when these viruses co-infected Spodoptera frugiperda larvae.
Spodoptera litura and Spodoptera frugiperda are polyphagous insect pests living on crops such as rice, corn, cotton and tobacco; they have been reported in subtropical locations in both the Old and New world, although cross migration of both insects has been reported [67]. S. litura have been recorded in 80 species of host plant [68] while S. frugiperda has been described in 186 such plants [69], many of them shared between both lepidoptera. Natural coinfection involving circulating variants of SpltNPV and SfMNPV could thus occur in the same host. Both species have similar sequences and genome organization, and it has been reported that SpltNPV can infect Spodoptera frugiperda-derived cells, such as Sf9 and Sf21 [65, 68]. The above and bioinformatics evidence provided here support the hypothesis that homologous recombination is used by baculoviruses in nature to acquire variability. The SfColA genome would thus seem to provide natural proof for affirming that horizontal gene transfer is exploited by organisms and viruses to increase their fitness and thus acquire a reproductive success ensuring their permanence in nature.
Acknowledgments
This work was supported by research funds provided by COLCIENCIAS (Colombia) and MinCyT (Ministry of Science and Technology; Argentina) Cooperation Program.
Additional files
Potentially expressed ORFs in the genome of SfMNPV ColA. In bold letter are indicated the core genes. (XLSX 32 kb)
Sequence comparisons and structural analysis of homologous regions. The palindromes contained into the homologous regions (hr) from SfMNPV ColA were analyzed and the consensus sequences represented by Sequence logos. Besides, predictions of the ssDNA secondary structures are shown. 1-A. hr-1 (28 palindromes). 1-B. hr-2 (4 palindromes). 1-C. hr-3 (3 palindromes). 1-D. hr-4 (24 palindromes). 1-E. hr-5 (14 palindromes). 1-F. hr-6 (4 palindromes). 1-G. hr-7 (24 palindromes). 1-H. hr-8 (14 palindromes). 1-I. All palindromes (115 repeats). 2. Largest A + T-rich region in the SfMNPV ColA genome. 2-A. Physical map of hr-1 and location of the largest A + T-rich region. 2-B. Nucleotide sequence detail. 2-C. Local ssDNA secondary structure. (TIF 3246 kb)
Footnotes
Gloria Patricia Barrera and Mariano Nicolás Belaich contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GPB carried out the genome isolation, participated in the molecular biology assays and bioinformatics studies and helped to draft the manuscript. MNB participated in the molecular biology assays, and conceived, designed and carried out some bioinformatics studies. Besides, MNB drafted the manuscript. MAP participated in the design of work, collaborated in the discussion of the results and helped to draft the manuscript. LFV conceived together with GPB the work and collaborated in discuss results and helped to draft the manuscript. PDG conceived and performed the most important bioinformatics studies and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ informations
GPB and LFV are researchers from CORPOICA (Corporación Colombiana de Investigación Agropecuaria; Colombia). MAP is a researcher from Fundación Instituto de Inmunología (Colombia) and professor at Universidad del Rosario, Bogotá, Colombia. PDG and MNB are members of the Research Career of CONICET (Consejo Nacional de Ciencia y Tecnología, Argentina) and professors of UNQ (Universidad Nacional de Quilmes, Bernal, Buenos Aires, Argentina).
Contributor Information
Gloria Patricia Barrera, Email: gbarrera@corpoica.org.co.
Mariano Nicolás Belaich, Email: mbelaich@unq.edu.ar.
Manuel Alfonso Patarroyo, Email: manuel.patarroyo@urosario.edu.co.
Laura Fernanda Villamizar, Email: lvillamizar@corpoica.org.co.
Pablo Daniel Ghiringhelli, Email: pdg@unq.edu.ar.
References
- 1.Jehle JA, Lange M, Wang HL, Hu ZH, Wang YJ, Hauschild W. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology. 2006;346:180–93. doi: 10.1016/j.virol.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 2.Herniou EA, Arif BM, Becnel JJ, Blissard GW, Bonning B, Harrison R, et al. Baculoviridae. In: King AMQ, Lefkowitz EJ, Adams MJ, Carstens EB, et al., editors. Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier Academic Press; 2011. pp. 163–73. [Google Scholar]
- 3.Rohrmann GF. Baculovirus Molecular Biology. Third. Bethesda (MD): National Center for Biotechnology Information (US); 2013. [PubMed] [Google Scholar]
- 4.Harrison RL, Herniou EA, Theilmann DA, Blissard GW, Becnel JJ, Arif BM, et al. Eight new species in the genus Alphabaculovirus. ICTV Official Taxonomy: Updates since the 8th report. 2012. [Google Scholar]
- 5.Clark PL, Molina-Ochoa J, Martinelli S, Skoda SR, Isenhour DI, Lee DJ, et al. Population variation of the fall armyworm, Spodoptera frugiperda, in western hemisphere. J Insect Sci. 2007;7:5. doi: 10.1673/031.007.0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Summers MD, Anderson DL. Characterization of nuclear polyhedrosis virus DNAs. J Virol. 1973;12(6):1336–46. doi: 10.1128/jvi.12.6.1336-1346.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knudson DL, Tinsley TW. Replication of a nuclear polyhedrosis virus in a continuous cell culture of Spodoptera frugiperda: purification, assay of infectivity, and growth characteristics of the virus. J Virol. 1974;14(4):934–44. doi: 10.1128/jvi.14.4.934-944.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knell JD, Summers MD. Investigation of genetic heterogeneity in wild isolates of Spodoptera frugiperda nuclear polyhedrosis virus by restriction endonuclease analysis of plaque-purified variants. Virology. 1981;112:190–7. doi: 10.1016/0042-6822(81)90624-3. [DOI] [PubMed] [Google Scholar]
- 9.Loh LC, Hamm JJ, Kawanishi C, Huang ES. Analysis of the Spodoptera frugiperda Nuclear Polyhedrosis Virus genome by restriction endonucleases and electron microscopy. J Virol. 1982;44(2):747–51. doi: 10.1128/jvi.44.2.747-751.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maruniak JE, Brown SE, Knudson DL. Physical maps of SfMNPV baculovirus DNA and its genomic variants. Virology. 1984;136:221–34. doi: 10.1016/0042-6822(84)90261-7. [DOI] [PubMed] [Google Scholar]
- 11.Gonzales MA, Smith GE, Summers MD. Insertion of the SfMNPV polyhedrin gene into an AcMNPV polyhedrin deletion mutant during viral infection. Virology. 1989;170:160–75. doi: 10.1016/0042-6822(89)90363-2. [DOI] [PubMed] [Google Scholar]
- 12.Liu JC, Maruniak JE. Nucleotide sequence and transcriptional analysis of the gp41 gene of Spodoptera frugiperda nuclear polyhedrosis virus. J Gen Virol. 1995;76:1443–50. doi: 10.1099/0022-1317-76-6-1443. [DOI] [PubMed] [Google Scholar]
- 13.Tumilasci VF, Leal E, Zanotto PM, Luque T, Wolff JL. Sequence analysis of a 5.1 kbp region of the Spodoptera frugiperda multicapsid nucleopolyhedrovirus genome that comprises a functional ecdysteroid UDP-glucosyltransferase (egt) gene. Virus Genes. 2003;27:137–44. doi: 10.1023/A:1025720425469. [DOI] [PubMed] [Google Scholar]
- 14.Simón O, Williams T, López-Ferber M, Caballero P. Genetic structure of a Spodoptera frugiperda nucleopolyhedrovirus population: High prevalence of deletion genotypes. Appl Environ Microbiol. 2004;70:5579–88. doi: 10.1128/AEM.70.9.5579-5588.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simón O, Williams T, López-Ferber M, Caballero P. Functional importance of deletion mutant genotypes in an insect nucleopolyhedrovirus population. Appl Environ Microbiol. 2005;71:4254–62. doi: 10.1128/AEM.71.8.4254-4262.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolff JL, Valicente FH, Martins R, Oliveira JV, Zanotto PM. Analysis of the genome of Spodoptera frugiperda nucleopolyhedrovirus (SfMNPV-19) and of the high genomic heterogeneity in group II nucleopolyhedroviruses. J Gen Virol. 2008;89:1202–11. doi: 10.1099/vir.0.83581-0. [DOI] [PubMed] [Google Scholar]
- 17.Harrison RL, Puttler B, Popham HJ. Genomic sequence analysis of a fast-killing isolate of Spodoptera frugiperda multiple nucleopolyhedrovirus. J Gen Virol. 2008;89:775–90. doi: 10.1099/vir.0.83566-0. [DOI] [PubMed] [Google Scholar]
- 18.Simón O, Palma L, Beperet I, Muñoz D, López-Ferber M, Caballero P, et al. Sequence comparison between three geographically distinct Spodoptera frugiperda multiple nucleopolyhedrovirus isolates: Detecting positively selected genes. J Invertebr Pathol. 2001;107:33–42. doi: 10.1016/j.jip.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Simón O, Palma L, Williams T, López-Ferber M, Caballero P. Analysis of a naturally-occurring deletion mutant of Spodoptera frugiperda multiple nucleopolyhedrovirus reveals sf58 as a new per os infectivity factor of lepidopteran-infecting baculoviruses. J Invertebr Pathol. 2012;109(1):117–26. doi: 10.1016/j.jip.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Barrera G, Simón O, Villamizar L, Williams T, Caballero P. Spodoptera frugiperda multiple nucleopolyhedrovirus as a potential biological insecticide: Genetic and phenotypic comparison of field isolates from Colombia. Biol Control. 2011;58:113–20. doi: 10.1016/j.biocontrol.2011.04.009. [DOI] [Google Scholar]
- 21.Barrera G, Williams T, Villamizar L, Caballero P, Simón O. Deletion genotypes reduce occlusion body potency but increase occlusion body production in a Colombian Spodoptera frugiperda nucleopolyhedrovirus population. PLoS One. 2013;8(10):e77271. doi: 10.1371/journal.pone.0077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajós JP, Pijnenburg J, Usmany M, Zuidema D, Závodszky P, Vlak JM. High frequency recombination between homologous baculoviruses in cell culture. Arch Virol. 2000;145:159–64. doi: 10.1007/s007050050012. [DOI] [PubMed] [Google Scholar]
- 23.Da Palma T, Doonan B, Tragerm N, Kasman L. A systematic approach to virus-virus interactions. Virus Res. 2010;149:1–9. doi: 10.1016/j.virusres.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrelli ML, Berretta MF, Belaich MN, Ghiringhelli PD, Sciocco-Cap A, Romanowski V. The Baculoviral Genome. In: García ML, Romanowski V, editors. Viral Genomes - Molecular Structure, Diversity, Gene Expression Mechanisms and Host-Virus InteractionsInTech. 2012. pp. 1–32. [Google Scholar]
- 25.Thézé J, Cabodevilla O, Palma L, Williams T, Caballero P, Herniou EA. Genomic diversity in European Spodoptera exigua multiple nucleopolyhedrovirus isolates. J Gen Virol. 2014;95(10):2297–09. doi: 10.1099/vir.0.064766-0. [DOI] [PubMed] [Google Scholar]
- 26.Gilbert C, Chateigner A, Ernenwein L, Barbe V, Bézier A, Herniou EA, et al. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nat Commun. 2014;5:3348. doi: 10.1038/ncomms4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erlandson MA. Genetic variation in field populations of baculoviruses: Mechanisms for generating variation and its potential role in baculovirus epizootiology. Virol Sin. 2009;24:458–69. doi: 10.1007/s12250-009-3052-1. [DOI] [Google Scholar]
- 28.Cory JS, Green BM, Paul RK, Hunter-Fujita F. Genotypic and phenotypic diversity of a baculovirus population within an individual insect host. J Invertbr Pathol. 2005;89:101–11. doi: 10.1016/j.jip.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 29.King LA, Possee RD. The baculovirus expression vector system: a laboratory guide. London: Chapman and Hall; 1992. [Google Scholar]
- 30.Carver T, Berriman M, Tivey A, Patel C, Böhme U, Barrell BG, et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–6. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–02. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;2:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuker M, Mathews DH, Turner DH. Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide. In: Barciszewski J, Clark BFC, editors. RNA Biochemistry and Biotechnology. The Netherlands: NATO ASI Series, Kluwer Academic Publishers;1999. p. 11–43.
- 36.Matzura O, Wennborg A. RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. Comput Appl Biosci. 1996;12(3):247–9. doi: 10.1093/bioinformatics/12.3.247. [DOI] [PubMed] [Google Scholar]
- 37.Cuartas PE, Barrera GP, Belaich MN, Barreto E, Ghiringhelli PD, Villamizar LF. The complete sequence of the first Spodoptera frugiperda Betabaculovirus genome: a natural multiple recombinant virus. Viruses. 2015;7(1):394–421. doi: 10.3390/v7010394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Wang J, Yao ZJ, Jin Q, Shen Y, Chen R. GenomeComp: a visualization tool for microbial genome comparison. J Microbiol Methods. 2003;54(3):423–6. doi: 10.1016/S0167-7012(03)00094-0. [DOI] [PubMed] [Google Scholar]
- 39.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salminen MO, Carr JK, Burke DS, McCutchan FE. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses. 1995;11:1423–5. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- 41.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1999;73(1):152–60. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–20. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 43.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 44.Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol. 1984;179:125–42. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- 45.Eisenberg D, Weiss RM, Terwilliger TC. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc Natl Acad Sci U S A. 1984;81:140–4. doi: 10.1073/pnas.81.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cornette JL, Kemp BC, Margalit H, Spouge JL, Berzofsky JA, DeLisi C. Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. J Mol Biol. 1987;195:659–85. doi: 10.1016/0022-2836(87)90189-6. [DOI] [PubMed] [Google Scholar]
- 47.Cornette JL, Margalit H, Berzofsky JA, DeLisi C. Periodic variation in side-chain polarities of T-cell antigenic peptides correlates with their structure and activity. Proc Natl Acad Sci U S A. 1995;92:8368–72. doi: 10.1073/pnas.92.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 49.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–60. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 50.Wu S, Zhang Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35:3375–82. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012;80(7):1715–35. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro, progress and status in 2005. Nucleic Acids Res. 2005;33:D201–5. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuzio J, Pearson MN, Harwood SH, Funk CJ, Evans JT, Slavicek JM, et al. Sequence and analysis of the genome of a baculovirus pathogenic for Lymantria dispar. Virology. 1999;253:17–34. doi: 10.1006/viro.1998.9469. [DOI] [PubMed] [Google Scholar]
- 55.Friesen PD. Regulation of baculovirus early gene expression. In: Miller LK, editor. The Baculoviruses. New York: Plenum Press/Plenum Publishing Corporation; 1997. pp. 141–70. [Google Scholar]
- 56.Lu A, Miller LK. Regulation of baculovirus late and very late gene expression. In: Miller LK, editor. The Baculoviruses. New York: Plenum Press/Plenum Publishing Corporation; 1997. pp. 193–16. [Google Scholar]
- 57.Miele SAB, Garavaglia MJ, Belaich MN, Ghiringhelli PD. Baculovirus: molecular insights on their diversity and conservation. Int J Evol Biol. 2011;2011:379424. doi: 10.4061/2011/379424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garavaglia MJ, Miele SAB, Iserte JA, Belaich MN, Ghiringhelli PD. The ac53, ac78, ac101 and ac103 genes are newly discovered core genes in the Family Baculoviridae. J Virol. 2012;86:12069–79. doi: 10.1128/JVI.01873-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meijer M, Karimi-Busheri F, Huang TY, Weinfeld M, Young D. Pnk1, a DNA kinase/phosphatase required for normal response to DNA damage by gamma-radiation or camptothecin in Schizosaccharomyces pombe. J Biol Chem. 2002;277:4050–5. doi: 10.1074/jbc.M109383200. [DOI] [PubMed] [Google Scholar]
- 60.Reddy VS, Shlykov MA, Castillo R, Sun EE, Saier MH., Jr The Major Facilitator Superfamily (MFS) revisited. FEBS J. 2012;279:2022–35. doi: 10.1111/j.1742-4658.2012.08588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng F, Wang R, Fang M, Jiang Y, Xu X, Wang H, et al. Proteomics analysis of Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus identified two new occlusion-derived virus-associated proteins, HA44 and HA100. J Virol. 2007;81(17):9377–85. doi: 10.1128/JVI.00632-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hou D, Zhang L, Deng F, Fang W, Wang R, Liu X, et al. Comparative proteomics reveal fundamental structural and functional differences between the two progeny phenotypes of a baculovirus. J Virol. 2013;87(2):829–39. doi: 10.1128/JVI.02329-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kondo A, Maeda S. Host range expansion by recombination of the baculoviruses Bombyx mori nuclear polyhedrosis virus and Autographa californica nuclear polyhedrosis virus. J Virol. 1991;65(7):3625–32. doi: 10.1128/jvi.65.7.3625-3632.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maeda S, Kamita SG, Kondo A. Host range expansion of Autographa californica nuclear polyhedrosis virus (NPV) following recombination of a 0.6-kilobase-pair DNA fragment originating from Bombyx mori NPV. J Virol. 1993;67(10):6234–8. doi: 10.1128/jvi.67.10.6234-6238.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woo SD, Kim WJ, Kim HS, Jin BR, Lee YH, Kang SK. The morphology of the polyhedra of a host range-expanded recombinant baculovirus and its parents. Arch Virol. 1998;143(6):1209–14. doi: 10.1007/s007050050368. [DOI] [PubMed] [Google Scholar]
- 66.Kamita SG, Maeda S, Hammock BD. High-frequency homologous recombination between baculoviruses involves DNA replication. J Virol. 2003;77(24):13053–61. doi: 10.1128/JVI.77.24.13053-13061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ellis SE. New Pest Response Guidelines: Spodoptera. USDA/APHIS/PPQ/PDMP. 2004. [Google Scholar]
- 68.Jain M, Das RH. Nucleotide sequence and molecular characterization of the structural glycoprotein gp41 gene homologue of Spodoptera litura nucleopolyhedrosis virus (SpltNPV-I) Mol Biol Rep. 2004;31(4):231–9. doi: 10.1007/s11033-005-1405-x. [DOI] [PubMed] [Google Scholar]
- 69.Casmuz A, Juárez ML, Socías MG, Murúa MG, Prieto S, Medina S, et al. Revisión de los hospederos del gusano cogollero del maíz, Spodoptera frugiperda (Lepidoptera: Noctuidae) Rev Soc Entomol Argent. 2010;69:209–31. [Google Scholar]