

Abstract
Incretin-based peptides are effective therapeutics for treating type 2 diabetes mellitus (T2DM). Oxyntomodulin (OXM), a dual agonist of GLP-1R and GCGR, has shown superior weight loss and glucose lowering effects, compared to single GLP-1R agonists. To overcome the short half-life and rapid renal clearance of OXM, which limit its therapeutic potential, both lipid and PEG modified OXM analogs have been reported. However, these approaches often result in reduced potency or PEG-associated toxicity. Herein we report a new class of cross-linked OXM analogs that show increased plasma stability and higher potency in activating both GLP-1R and GCGR. Moreover, the extended in vivo half-life results in superior anti-hyperglycemic activity in mice compared to the wild-type OXM.
Obesity is a major risk factor for developing type 2 diabetes mellitus (T2DM). Oxyntomodulin (OXM), a 37-amino acid peptide hormone derived from proglucagon, is an attractive potential therapy for treatment of T2DM due to its multifaceted effects on glucose homeostasis, food intake and energy expenditure.(1) Remarkably, the weight loss and glucose lowering effects of OXM were found to be superior to those of the glucagon-like peptide-1 (GLP-1) receptor only agonists after infusion in preclinical models.(2, 3) However, the clinical application of OXM is limited by its short circulatory half-life; hence, PEG and lipid modified OXM analogs have been explored. While these conjugates have shown significantly longer circulatory half-lives, they often exhibit considerably reduced potency; as a result, relatively large quantities of the modified peptides are injected during their administration. These large doses have limited the exploration of alternative delivery technologies such as microneedles or nanoparticles.(4, 5)
Since incretin peptides generally bind to their cognate receptors in an α-helical conformation, modifications that stabilize α-helix should increase binding affinity to its receptors. Moreover, proteolytic stability may also be enhanced in a helical rather than an extended conformation. Indeed, a number of helix stabilization strategies have been developed (6–12) and successfully applied to various classes of bioactive peptides. We have previously developed simple disulfide- and biaryl-based cross-linking chemistries that span two turns of an α-helix (13). The resulting cross-linked peptides showed higher α-helicity and in the biaryl case, increased proteolytic stability and cell-permeability.(14–16) Herein, we report the application of this latter cross-linking chemistry to OXM to increase both its circulatory half-life and its potency toward GLP1R and GCGR.
OXM contains a C-terminal extension of glucagon and also exhibits high sequence homology with the incretin peptide, GLP-1 (Figure 1a). In order to facilitate biaryl cross-linking without disrupting receptor binding, residues that are solvent exposed and oriented on the same side of the helix, i.e., residues at i and i+7 positions, were replaced with D- and L-cysteine, respectively. Since the N-termini of these peptides are highly conserved and play a crucial role in activation of GLP-1 receptor (GLP-1R),(17–20) we focused on Cys substitutions at the C-terminus of OXM. Because neither structure-activity studies of alanine-substituted OXM variants, nor the structure of OXM bound to its receptor have been reported, we designed our cysteine mutants based on glucagon, which has been extensively studied by both mutagenesis and structural methods.(21) To predict which residues are solvent exposed, glucagon was superimposed with GLP-1 in the binding pocket of GLP-1R (Fig. 1b). Because of its high sequence identity with glucagon, we predicted that the N-terminus of OXM, OXM (1–29), will adopt the same bound conformation as the full-length glucagon. We also hypothesized that the octapeptide extension of OXM, OXM (30–37), will exhibit a type II β-helical turn.(22) Based on this homology model, we generated two OXM mutants with pairs of solvent-exposed D- and L-cysteine residues that are separated by 6 residues (R17 and Q24; D21 and N28), and which most likely not involved in direct binding to the extracellular domain of GLP-1R and GCGR.
Figure 1.

(a) Sequences of oxyntomodulin, glucagon, GLP-1 and exendin-4. The conserved N-terminal residues crucial for receptor activation are underlined. The sites for cysteine substitution and subsequent cross-linking on oxyntomodulin are colored. (b) Structural model of oxyntomodulin bound to the extracellular domain of GLP-1R (PDB code 3C59), with the cross-linking sites colored. The oxyntomodulin structure was modeled after the crystal structure of glucagon (PDB code: 1GCN) with the octapeptide extension shown as a dotted type II β-helical turn.(22)
We used the DPP-IV resistant OXM, OXM-1 (OXM with D-serine at 2nd position), as a template for cysteine substitution and subsequent sidechain cross-linking.(23) To assess how chemical modification affects receptor activation, we developed a cAMP response element (CRE) driven luciferase reporter for HEK293 cells overexpressing GLP-1R and GCGR receptors. The EC50 values of OXM-1 for GLP-1R and GCGR were 10 nM and 3 nM, respectively (Table 1), matching closely the reported values.(20) We then tested the activity of the linear di-cysteine substituted peptides 1 (R17 and Q24 are substituted with D and L-cysteine, respectively) and 2 (D21 and N28 are substituted with D and L-cysteine, respectively). Both 1 and 2 showed reduced potency for GLP-1R activation, but peptide 1 activated GCGR more potently than peptide 2. Hence, we proceeded to cross-link 1 with 4,4′-bis(bromomethyl)biphenyl (Bph), to generate peptide 3. Although cross-linking increased the activities toward both GCGR (16-fold) and GLP-1R (2-fold) compared to its linear counterpart, peptide 3 still exhibited substantially reduced activity compared to OXM-1. Since a D-amino acid in the middle of a helical peptide can cause helix distortion,(24) we substituted R17 with L-cysteine and generated peptide 4 (both R17 and Q24 are substituted with L-cysteine). Although we anticipated that L-Cys17 might have a less favorable geometry for crosslink formation, peptide 4 showed roughly 2-fold increase in GLP-1R and GCGR activities compared to its D,L counterpart, peptide 1.
Table 1.
Sequences of the modified oxyntomodulin analogs and their agonist activities in the activation of GLP-1R and GCGR using the cell-based luciferase reporter assaya
| Name | Sequence | Agonist activity (nM)
|
|
|---|---|---|---|
| GLP-1R | GCGR | ||
| OXM-1 | HsQGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA | 10 | 3 |
| 1 | HsQGTFTSDYSKYLDScRAQDFVCWLMNTKRNRNNIA | 1000 | 50 |
| 2 | HsQGTFTSDYSKYLDSRRAQcFVQWLMCTKRNRNNIA | >2000 | >1000 |
| 3 | HsQGTFTSDYSKYLDSc′RAQDFVC′WLMNTKRNRNNIAb | ~500 | 3 |
| 4 | HsQGTFTSDYSKYLDSCRAQDFVCWLMNTKRNRNNIA | ~500 | 30 |
| 5 | HsQGTFTSDYSKYLDECRAQDFVCWLMNTKRNRNNIA | ~400 | 20 |
| 6 | HsQGTFTSDYSKYLDEC′RAQDFVC′WLMNTKRNRNNIA | ~100 | 0.8 |
| 7 | HsQGTFTSDYSKYLDECAAKEFICWLMNTKRNRNNIA | 17 | 30 |
| 8 | HsQGTFTSDYSKYLDECAVRLFICWLMNTKRNRNNIA | 1 | 1000 |
| 9 | HsQGTFTSDYSKYLDEC′AAKEFIC′WLMNTKRNRNNIA | 0.2 | 0.7 |
| 10 | HsQGTFTSDYSKYLDEC′AVRLFIC′WLMNTKRNRNNIA | 56 | ~200 |
All peptides contain the unmodified N-termini and the amidated C-termini.
c′ and C′ denote the Bph-cross-linked D-cysteine and L-cysteine, respectively.
To further increase receptor activity, we next generated a chimera OXM by incorporating key binding residues from the GLP-1 agonists GLP-1 and exendin-4 (Ex-4). Since substitution of glucagon-Ser16 by Glu was reported to enhance GLP-1R activity without altering GCGR activity,(25) we synthesized peptide 5 by replacing Ser16 with Glu in peptide 4, and found modest enhancement in the GLP-1R and GCGR activities. Because incorporation of residues from exendin-4 or GLP-1 in the middle of the glucagon sequence significantly increases GLP-1R activity,(18) we substituted GLP-1 residues (AAKEFI) and exendin-4 residues (AVRLFI) in the middle of peptide 5 (RAQDFV) to afford peptides 7 and 8, respectively (Table 1). To our delight, peptides 7 and 8 showed >80-fold higher potency in GLP-1R activation, compared to peptide 5, although peptide 8 was less potent in GCGR activation (Table 1). Cross-linking of 7 led to 80- and 40-fold increases in activity for GLP-1R and GCGR, respectively, giving rise to a balanced subnanomolar dual agonist peptide 9 for potent activation of both receptors (EC50 = 0.2 nM for GLP-1R, and 0.7 nM for GCGR; Table 1).
Having identified the optimal di-cysteine-containing OXM sequence, we next modified peptide 7 using a panel of cysteine-reactive cross-linkers: Bpy (CL-2), Alk (CL-3), Phe (CL-4) and mBph (CL-5) (see Table 2 for structures).(26) To our satisfaction, cross-linked peptide 11 showed even more potent agonist activities in dual activation of GLP-1R and GCGR with EC50 values of 0.07 nM and 0.18 nM, respectively (Figure S2).
Table 2.
Structures and agonist activities of the various cross-linked analogs of peptide 7.
| OXM Sequences | Cross-linker structure | Agonist activity (nM)a
|
|
|---|---|---|---|
| GLP-1R | GCGR | ||
| 9 |
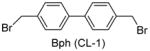
|
0.20 ± 0.05 | 0.74 ± 0.30 |
| 11 |
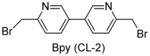
|
0.07 ± 0.01 | 0.18 ± 0.02 |
| 12 |
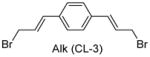
|
0.45 ± 0.03 | 1.00 ± 0.12 |
| 13 |
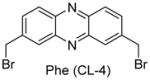
|
0.15 ± 0.02 | 0.24 ± 0.12 |
| 14 |
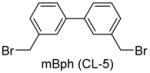
|
0.40 ± 0.02 | 0.84 ± 0.15 |
Luciferase reporter assay was performed three times to derive mean IC50 values and standard deviations.
To gain insight into structural basis for the increased activity after cross-linking, we compared the far-UV circular dichroism spectra of 9 and 11 to that of OXM-1. All three spectra showed local minima at 208 and 222 nm, indicating the presence of an α-helix (Figure S3a).(27) The percent helicity, ca. in the range of 20–23%, was found to be similar based on the [θ]222 values (Figure S3b), suggesting that the differences in agonist activity cannot be explained by percent helicity.(28) However, we observed a significant difference in the ratio of [θ]222/[θ]208, a measure of the relative amounts of 310- and α-helix in the conformational ensemble, as 9 and 11 showed ratios close to 1.0 indicating ideal α-helices (Figure S3b) whereas OXM-1 showed a ratio of 0.68.(29) Assuming the 310–helix represents an unproductive conformation, the lack of 310–helix from the conformational ensemble may be beneficial to the receptor binding, although other factors other than secondary structures may also affect the binding.(26, 30) For example, despite having similar CD spectra, peptide 11 has 3–4 fold greater efficacy in receptor activation than peptide 9 (Table 1). This difference can be explained by the presence of the pyridyl nitrogen in the Bpy structure, which may form a hydrogen bond with Glu-128 of the extracellular domain of GLP-1R (Figure S4).
To determine the half-lives of the OXM peptides, we performed pharmacokinetic (PK) studies in mice by injecting the peptides through either subcutaneous (s.c) or intravenous (i.v) routes. The peptide concentrations in the plasma at different time points were measured indirectly using the cell-based luciferase reporter assay (the detection limit for peptide concentration in mouse serum is ~ 10 nM). We attempted to use a LC-MS based method for analysis, but the peptide concentrations in mouse serum were below the limits of detection. The concentration of OXM-1 was negligible after 1 h when injected either i.v or s.c., as evidenced by no receptor activation (< 10 nM). The elimination half-lives of the intravenously injected peptides were found to be 0.1 h, 1.2 h and 1.4 h for OXM-1, 9 and 11, respectively (Figure 2a). When administered subcutaneously, the half-lives of OXM-1, 9 and 11 were found to be 0.6 h, 1.9 h and 1.9 h, respectively (Figure 2b). When the cell-based reporter assay was employed to determine the half-life of the peptide in vitro after incubation with freshly isolated mouse serum, a similar trend was observed (t1/2 = 5.4 h for OXM-1 vs. ≈ 13 h for 9 and 11) (Figure S5). The greater half-lives observed for the cross-linked OXMs in vivo may result from enhanced serum albumin binding by the cross-linked peptides as we have observed previously.(14) We suspect that the in vivo half-lives can be further improved by modifying the aryl cross-linker with a short PEG-fatty acid moiety.(31, 32)
Figure 2.

Chemical cross-linking extends OXM half-life and efficacy. In vivo stability of the OXM analogs after intravenous (a) and subcutaneous (b) injection of the peptides into mice (n = 3). The peptide concentrations in mouse plasma at the various times were determined using the GLP-1R activation assay. Assay was performed in triplicate. Half-lives of the OXM analogs were calculated by fitting the curve to either two-phase exponential decay (i.v) or one-phase exponential decay (s.c) in Prism 6.0. *No clear distinction was detected between the signal and the background. Cross-linked peptides 9 and 11 show greater activity in the oral glucose tolerance test in mice (n = 4). Mice were injected with the peptides (10 μg/mice) subcutaneously 4 hours prior to the glucose challenge. (c) The glucose concentrations in mouse blood were monitored for up to 150 minutes. (d) Bar graph showing the total amount of glucose in the mice obtained by measuring the area under curve (AUC).
Since OXM is known to reduce blood glucose levels in diabetic patients,(3) we evaluated the efficacy of the cross-linked OXM analogs in an oral glucose tolerance test (OGTT). As a positive control, Ex-4 significantly decreased the blood glucose level during the entire monitoring period (Figure 2c) and the area under curve (AUC) by 30% (Figure 2d), whereas OXM-1 did not exhibit any improvement over the vehicle. Peptides 9 and 11 significantly decreased blood glucose levels to 40 and 45%, respectively, which are greater than that of Ex4 (Figure 2c, 2d). The increased in vivo efficacies of the cross-linked peptides observed here likely result from both higher dual-agonist activities (Tables 1 and 2) and the extended in vivo half-lives (Figure 2a, 2b).
In summary, we have designed a class of chemically cross-linked OXM analogs that show balanced, sub-nanomolar activities in activating the GLP-1 and glucagon receptors. While the cross-linking only marginally increased peptide helicity, substantial improvements in dual-agonist activity as well as in vivo stability were obtained, which makes this approach appealing as other chemical modification strategies such as PEGylation can result in reduced OXM potency.(33) The combined high potency and enhanced in vivo stability resulted in greater efficacy in the oral glucose tolerance test, even at very low dosages (10 μg/mice), highlighting the potential of this technology to obviate the need for high-dose administration of the peptide drugs. We are currently evaluating the cross-linked OXM analogs in diet induced obesity models as well as exploring additional modification of the cross-linkers with lipid moieties to further enhance in vivo half-life.
Supplementary Material
Acknowledgments
This work was partially supported by the Pardee Foundation, the Oishei Foundation, and the National Institutes of Health (GM 085092) (to Q.L.).
Contributor Information
Peter G Schultz, Email: schultz@scripps.edu.
Qing Lin, Email: qinglin@buffalo.edu.
Weijun Shen, Email: wshen@calibr.org.
References
- 1.Kerr BD, Flatt PR, Gault VA. (d-Ser2)Oxm[mPEG-PAL]: A novel chemically modified analogue of oxyntomodulin with antihyperglycaemic, insulinotropic and anorexigenic actions. Biochem Pharmacol. 2010;80:1727–1735. doi: 10.1016/j.bcp.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Du X, Kosinski JR, Lao J, Shen X, Petrov A, Chicchi GG, Eiermann GJ, Pocai A. Differential effects of oxyntomodulin and GLP-1 on glucose metabolism. Am J Physiol Endocrinol Metab. 2012;303:E265–271. doi: 10.1152/ajpendo.00142.2012. [DOI] [PubMed] [Google Scholar]
- 3.Pocai A. Action and therapeutic potential of oxyntomodulin. Mol Metab. 2014;3:241–251. doi: 10.1016/j.molmet.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Della Rocca J, Liu D, Lin W. Are high drug loading nanoparticles the next step forward for chemotherapy? Nanomedicine. 2012;7:303–305. doi: 10.2217/nnm.11.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai B, Xia W, Bredenberg S, Engqvist H. Self-setting bioceramic microscopic protrusions for transdermal drug delivery. J Mater Chem B. 2014;2:5992–5998. doi: 10.1039/c4tb00764f. [DOI] [PubMed] [Google Scholar]
- 6.Houston ME, Jr, Gannon CL, Kay CM, Hodges RS. Lactam bridge stabilization of alpha-helical peptides: ring size, orientation and positional effects. J Pept Sci. 1995;1:274–282. doi: 10.1002/psc.310010408. [DOI] [PubMed] [Google Scholar]
- 7.Schafmeister CE, Po J, Verdine GL. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 8.Kawamoto SA, Coleska A, Ran X, Yi H, Yang CY, Wang S. Design of triazole-stapled BCL9 alpha-helical peptides to target the beta-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. Journal of medicinal chemistry. 2012;55:1137–1146. doi: 10.1021/jm201125d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson DY, King DS, Chmielewski J, Singh S, Schultz PG. General approach to the synthesis of short .alpha.-helical peptides. J Am Chem Soc. 1991;113:9391–9392. [Google Scholar]
- 10.Haney CM, Loch MT, Horne WS. Promoting peptide alpha-helix formation with dynamic covalent oxime side-chain cross-links. Chem Commun (Camb) 2011;47:10915–10917. doi: 10.1039/c1cc12010g. [DOI] [PubMed] [Google Scholar]
- 11.Jo H, Meinhardt N, Wu Y, Kulkarni S, Hu X, Low KE, Davies PL, DeGrado WF, Greenbaum DC. Development of alpha-helical calpain probes by mimicking a natural protein-protein interaction. J Am Chem Soc. 2012;134:17704–17713. doi: 10.1021/ja307599z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson LM, Barrick S, Hager MV, McFedries A, Homan EA, Rabaglia ME, Keller MP, Attie AD, Saghatelian A, Bisello A, Gellman SH. A Potent α/β-Peptide Analogue of GLP-1 with Prolonged Action in Vivo. J Am Chem Soc. 2014;136:12848–12851. doi: 10.1021/ja507168t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.David Y, Jackson DSK, Chmielewski Jean, Sunil Singh PGS. General Approach to the Synthesis of Short a-Helical Peptides. J Am Chem Soc. 1991;113:9391–9392. [Google Scholar]
- 14.Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang HG, Lin Q. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J Am Chem Soc. 2012;134:14734–14737. doi: 10.1021/ja306864v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muppidi A, Wang Z, Li X, Chen J, Lin Q. Achieving cell penetration with distance-matching cysteine cross-linkers: a facile route to cell-permeable peptide dual inhibitors of Mdm2/Mdmx. Chem Commun (Camb) 2011;47:9396–9398. doi: 10.1039/c1cc13320a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muppidi A, Zhang H, Curreli F, Li N, Debnath AK, Lin Q. Design of antiviral stapled peptides containing a biphenyl cross-linker. Bioorg Med Chem Lett. 2014;24:1748–1751. doi: 10.1016/j.bmcl.2014.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Druce MR, Minnion JS, Field BC, Patel SR, Shillito JC, Tilby M, Beale KE, Murphy KG, Ghatei MA, Bloom SR. Investigation of structure-activity relationships of Oxyntomodulin (Oxm) using Oxm analogs. Endocrinology. 2009;150:1712–1722. doi: 10.1210/en.2008-0828. [DOI] [PubMed] [Google Scholar]
- 18.Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, Findeisen H, Bruemmer D, Drucker DJ, Chaudhary N, Holland J, Hembree J, Abplanalp W, Grant E, Ruehl J, Wilson H, Kirchner H, Lockie SH, Hofmann S, Woods SC, Nogueiras R, Pfluger PT, Perez-Tilve D, DiMarchi R, Tschop MH. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5:749–757. doi: 10.1038/nchembio.209. [DOI] [PubMed] [Google Scholar]
- 19.Al-Sabah S, Donnelly D. A model for receptor–peptide binding at the glucagon-like peptide-1 (GLP-1) receptor through the analysis of truncated ligands and receptors. Br J Pharmacol. 2003;140:339–346. doi: 10.1038/sj.bjp.0705453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pocai A, Carrington PE, Adams JR, Wright M, Eiermann G, Zhu L, Du X, Petrov A, Lassman ME, Jiang G, Liu F, Miller C, Tota LM, Zhou G, Zhang X, Sountis MM, Santoprete A, Capito E, Chicchi GG, Thornberry N, Bianchi E, Pessi A, Marsh DJ, SinhaRoy R. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58:2258–2266. doi: 10.2337/db09-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasaki K, Dockerill S, Adamiak DA, Tickle IJ, Blundell T. X-ray analysis of glucagon and its relationship to receptor binding. Nature. 1975;257:751–757. doi: 10.1038/257751a0. [DOI] [PubMed] [Google Scholar]
- 22.Aumelas A, Audousset-Puech MP, Heitz A, Bataille D, Martinez J. 1H n.m.r. conformational studies on the C-terminal octapeptide of oxyntomodulin, a beta-turn locked by a salt bridge. International journal of peptide and protein research. 1989;34:268–276. doi: 10.1111/j.1399-3011.1989.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 23.Santoprete A, Capito E, Carrington PE, Pocai A, Finotto M, Langella A, Ingallinella P, Zytko K, Bufali S, Cianetti S, Veneziano M, Bonelli F, Zhu L, Monteagudo E, Marsh DJ, Sinharoy R, Bianchi E, Pessi A. DPP-IV-resistant, long-acting oxyntomodulin derivatives. J Pept Sci. 2011;17:270–280. doi: 10.1002/psc.1328. [DOI] [PubMed] [Google Scholar]
- 24.Hong SY, Oh JE, Lee KH. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem Pharmacol. 1999;58:1775–1780. doi: 10.1016/s0006-2952(99)00259-2. [DOI] [PubMed] [Google Scholar]
- 25.Patterson JT, Day JW, Gelfanov VM, DiMarchi RD. Functional association of the N-terminal residues with the central region in glucagon-related peptides. J Pept Sci. 2011;17:659–666. doi: 10.1002/psc.1385. [DOI] [PubMed] [Google Scholar]
- 26.Muppidi A, Doi K, Ramil CP, Wang HG, Lin Q. Synthesis of cell-permeable stapled BH3 peptide-based Mcl-1 inhibitors containing simple aryl and vinylaryl cross-linkers. Tetrahedron. 2014;70:7740–7745. doi: 10.1016/j.tet.2014.05.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JP, Liu C, Li T, Zhu G, Li X. Development of stapled helical peptides to perturb the Cdt1–Mcm6 interaction. J Pept Sci. 2015;21:593–598. doi: 10.1002/psc.2779. [DOI] [PubMed] [Google Scholar]
- 29.Toniolo C, Polese A, Formaggio F, Crisma M, Kamphuis J. Circular Dichroism Spectrum of a Peptide 310-Helix. J Am Chem Soc. 1996;118:2744–2745. [Google Scholar]
- 30.Bezer S, Matsumoto M, Lodewyk MW, Lee SJ, Tantillo DJ, Gagne MR, Waters ML. Identification and optimization of short helical peptides with novel reactive functionality as catalysts for acyl transfer by reactive tagging. Org Biomol Chem. 2014;12:1488–1494. doi: 10.1039/c3ob41421c. [DOI] [PubMed] [Google Scholar]
- 31.Pollaro L, Heinis C. Strategies to prolong the plasma residence time of peptide drugs. MedChemComm. 2010;1:319–324. [Google Scholar]
- 32.Fishburn CS. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97:4167–4183. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- 33.Bianchi E, Carrington PE, Ingallinella P, Finotto M, Santoprete A, Petrov A, Eiermann G, Kosinski J, Marsh DJ, Pocai A, SinhaRoy R, Pessi A. A PEGylated analog of the gut hormone oxyntomodulin with long-lasting antihyperglycemic, insulinotropic and anorexigenic activity. Bioorg Med Chem. 2013;21:7064–7073. doi: 10.1016/j.bmc.2013.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.