

Abstract
Heat shock protein 90 (HSP90) is a molecular chaperone that is up-regulated in cancer and is required for the folding of numerous signaling proteins. Consequently, HSP90 represents an ideal target for the development of new anti-cancer agents. The human HSP90 isoform, glucose-regulated protein 94 (GRP94), resides in the endoplasmic reticulum and regulates secretory pathways, integrins, and Toll-like receptors, which contribute to regulating immunity and metastasis. However, the cellular function of GRP94 remains underinvestigated. We report that GRP94 knockdown cells are defective in intracellular transport and, consequently, negatively impact the trafficking of F-actin toward the cellular cortex, integrin α2 and integrin αL toward the cell membrane and filopodia, and secretory vesicles containing the HSP90α-AHA1-survivin complex toward the leading edge. As a result, GRP94 knockdown cells form a multipolar spindle instead of bipolar morphology and consequently manifest a defect in cell migration and adhesion.
Keywords: actin, cell polarity, heat shock protein 90 (HSP90), intracellular trafficking, survivin, aha1, grp94, integrin
Introduction
Cell migration is the process by which cells move during developmental morphogenesis, tissue repair and regeneration, and cancer metastasis. During metastasis, cancer cells polarize in response to chemokines or environmental cues originating from the extracellular matrix (ECM).2 With the assistance of microtubules, the microtubule-organizing center and the Golgi apparatus orient toward the direction of migration to aid vesicular transport toward the leading edge (1, 2). At the leading edge of the polarized cell, actin polymerization occurs rapidly to form lamellipodia, from which slender cytoplasmic projections called filopodia develop (3–5). These filopodia play roles in sensing, migration, cell-cell interactions, and adhesion (6). Adhesion of filopodia and lamellipodia to the ECM occurs through integrin receptors. Integrin receptors are a large superfamily of heterodimeric receptors that bind ECM ligands or cognate ligands on adjacent cells. The macromolecular assembly through which mechanical force and regulatory signals are transmitted between the ECM and a neighboring cell is described as a focal adhesion, which is composed of integrins, adapter proteins, and signaling molecules (7). The focal adhesion complex undergoes endo-exocytic cycles, and disruption of the endosomal transport or exocytic fusion of recycling vesicles affects cell polarity and migration (8, 9).
The endoplasmic reticulum (ER) is the manufacturing site of newly synthesized proteins that are folded and targeted to their destination. The ER works continuously with the outer layer of the nuclear envelope but separate from the Golgi apparatus. Protein transport from the ER to Golgi occurs through coatomer I- and II-coated vesicles, with final transport to the plasma membrane occurring via endosomes. Cell migration requires the transport of proteins to the leading edge to establish cell polarity, filopodia and lamellipodia formation, adhesion, signaling, and translocation. In addition, several secretory proteins are required at the leading edge to break down the ECM that aids in cell motility. GRP94 is the ER-resident HSP90 molecular chaperone that modulates protein folding, processing, and secretion. Because protein folding in the ER is an essential process, loss of GRP94 function is essential to embryonic development (10). GRP94 deletion also affects the trafficking of Toll-like receptors and integrin signaling in immune cells (11, 12). More recently, studies in liver-specific GRP94 knock-out mice revealed that GRP94 is a pro-oncogenic chaperone (13). Although there are several reports that highlight the biological functions of GRP94 using organ-specific knock-out mice, the role of GRP94 in cellular biology remains relatively underinvestigated.
In this study, the effect of GRP94 on cell migration was assessed using a stable GRP94 knockdown in the highly metastatic prostate cancer cell line, PC3-MM2. GRP94 knockdown cells were defective in cell migration due to a deficit in migration directionality. The GRP94 knockdown cells manifested an unusual morphology that formed multipolar spindles instead of bipolar spindles and were unable to form filopodia. Proteomic analysis revealed that GRP94 knockdown resulted in a deficit in proteins that regulate cellular transport processes. Further investigation determined that GRP94 is required for the transport of F-actin, fascin, integrin α2, and integrin αL toward the cell surface. The GRP94 knockdown cells were also defective in the transport of secretory vesicles containing HSP90α-AHA1-survivin to the leading edge of the migrating cells. Consequently, the GRP94 knockdown cells migrated randomly, were deficient in cell-matrix adhesion, and were unable to form three-dimensional spheroidal structures in vitro. Collectively, this work provides novel mechanistic insight into the importance of GRP94 in regulating cancer cell migration.
Experimental Procedures
Chemicals, Reagents, and Plasmids
The novobiocin analogue, KU-135, was synthesized as described previously, and purity was verified as >95% by NMR and mass spectrometry (14). Recombinant human soluble TRAIL was a kind gift from Dr. Thomas Sayers (National Institutes of Health). Staurosporine was obtained from Sigma. Staurosporine, KU-135, and TRAIL were dissolved in DMSO and stored at −20 °C until use. AHA1-GFP plasmid was obtained from Origene (Rockville, MD) (catalogue no. RG201782). Canine GRP94-KDEL-GFP plasmid was a kind gift from Dr. Yair Argon (15). The plasmids were transfected into PC3-MM2 wild type and GRP94 knockdown cell lines using Lipofectamine 2000 as described by the manufacturer. The GRP94 shRNA (RHS4696-99638599), (5′-GGAAAGAATCAAGGAGG-3′) and scrambled shRNA plasmids were obtained from Open Biosystems.
Antibodies
The following antibodies were used for Western blotting and/or co-immunoprecipitation: rabbit anti-HSP90α RB-119-P (Neomarkers/Thermo Scientific, Waltham, MA); rabbit anti-phospho-HSP90α Thr-5/7 (catalog no. 3488) and rabbit anti-survivin (catalog no. 2808) (Cell Signaling Technology, Danvers, MA); goat anti-HSP90β (catalog no. sc1057), rat anti-GRP94 (catalog no. sc32249), goat anti-integrin αL (catalog no. sc6609), rabbit anti-SYNE2 (catalog no. sc99066), and rabbit anti-actin (catalog no. sc1616-R) (Santa Cruz Biotechnology, Inc.); mouse anti-TRAP1 (catalog no. 612344) (BD Biosciences); rabbit anti-AHA1 (catalog no. ab83036), mouse anti-AHA1 (catalog no. ab56721), mouse anti-RAC (catalog no. ab13048) (detects RAC1 with slight cross-reactivity with RAC2), and rabbit anti-integrin α2 (catalog no. ab181548) (Abcam, Cambridge, MA); rabbit anti-RAB3GAP1 (catalog no. SAB4500914) (Sigma); phalloidin 647 (catalog no. A22287) (Invitrogen); mouse anti-fascin (catalog no. MAB3582) (Millipore, Billerica, MA); and Oregon green wheat germ agglutinin (catalog no. W6748) (Life Technologies, Inc.).
Cell Culture and Stable Cell Lines
The GRP94 shRNA construct along with the control (nonspecific shRNA) were obtained as glycerol stocks, and the plasmids were recovered with a Qiagen plasmid kit (MIDI prep) using the manufacturer's protocol. The plasmids containing the nonspecific and GRP94-specific shRNA were packaged into third generation lentivirus particles (Clontech) and concentrated using the Lenti-X concentrator kit. The concentrated lentiviral particles were transduced according to the protocol mentioned in the Open Biosystems manual and then selected for puromycin resistance. The puromycin-resistant control and GRP94 knockdown cells were monitored by the TurboRFP signal, and knockdown was verified by Western blotting analysis. PC3-MM2 cell lines with a stable knockdown of HSP90α and HSP90β have been reported earlier (16). The PC3-MM2 cell lines were maintained in DMEM supplemented with 10% FBS, streptomycin, and penicillin at 37 °C, 5% CO2. The stable HSP90 isoform-specific knockdown cells were cultured as above but with the addition of 5 μg/ml puromycin.
Western Blotting Analysis
The various cell lines were harvested in cold phosphate-buffered saline (PBS) and lysed on ice for 1 h in mammalian protein extraction reagent (Pierce) containing protease and phosphatase inhibitor mixtures. Cancer patients' tissue samples were obtained from the University of Kansas Medical Center. Approximately 5 mg of the tissue samples were homogenized in ∼300 μl of mammalian protein extraction reagent lysis buffer, and the lysates were clarified at 14,000 × g for 10 min at 4 °C. Protein concentrations were determined using the Pierce BCA protein assay kit per the manufacturer's instructions. Equal amounts of protein (2.5–20 μg) were electrophoresed under reducing conditions (8% polyacrylamide gel), transferred to a PVDF membrane, and immunoblotted with the corresponding specific antibodies. Membranes were incubated with an appropriate horseradish peroxidase-labeled secondary antibody, developed with a chemiluminescent substrate and visualized.
Co-immunoprecipitation
PC3-MM2 cell lines were plated in 10-cm cell culture dishes or T25 flasks and allowed to grow to ∼80% confluence. PC3-MM2 cell lines were untreated or received DMSO (0.1%) or the indicated drug treatment. After drug treatment, the PC3-MM2 cell lines were harvested in lysis buffer containing 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.1% Nonidet P-40, and protease and phosphatase inhibitor mixtures. Lysates were clarified by centrifugation at 10,000 × g, and protein concentration was determined using the BCA assay. For co-immunoprecipitation, 500 μg of total protein was diluted to a 500-μl total volume in lysis buffer and incubated with 10 μl of primary antibody overnight at 4 °C with rocking. Immune complexes were captured with 30 μl of DynaBeads Protein G (Invitrogen) for 3 h with rocking at 4 °C. Protein G bead complexes were washed three times with ice-cold lysis buffer, and the samples were boiled and subjected to SDS-PAGE and Western blotting analysis.
Immunofluorescence Analysis
For cell imaging, 1-μm Slide 8-well ibidiTreat IBIDITM glass slides were used. PC3-MM2 cells were fixed with freshly made 4% (w/w) paraformaldehyde in PBS for 15 min, permeabilized with 0.1% (w/w) Tween 20 in PBS for 5 min, and quenched with 0.1% (w/w) sodium borohydride for 5 min. The sections were blocked with 3% (w/w) BSA in PBS for 1 h and incubated with the primary antibody at a 1:100 concentration in 1% BSA in PBS overnight, prior to incubation with secondary antibody conjugated with Alexa Fluor 488 or 568 for 3 h. The sections were counterstained with DAPI and/or with phalloidin to visualize DNA and F-actin, respectively. The wells were washed three times with PBS after each step. Confocal images were acquired using a custom epifluorescent/confocal microscope composed of the following components: an Olympus IX81 inverted spinning disc confocal microscope base (Olympus America, Center Valley, PA), a Prior microscope stage for automated image acquisition (Prior Scientific, Rockland, MA), an Olympus ×60 oil immersion objective for confocal images (Olympus) and a Hamamatsu (Hamamatsu, Japan) electron-multiplying charge-coupled device camera. Images were captured using the SlideBook acquisition and analysis software (Intelligent Imaging Innovations, Denver, CO). Images were collected with 8–10 image stacks with a 0.3-μm step size through the cells. Images were processed using ImageJ software (National Institutes of Health, Bethesda, MD).
Cell Migration Assay
The cells were seeded in a 24-well plate in complete medium and allowed to form a monolayer. After monolayer formation, a scratch was introduced with a sterile 0.1–10-μl pipette tip. The medium was replaced with fresh medium in the absence or presence of the indicated drug concentrations. Photomicrographs were taken at different time points with an Olympus IX71 microscope using a ×10 air lens and CellSans Dimensions software. The images were processed with ImageJ software. The average horizontal distance between the scratches was measured at various time points.
Live Cell Migration Assay
The control (nonspecific shRNA), GRP94 knockdown, or AHA1-GFP transfected cells were seeded in 1-μm Slide 8-well ibidiTreat IBIDI glass slides to form a monolayer. After monolayer formation, the scratch was introduced with a sterile 0.1–10-μl pipette tip. Live cell images were acquired using a custom epifluorescent microscope composed of the following components: an Olympus IX81 inverted base (Olympus America, Center Valley, PA), a Prior microscope stage for automated image acquisition, a Sutter automated optical filter wheel (Sutter Instrument, Novato, CA), an Olympus ×40 long working distance air objective (Olympus), and a Hamamatsu ORCA-Flash version 4.0 complementary metal-oxide semiconductor camera.
Image Quantification
Image Tracking Analysis
Images were captured and processed using the SlideBook acquisition and analysis software. The image tracking analysis was performed by using the manual tracking plugin in ImageJ. The ImageJ manual tracking data were then used in the IBIDI Chemotaxis and Migration Tool to generate the graphs.
Cell Morphology Analysis
The morphology of the cells was measured quantitatively using the CellProfiler program. The live cell bright field images of the control and GRP94 knockdown cells were used to plug in to the CellProfiler software, and the characteristics of the cell phenotypes were measured according to previously published literature (17).
Co-localization Analysis
The co-localization of two proteins in immunofluorescent images were measured by using JACoP (Just Another Co-localization Plugin) in the ImageJ software, and Pearson's co-efficient (r) was measured (18).
Cell Fractionation for Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) Analysis
Lysine- and arginine-deficient DMEM was purchased from Thermo Fisher Scientific. Isotopically enriched (>98%) l-Lys (12C6,14N2,2H4 (K4) and 13C6,15N2 (K8)) and l-Arg (13C6,14N4) (R6) and 13C6,15N4 (R10)) were obtained from Sigma-Isotec. For proteomic analysis, the control cells, HSP90β knockdown cells, and GRP94 knockdown cells were grown in DMEM SILAC medium supplemented with 10% dialyzed FBS, streptomycin, penicillin, and light (125 mg/liter K0, 84 mg/liter R0), medium (K4/R6), or heavy (K8/R10) labeled medium, respectively. The cells were passaged at least 5 times to ensure complete incorporation of labeled amino acids into the cells. Five 15-cm plates of each cell line were expanded in K0/R0-, K4/R6-, or K6/R10-containing medium before harvesting.
The cells were trypsinized, washed twice with ice-cold PBS, and resuspended in 10 ml of isolation buffer containing 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 0.2 m d-mannitol, 0.05 m sucrose, 0.5 mm sodium orthovanadate, 1 mm sodium fluoride, and protease inhibitor mixture (19). The cells were homogenized with the aid of a Teflon pestle, and lysis was confirmed microscopically. After sedimenting the cell debris, the protein concentration of each lysate was measured in quadruplicate using the BCA protein assay and bovine serum albumin as the standard. The coefficient of variation was determined for each set of quadruplicate measures, and if the variability exceeded 5%, the protein assay was repeated for that set of samples. The samples were then mixed together in a 1:1:1 mass ratio, yielding 24–30 mg of protein.
The cell lysates were centrifuged at 8000 × g for 10 min, and the crude mitochondrial pellet was washed twice in isolation buffer and frozen. The reserved supernatant was centrifuged at 14,000 × g to isolate a microsomal fraction, and the remaining supernatant was concentrated overnight by TCA precipitation; the resulting protein fraction was frozen. The microsomal pellet was washed as above and dissolved in a minimum amount of isolation buffer and subjected to SDS-PAGE.
Mass Spectrometry and Protein Identification
Proteins were identified following one-dimensional SDS-PAGE coupled to reverse phase HPLC with introduction of the effluent into a linear quadrupole ion trap Fourier transform ion cyclotron resonance tandem mass spectrometer (20).
About 75–100 μg of protein was fractionated by SDS-PAGE, the proteins were visualized by staining the gel, and the lane was cut into 13 sections for in-gel tryptic digestion. The gel pieces were placed in silanized microcentrifuge tubes and destained with 100 mm ammonium bicarbonate in 50% acetonitrile (21). Following reduction (10 mm dithiothreitol at 55 °C for 1 h) and alkylation (55 mm iodoacetamide for 30 min in the dark at room temperature), the gel pieces were washed with 100 mm ammonium bicarbonate in 50% acetonitrile, dehydrated with 100% acetonitrile, and dried. The gel pieces were rehydrated on ice in a minimal volume of 25 mm ammonium bicarbonate, pH 7.5, containing 12.5 ng/μl Trypsin Gold (Promega Corp., Madison, WI), covered with a sufficient volume of 25 mm ammonium bicarbonate, pH 7.5, and the proteins were digested overnight at 37 °C. The samples were briefly centrifuged, the trypsin was inactivated by the addition of formic acid to a final concentration of 5%, and the supernatant was used to provide two technical duplicates for LC-MS/MS analysis.
The chromatographic and mass spectrometric conditions were as described in our previous reports (22, 23). Raw data were processed using MaxQuant software, version 1.5.0.12 (Max Planck Institute of Biochemistry) (24) with an integrated Andromeda algorithm for protein identification (25). Spectra were searched against a concatenated forward-reverse protein sequence database composed from Uniprot Human_2013_01 (68,079 sequences) appended with common contaminants. See supplemental Table 1 to view all of the identification and quantification parameters used in the analysis. The isotopic multiplicity was set to consider the K4/R6 and K8/R10 isotope combinations, and quantification required at least two peptides.
Three-dimensional Cell Culture Assay
PC3-MM2 cells pretreated for 24 h with DMSO, or the drugs were used for the three-dimensional cell culture assays. Three-dimensional gel chambers were made using Matrigel (BD Bioscience) in DMEM supplemented with 10% FBS, streptomycin, and penicillin, with or without the drugs dissolved in DMSO. Approximately 5000 PC3-MM2 drug-treated cells were added in each well in overlay medium containing 2% Matrigel and 5 ng/ml EGF (Promega) and incubated for 5 days. Photomicrographs were taken after 5 days with an Olympus IX71 microscope using a ×10 air lens with CellSans Dimensions software. The images were processed with ImageJ software.
Results
GRP94 Knockdown Cells Are Defective in Cell Migration Due to a Lack of Migration Directionality
Cell motility/migration is critical to metastasis. In vitro cell migration assays and live cell microscopy with both control and GRP94 knockdown cells were performed to investigate the role of GRP94 in the cell migration process. Whereas wild type and control cells migrated toward the wound and closed the gap within 24 h, the GRP94 knockdown cells manifested a defect in cell migration and were unable to move efficiently toward the wound (Fig. 1A). Supplemental Videos 1 and 2 and Fig. 1A show that the control cells exhibit a bipolar morphology and formed filopodia toward the direction of migration. However, the GRP94 knockdown cells exhibited a round morphology during translocation/retraction and produced shorter projections throughout the cell membrane.
FIGURE 1.

Cell migration and migration behavior pattern of GRP94 knockdown cells. A, cell migration montage of the control and GRP94 KD cells. Fluorescent photomicrographs of the control and GRP94 KD cells were taken at 15-min intervals for 19 h of the cell migration/wound healing assay. Results are representative of three independent biological replicates of wound healing assays per cell line. Time stamp, hh:mm:ss; scale bar, 50 μm. B, aggregated trajectories of individual control and GRP94 KD cells migrating for 5 h toward the wound center. Cell tracking analyses were performed by using 20 cells from each condition. C, Accumulated distance, total distance traveled; Euclidean distance, ordinary/straight line distance between the starting and the final distance of individual control and GRP94 KD cells. D, live cell imaging of GRP94 KD and GRP94-KDEL-GFP (GRP94 revertant) cells. E, cell migration assay of the control (RFP), GRP94 KD (RFP), and GRP94 revertant (RFP and GFP) cells. Representative fluorescent live cell images at the 0 and 24 h time points of three independent biological replicates of cell migration assay are shown.
The whole-cell motility behaviors were analyzed from long time lapse videos by tracking the movement of individual control or GRP94 knockdown cells toward the wound. Only independent cells moving into the wound were tracked because interactions with neighboring cells could complicate motility. Tracking studies yielded data for accumulated (total distance traveled) and Euclidean distance (straight line distance between the two points) (Fig. 1B). Comparisons of these data indicate that the GRP94 knockdown cells traveled ∼3-fold less total distance (Fig. 1B), but the Euclidean distance was ∼21-fold less than control cells during the same time. The control and GRP94 knockdown cells were similarly motile, and the trajectory plot shows that control cells moved toward the scratch. However, GRP94 knockdown cells moved in random directions and consequently did not travel toward the wound (Fig. 1C). These data suggest that the GRP94 knockdown cells are unable to sustain the directionality of movement. Consistent with the quantitative analyses, video observations of cell movement showed that the mutant cells exhibit a reduced ability to maintain the leading edge.
Rescue experiments were performed to validate the effects of the GRP94 knockdown by introducing a canine GRP94-GFP-KDEL plasmid into the GRP94 knockdown cells. Fig. 1D is a fluorescent live cell image of a GRP94 knockdown cell expressing the GRP94-GFP with the ER marker KDEL (15). The rescue strain along with the control and GRP94 knockdown cells were used in the cell migration assay, and expression of GRP94-KDEL-GFP enabled the cells to migrate into the wound and completely close the wound gap in 24 h, similar to the control cells. As expected, the non-transfected GRP94 knockdown cells remained defective in cell migration (Fig. 1E).
GRP94 Knockdown Cells Are Defective in Bipolar Spindle-shaped Morphology
The morphology of control and GRP94 knockdown cells was studied by immunostaining with wheat germ agglutinin and live cell imaging. Control cells were mainly bipolar and produced a spindle-shaped morphology, whereas GRP94 knockdown cells exhibited a multipolar morphology with multiple edges (Fig. 2A). The morphological characteristics of the cells were measured using CellProfiler software. The compactness, minimum Feret diameter, and mean radius of GRP94 knockdown cells were significantly different from the control cells. The compactness of a cell is the variance of the radial distance of the object's pixels from the centroid divided by the area; a rounder cell is more compact. GRP94 knockdown cells are more compact than the control cells. Similarly, minimum Feret diameter is the minimum distance between two parallel lines tangent on either side of the object. A bipolar spindle-shaped cell will have lower minimum Feret diameter than a multipolar round shaped cell. Control cells had a lower minimum Feret diameter than the GRP94 knockdown cells. The mean radius is the mean distance of any pixel in the object to the closest pixel outside of the object. The mean radius of a circular object will be higher than a spindle-shaped cell. Quantification of the morphology of the control and GRP94 knockdown cells supports our observation that the control cells are bipolar and longer, whereas the GRP94 knockdown cells are multipolar and rounder (Fig. 2B).
FIGURE 2.

Morphology of GRP94 KD cells. A, GRP94 KD cells form multipolar spindles. Shown are representative bright field live cell and immunofluorescent images of the control and GRP94 KD cells. Three independent biological replicates of the cell spreading assay were performed for each condition. Control and GRP94 KD cells were fixed and stained with Oregon green wheat germ agglutinin. B, the morphology of the cell shape was measured by CellProfiler analysis, and the compactness, minimum Feret diameter, and mean radius of the control and GRP94 KD cells were quantified. At least 100 cells from each condition were used for CellProfiler analysis. C, live cell imaging (bright field and GFP) of the GRP94 revertant cells. The bipolar morphology of the GRP94 revertant is shown by the white arrow. D, time lapse montage showing the spreading morphology of control, GRP94 KD, and GRP94 revertant cells on IBIDI-coated μ-slides. Photomicrographs of the control and GRP94 KD cells were taken at 5-min intervals for 10 h. Time stamp, hh:mm:ss; scale bar, 10 μm. At least three independent technical replicates of time lapse microscopy of cell spreading assays were used for analysis.
The canine GRP94-KDEL-GFP plasmids were transfected in the GRP94 knockdown cells, and the morphology was determined. The GFP-positive cells were bipolar, whereas the GFP-negative cells were multipolar/rounder in shape, clearly demonstrating the impact of GRP94 on cell morphology (Fig. 2C).
Because the morphologies of the cells are heterogeneous and depend on whether the cells are actively moving, spreading cell assays were performed to investigate formation of the bipolar morphology. Cells were detached by trypsinization and allowed to respread on IBIDI-treated 15-μm slide wells, and cell morphology was recorded by time lapse imaging (Fig. 2D and supplemental Videos 3–5). The videos and the time lapse images of representative control, GRP94 knockdown, and GRP94 knockdown + GRP94-KDEL-GFP cells showed the control and revertant cells to form filopodia, lamellipodia, and a bipolar spindle. However, GRP94 knockdown cells formed shorter projections throughout the cell membrane and were unable to form a bipolar morphology (Fig. 2D and supplemental Videos 3–5).
GRP94 Knockdown Cells Are Defective in Protein Transport Process
The above results demonstrate that GRP94 is required for cell migration and maintaining a bipolar spindle shape morphology. Because GRP94 is an ER-resident HSP90 isoform and affects mainly secretory pathways, the cells were fractionated, and the microsomal fraction was investigated by quantitative proteomic analysis (23). HSP90β is the cytosolic human HSP90 isoform that is constitutively expressed and is responsible for the maturation and degradation of proteins required for normal cellular maintenance (26). Therefore, the HSP90β KD cells were used as a positive control to identify selective effects of GRP94 knockdown. Purity of the cell organelle fractionation was assessed by Western blotting analysis. As expected, HSP90α and HSP90β were detected in the cytosolic fraction; GRP94 and TRAP1 were primarily in the microsomal and mitochondrial fractions, respectively (Fig. 3B).
FIGURE 3.

Proteomic analysis of GRP94 KD cells. A, overrepresented pathways of the significantly down-regulated proteins in HSP90β or GRP94 knockdown cells compared with the control cells (corrected p value <0.0001). Two technical replicates were used for the proteomic analysis. B, representative Western blots of HSP90α, HSP90β, GRP94, TRAP-1, integrin α2, integrin αL, RAC1, LAMP1, RAB2, SYNE2, and VAMP2. Actin was used as loading control. Two independent biological replicates were used for the Western blotting analyses. C, the control and GRP94 KD cells were divided into microsomal (MIC), mitochondrial (Mito), and cytoplasmic (Cyto) fractions, and Western blotting analysis was performed. Representative Western blots show the levels of HSP90α, HSP90β, GRP94, TRAP-1, RAC1, VAMP2, LAMP1, RAB2, SYNE2, and actin. The level of HSP90β serves as the control. Three independent biological replicates were used to verify the proteomics data. D, association of GRP94 with RAC1, VAMP2, LAMP1, RAB2, and SYNE2. GRP94 was co-immunoprecipitated from the wild type PC3-MM2 cells, and the levels of GRP94, RAC1, VAMP2, LAMP1, RAB2, and SYNE2 were analyzed. Two independent biological replicates were used to verify the protein-protein interaction.
The proteomic analysis identified a total of 527 proteins in the microsomal fraction, of which 409 were quantified by two or more peptides (supplemental Table 1). As anticipated, GRP94 was down-regulated by ∼80% in the microsomes of GRP94 KD cells, whereas in the HSP90β KD cells, this protein was not decreased in the microsomal fraction. Similarly, HSP90β was down-regulated in the HSP90β KD cells while unaffected in the GRP94 KD cells, validating selectivity of the knockdown (supplemental Table 1). The normalized protein ratios (HSP90β KD (M)/control (L) or GRP94 knockdown H)/control (L)) were log 2-transformed, and proteins that had a log 2 ratio <−1 were considered significantly down-regulated (Table 1) and were submitted to Ingenuity Pathway Analysis to determine overrepresented pathways. Comparison of the two cell lines found that pathways linked to integrin, Rac, paxillin, and Pak signaling as well as actin motility and actin regulation by Rho family of GTPases were more affected (corrected p value <0.0001) in GRP94 KD cells compared with the HSP90β KD cells (Fig. 3A). Gene ontology analysis further revealed that of 45 down-regulated proteins in GRP94 KD cells, 25 represented cellular localization processes, and 20 represented protein transport processes (Table 1).
TABLE 1.
Down-regulated proteins in the microsomal fraction of the Grp94 KD cells
Red type, protein transport; green type, cellular localization; purple boldface type, both transport and cellular localization.
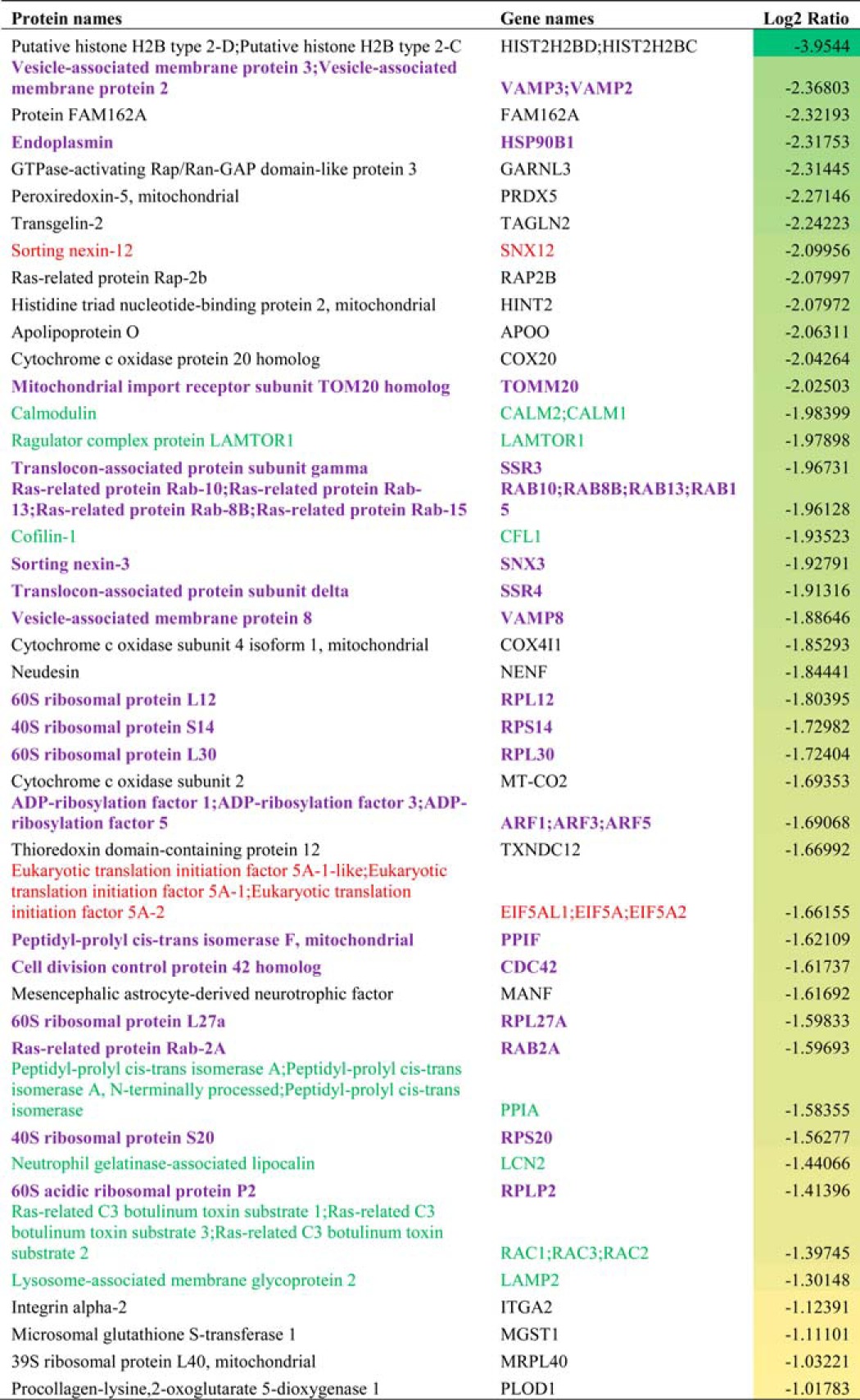
Next, the proteomic results were verified by Western blotting analysis. Integrin α2, RAC1, VAMP2, LAMP1, RAB2, and SYNE2 were chosen because they affect protein trafficking pathways and cancer directly (27–34). SYNE2 was detected in the control but not in the GRP94 KD population; hence, it was not present in the initial analysis (supplemental Table 1). The whole-cell lysate analysis shows that the levels of these proteins were not down-regulated by the GRP94 knockdown cells. In fact, some of these proteins, such as RAC1, RAB2, VAMP2, and SYNE2, were dependent on HSP90α and HSP90β but not on GRP94 (Fig. 3B). Although the total levels of the membrane proteins were not altered, their level in the Mic fraction was less in the GRP94 knockdown cells. Consistent with the proteomic data, the levels of RAC1, VAMP2, LAMP1, RAB2, and SYNE2 were all down-regulated in the Mic fraction of GRP94 knockdown cells. Cytosolic HSP90β levels were used as a loading control (Fig. 3C).
Co-immunoprecipitation of GRP94 was carried out to study whether the identified GRP94-regulated proteins interact with GRP94. Although RAB2 did not interact with GRP94, it did associate with RAC1, VAMP2, LAMP1, and SYNE2, suggesting that these are GRP94-dependent client proteins (Fig. 3D). Overall, the proteomic and Western blotting analyses suggest that GRP94 is responsible for localization of the membrane-bound proteins in cellular trafficking pathways that affect cell motility.
GRP94 Knockdown Cells Are Defective in Filamentous Actin Transport to the Cellular Cortex
Rho family GTPase members, such as RAC and CDC42, control the actin cytoskeleton at the cellular cortex region (35–37). Actin polymerization occurs at the leading edge of a migratory cell in response to Rho GTPase signaling. SYNE2 (Nesprin 2) is a multi-isomeric modular protein that binds filamentous actin to the nuclear envelope LINC complex comprising SUN and KASH proteins (Fig. 4A). Defects in tethering of the nucleus to the cytoskeleton can lead to age-related diseases, such as muscular dystrophy, nuclear envelopathy, and cancer (38–41). Overall, Rho family GTPases organize filamentous actin at the filopodia and lamellipodia and SYNE2 organizes it at the outer surface. We hypothesized that the GRP94 knockdown cells would be defective in filamentous actin organization because our results show that GRP94 is required to fold RAC, CDC42, and SYNE2, which are linked with remodeling of the actin cytoskeleton (42). Because the GRP94 knockdown cells lacked directionality during cell migration, we determined whether they were defective in lamellipodia and/or filopodia formation. Fascin is responsible for the organization of actin filament bundles, formation of membrane ruffles (lamellipodia), and protrusion of filopodia (43). Control and GRP94 knockdown cells were stained with fascin and phalloidin to study lamellipodia and filopodia formation. The phalloidin stain of control cells exhibited typical actin organization with filamentous actin enriched at both the cortex and filopodia regions, along with some actin patches in the cytoplasm (Fig. 4B). However, the GRP94 knockdown cells were defective in filamentous actin localization (Fig. 4B). The actin filaments formed in these cells were present throughout the cytoplasm, suggesting that GRP94 knockdown cells are defective in the transportation of actin filaments. However, introduction of GRP94-KDEL-GFP into the GRP94 knockdown cells promoted the relocalization of F-actin to the cellular cortex. Similarly, fascin was localized in the cortex region and at the filopodia in the control cells, but fascin was localized as patches in the cytoplasm and away from the cell cortex in the GRP94 knockdown cells (Fig. 4B). Control cells formed filopodia marked with phalloidin and fascin, but GRP94 knockdown cells formed short and thick pseudopodia instead of long and slender filopodia throughout the cell surface (Fig. 4B). Overall, these data suggest that GRP94 knockdown cells are defective in the transport of actin filaments. Consequently, these cells form shorter filopodia throughout the cell membrane (Figs. 4B and 6B) and produce a multipolar morphology (Fig. 2A), which together results in random cell motility (Fig. 1, B and C).
FIGURE 4.

GRP94 knockdown alters the localization of fascin and F-actin. A, schematic of the tethering of nucleus to the actin cytoskeleton. Nuclear lamins bind the inner nuclear protein SUN that forms a complex with the outer nuclear membrane protein KASH called LINC (linker of nucleoskeleton and cytoskeleton) complex. The LINC complex tethers the nucleus to the cytoskeleton. B, control, GRP94 KD, and GRP94 revertant cells were fixed and stained with AF647 phalloidin (gray), fascin (green), and DAPI (blue). Scale bar, 10 μm. Fluorescent images are representative of three independent biological replicates. INM, inner nuclear membrane; ONM, outer nuclear membrane.
FIGURE 6.

Localization of AHA1 in migrating cells. A, localization of HSP90α, HSP90β, RAB3GAP1, and AHA1 in PC3-MM2 cells. Wild type PC3-MM2 cells were fixed and stained for HSP90α and AHA1, RAB3GAP1, and AHA1 or HSP90β. Nuclei were stained with DAPI (blue). Scale bar, 10 μm. The representative images show that AHA1 co-localizes with HSP90α and RAB3GAP1. RAB3GAP1 is a marker for secretory vesicles. The secretory vesicles containing AHA1 and HSP90α or RAB3GAP1 are marked with white arrows. HSP90β is localized in the cytoplasm and the cell cortex region. At least 25 images were processed for the co-localization assay. B, heat map of AHA1-GFP in control and GRP94 KD cells in a cell migration assay. Cell migration montage of the control and GRP94 KD cells transfected with AHA1-GFP plasmid. Fluorescent photomicrographs of the control and GRP94 KD cells were taken at 15-min intervals for 9 h of the cell migration/wound healing assay. Results are representative of three independent wound healing assays per cell line. Time stamp, hh:mm:ss; scale bar, 10 μm. The white arrows in the control cells indicate AHA1-GFP localization at the tip of the filopodia in the direction of the migration. The white arrows in the GRP94 KD cells also indicate the AHA1-GFP localization, but instead of filopodia, thick and short pseudopodia were observed throughout the cell membrane. C, localization of AHA1 in the control, GRP94 KD, and GRP94 revertant cells. Control, GRP94 KD, and GRP94 revertant cells were fixed and stained for AHA1 and DAPI (blue). Scale bar, 10 μm. The white arrows in the control cells indicate AHA1 localization in secretory vesicles, but the staining is evident in the endoplasmic reticulum in GRP94 KD cells. Fluorescent images are representative of three independent biological replicates.
GRP94 Is Required for Integrin Transport toward the Cell Surface
Integrins are located on the cell surface and are well known cell adhesion molecules that mediate cell-cell and cell-matrix interactions during metastasis (44). The integrin complexes attach to the intracellular actin cytoskeleton and ECM and link these networks through the cell membrane (45). Integrin molecules undergo endocytosis and are sent to late endosomes and lysosomes for degradation. Alternatively, integrins may also enter a RAB-dependent vesicle transport pathway for recycling (46, 47). VAMP2 localizes mainly to endosomes and takes part in membrane docking and fusion. VAMP2 has also been reported to mediate trafficking of the integrin α5-β1 complex to the cell membrane (48).
GRP94 affected protein transport processes by regulating the endocytic proteins VAMP2 and LAMP2 and the intracellular trafficking protein, RAB2 (Table 1 and Fig. 3). Consistent with these data, our proteomic analysis revealed that the levels of integrin α2 were dependent on GRP94 (Table 1 and Fig. 3). To determine whether integrin molecules are dependent upon GRP94 or other HSP90 isoforms, GRP94, HSP90α, and HSP90β knockdown cells were constructed (16, 49). Integrin αL was included in the Western blotting analysis because it was previously reported to be dependent on GRP94 (11). Although the levels of both integrin α2 and integrin αL were down-regulated in HSP90α and HSP90β knockdown cells, the levels were significantly reduced upon GRP94 knockdown (Fig. 3B).
Co-immunoprecipitation studies were performed to further investigate whether integrin α2 and integrin αL are client proteins of HSP90α, HSP90β, or GRP94. The co-immunoprecipitation results show that both integrin α2 and integrin αL bind GRP94 and HSP90β but interact less with HSP90α and do not bind the mitochondrial HSP90, TRAP-1 (Fig. 5A). Binding of integrin α2 and integrin αL with GRP94 (Fig. 5A) and the reduced integrin levels in GRP94 knockdown cells (Fig. 3B) suggest that the integrin maturation process depends upon GRP94.
FIGURE 5.

Dependence of integrin α2 and integrin αL on HSP90 isoforms. A, association of integrin α2 and integrin αL with HSP90α, HSP90β, and GRP94. Integrin α2 and integrin αL were co-immunoprecipitated (IP) from wild type PC3-MM2 cells, and the levels of integrin α2, integrin αL, HSP90α, HSP90β, GRP94, and TRAP-1 were analyzed. Actin was used as a loading control. Two independent biological replicates were used to verify the protein-protein interaction. B and C, localization of integrin α2 and integrin αL in the control, GRP94 KD, and GRP94 revertant cells. Control and GRP94 knockdown cells were fixed and stained with integrin α2 and integrin αL, and the nucleus was stained with DAPI (blue). Scale bar, 10 μm. Fluorescent images are representative of three independent biological replicates.
To better understand the role of GRP94 in integrin trafficking, integrin α2 and integrin αL localization was determined in the control and GRP94 knockdown cells. Integrin α2 and integrin αL both localized at the cortex, cell surface, and leading edge in control cells. In contrast, integrin α2 and integrin αL were not localized at the cortex, cell surface, or filopodia region in GRP94 knockdown cells. Instead, they localized to the cytoplasm at much lower levels. Expression of GRP94-KDEL-GFP in the GRP94 knockdown cells re-established localization of integrin α2 and integrin αL at the cellular cortex region (Fig. 5, B and C). HSP90α was expressed at the leading edge, whereas HSP90β localized to the cell cortex regions (Fig. 6A). These data suggest that the integrin molecules present at the leading edge and cell cortex are recycled by HSP90α and HSP90β, respectively.
GRP94 Knockdown Cells Are Defective in the Transport of Secretory Vesicles Containing the HSP90α-AHA1 Complex
An important characteristic of cell migration and metastasis is the secretion of proteins toward the leading edge that is responsible for breaking down the ECM and providing directional control during cell movement (50). Previously, we reported that HSP90α forms a complex with the late phase co-chaperone, AHA1, and that the HSP90α-AHA1 complex is transported toward the leading edge of a migrating cell in secretory vesicles (Fig. 6A) (49). Our cell organelle fractionation studies also revealed that cytosolic HSP90α levels were dependent upon GRP94 (Fig. 3C). Localization of the HSP90α-AHA1 complex in the secretory vesicle was determined by co-localization and co-immunoprecipitation with a secretory vesicle marker protein, RAB3GAP1 (Fig. 6A) (49). Strongly supporting the co-localization of HSP90α, AHA1, and RAB3GAP1, image quantification showed that Pearson's co-efficient (r) was 0.92 ± 0.009 (n = 25, p < 0.001) for HSP90α and AHA1 and 0.95 ± 0.003 (n = 25, p < 0.001) for RAB3GAP1 and AHA1. Consequently, localization of AHA1 was studied in GRP94 knockdown cells. The control and GRP94 knockdown cells were transfected with an AHA1-GFP plasmid, and AHA1-GFP expression was observed in live cell migration assays for 9 h (Fig. 6B and supplemental Videos 6 and 7). Because these cells overexpress AHA1-GFP, the GFP signal was observed throughout the cytoplasm. The heat map of AHA1-GFP revealed the concentration of AHA1 during cell migration. The AHA1-GFP heat map in supplemental Video 6 and Fig. 6B shows concentrated AHA1 globules at the filopodia of migrating cells. In fact, some of the AHA1-GFP globules were observed outside of the cells in supplemental Video 6. AHA1 was also detected in the extracellular medium, but the secretion of AHA1 was not defective in GRP94 knockdown cells (data not shown). The GRP94 knockdown cells migrated randomly, and filopodia were not formed. In contrast, pseudopodia-like organelles were observed throughout the cell surface, and the AHA1-GFP globules were visualized in the pseudopodia-like organelles (Fig. 6B and supplemental Video 7). These data suggest that secretory vesicles are not transported toward the direction of cell migration in GRP94 knockdown cells. AHA1 localization was also performed in control, GRP94 knockdown, and GRP94 revertant cells. AHA1 localized primarily to the ER in the GRP94 knockdown cells (Fig. 6C) but was observed primarily in the cytoplasm and secretory vesicles of control and GRP94 revertant cells (Fig. 6C).
Survivin Is a Part of the Secretory Vesicle Containing the HSP90α-AHA1 Complex
From our data, it was hypothesized that the HSP90α-AHA1 complex is responsible for folding/maturing proteins present in the polar region that are important for establishing and maintaining cell polarity. Survivin is a multifunctional protein that has independent roles in apoptosis, mitosis, and metastasis (51, 52) and is also found in the exosome (53). Therefore, we determined whether survivin is part of the secretory vesicle that contains the HSP90α-AHA1 complex. During interphase, survivin strongly co-localized with AHA1 at the polar region (Fig. 7A). Quantification of the co-localization showed that Pearson's co-efficient (r) was 0.77 ± 0.02 (n = 25, p < 0.001) for survivin and AHA1. Moreover, HSP90α, RAB3GAP1, and survivin co-localized with AHA1 (Figs. 6A and 7A), which suggests that the secretory vesicles also contain survivin, along with the HSP90α-AHA1 complex in cancer cells.
FIGURE 7.

Association of AHA1 and survivin in PC3-MM2 cells. A, co-localization of AHA1 and survivin in the wild type PC3-MM2 cells. Wild type PC3-MM2 cells were fixed and stained for AHA1, survivin, and DAPI (blue). Scale bar, 10 μm. The white arrow indicates that both AHA1 and survivin co-localized at the leading edge of a migrating interphase cell. Fluorescent images are representative of three independent biological replicates. At least 25 images were processed for the co-localization assay. B, survivin binds AHA1 and HSP90α but no other HSP90 isoforms under normal and apoptotic conditions. Survivin was co-immunoprecipitated (IP) from PC3-MM2 cells treated with nothing, DMSO (0.1%), staurosporine (1 μm), KU-135 (1 μm), or TRAIL (1 μm) for 24 h, and HSP90α, phospho-HSP90α (Thr-5/7), HSP90β, GRP94, TRAP1, AHA1, and survivin were detected by Western blotting analysis. Actin was used as a loading control. Two independent biological replicates were used to verify the protein-protein interaction.
Next, co-immunoprecipitation studies were performed to confirm the microscopy results. Because survivin is an anti-apoptotic protein, cells were treated with various agents to induce apoptosis, including staurosporine, KU-135 (54), and TRAIL. The induction of apoptosis was verified by the presence of phosphorylated HSP90α at threonines 5/7 (55). Co-immunoprecipitation results confirmed that survivin associated with both HSP90α and AHA1 under normal and apoptotic conditions (Fig. 7B). Taken together, these data suggest that survivin is a client protein of the HSP90α-AHA1 complex that is present in the secretory vesicle of a migrating cell and that the GRP94 knockdown cells are unable to secrete these vesicles toward the direction of motility.
GRP94 Promotes Tumorigenesis
Previous reports have indicated that GRP94 is a pro-oncogenic chaperone (13) that promotes cell adhesion and causes tumorigenesis and metastasis in various organs (56, 57). A three-dimensional in vitro tumor formation assay was used to investigate whether the defects in GRP94 knockdown cells would translate into defects in tumor formation and metastasis. Multicellular spheroid and satellite formation in three-dimensional in vitro tumor models are indicative of cell-cell and cell-matrix interactions and represent an attractive system to evaluate the tumorigenic and metastatic properties of cells (58). Whereas wild type and control cells formed spheroids and satellites, the GRP94 knockdown cells were unable to form spheroids or satellites (Fig. 8A). In fact, the GRP94 cells were unable to form aggregates (Fig. 8A), suggesting that a loss of GRP94 results in a deficiency in cell-cell and cell-matrix interactions necessary for tumorigenesis.
FIGURE 8.

GRP94 mediates tumor formation. A, three-dimensional tumor formation assay; bright field and fluorescent images of a three-dimensional in vitro tumor formation assay of the wild type, control, and GRP94 KD cells. After 6 days of three-dimensional culture, the wild type and control cells formed spheroids (black arrow) and satellites (black arrowhead), characteristics of tumorigenesis and metastasis. In contrast, the GRP94 KD cells remained separated and failed to form spheroids and satellites. Bright field images are representative of three independent biological replicates. B, expression of HSP90 isoforms in clinical samples. Representative Western blots show the levels of HSP90α, HSP90β, GRP94, TRAP-1, and AHA1 in tissue samples from various cancer patients. Actin was used as loading control. Lane 1, normal prostate; lanes 2 and 3, prostate cancer tissue from two sites from the same individual; lanes 4 and 5, normal liver and breast cancer metastatic to liver from the same individual; lanes 6 and 7, normal lung and tumorous lung tissues from the same individual. Two technical replicates were performed for each clinical sample.
The expression of GRP94 and other HSP90 isoform levels were also evaluated in clinical samples of cancer patients. HSP90 isoform levels were measured in a panel of human tumors along with normal adjacent tissue from the same patient. In a normal cell adjacent to the tumor, HSP90 is not detected by Western blotting analysis, whereas in tumor tissues, HSP90 isoforms were differentially overexpressed (Fig. 8B). This suggests that cancers activate different signaling pathways that rely upon various HSP90 isoforms, including GRP94, and further supports the role of GRP94 in tumorigenesis.
Discussion
HSP90 is an evolutionarily conserved molecular chaperone that in the presence of co-chaperones can fold nascent polypeptides and stabilize/activate some receptors. In Saccharomyces cerevisiae, HSP90 exists as two isoforms, namely Hsc82 and HSP82, which are constitutively or inducibly expressed, respectively (59). In higher eukaryotes, HSP90 exists as four isoforms: HSP90α, HSP90β, GRP94, and tumor necrosis factor receptor-associated protein 1 (TRAP1). HSP90α and HSP90β are cytosolic, GRP94 resides in the ER, and TRAP1 localizes to the mitochondria. Cancer cells manipulate the HSP90 chaperone machinery to fold mutated or overexpressed oncoproteins, which aids progression of malignant phenotypes (60). In this study, it was shown that distinct HSP90 isoforms are up-regulated in different cancers. Therefore, knowledge of the biological function manifested by each HSP90 isoform is required to determine which target is contributing to tumor formation, progression, and/or metastasis.
Toward this goal, we performed the first systematic analysis of GRP94 KD cells to investigate its cellular function. GRP94 KD cells were defective in cell motility and the ability to form a spindle-shaped morphology. Proteomic studies revealed that the GRP94 KD cells are defective in protein transport processes affecting actin-based motility. Further studies verified that the GRP94 KD cells are also defective in F-actin transport processes. Cell migration requires protein transport toward the leading edge for the formation of filopodia, lamellipodia, and focal adhesion complexes. Because the GRP94 KD cells are defective in protein transport processes, the cells are also defective in filopodia formation. Instead of forming filopodia toward the direction of migration, these cells formed filopodia throughout the membrane, causing the cells to move randomly. Additionally, the GRP94 KD cells down-regulated focal adhesion complex proteins and integrins, causing defects in cell-cell and cell-matrix interactions. Last, the GRP94 KD cells were defective in the transport of secretory vesicles or exosomes containing HSP90α-AHA1-survivin toward the leading edge because the HSP90α-AHA1-survivin complex was dispersed throughout the cell membrane (Fig. 9).
FIGURE 9.
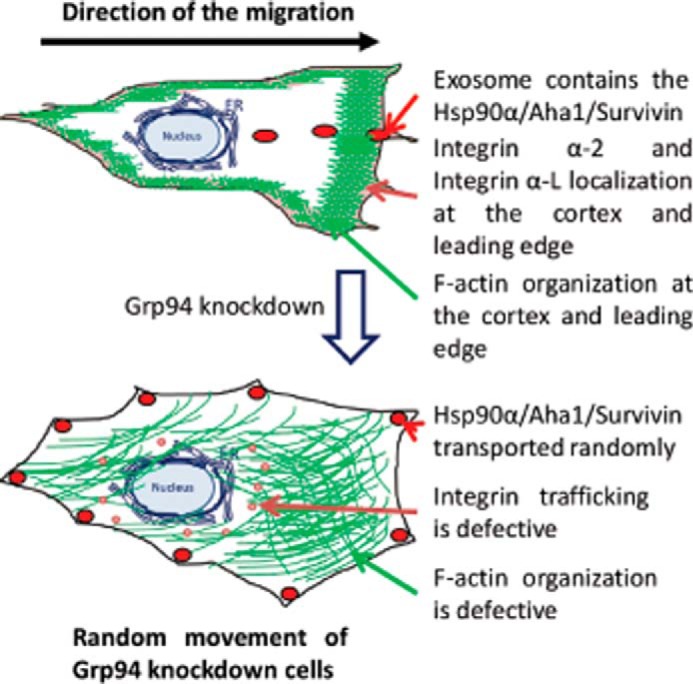
Summary figure. The wild type cancer cells migrate toward the direction of the wound, whereas the GRP94 KD cells are defective in cellular migration.
GRP94 has been shown to be expressed in oocytes during early embryonic development and at later stages of organogenesis and is lowered to a basal level in adults (61–63). During tumorigenesis and metastasis, additional proteins are synthesized, which appear to correlate with increased expression of GRP94. Because GRP94 is essential for mesoderm induction, global deletion of GRP94 in mice is embryonic lethal (10). Although GRP94 was shown to be essential in the development of mouse embryos, a homolog of GRP94 is not detected in S. cerevisiae. In fact, ScHsc82 and ScHSP82 are sufficient for responding to all cellular stress. In subsequent studies, GRP94−/− embryonic stem cells were able to differentiate in culture (10); however, these cells were unable to differentiate into muscle lineages and did not produce insulin growth factor II (10, 64). In this study, GRP94-specific shRNA was used to obtain a stable GRP94 knockdown cell line. The GRP94 knockdown cells were able to differentiate, which indicates that although GRP94 is required for embryonic development, it is not essential for other processes.
Previous reports indicated that only a few integrin molecules, such as integrin αL (CD11a), integrin α4 (CD49d), and integrin β2 (CD18), were defective in intracellular transport in the absence of GRP94 in murine B-cells (11, 12). Other integrin molecules did not appear to be dependent upon GRP94. This study reports for the first time that only the levels of integrin αL (CD11a) and integrin α2 (CD49b) are dependent on GRP94 in human PC3-MM2 metastatic prostate cancer cells. In contrast, the expression of integrin β2 (CD18) was not dependent upon GRP94; it was only down-regulated ∼30% in the GRP94 KD cells (supplemental Table 1). Additionally, it was shown that integrin αL and integrin α2 bound GRP94 as well as HSP90α and HSP90β. From microscopic examinations, it appears that the integrin molecules present in the filopodia and at the leading edge of a migratory cell are modulated by HSP90α, and those at the cellular cortex region are modulated by HSP90β.
Recently, it was demonstrated that HSP90α and the co-chaperone, AHA1, readily form a complex that localizes to secretory vesicles at the leading edge of a migratory cell (49). This HSP90α-AHA1 complex is required for RAC1 maturation and cell migration. Here, it is reported that the secretory vesicle also contains the well known protein, survivin, which is a client of the HSP90α-AHA1 complex. Survivin is a multifunctional protein that forms the chromosomal passenger complex along with borealin and INCENP and plays multiple roles during mitosis (65). Survivin is also a member of the IAP family of anti-apoptotic proteins that inhibits caspase activation (66). Recently, Altieri and co-workers (51) showed that survivin along with other members of the IAP family of anti-apoptotic proteins stimulate cell motility and metastasis by activating the NF-κB signaling pathway that is independent of IAP inhibition of cell death. It has been reported that directional cell motility is controlled by exosomes that contain fibronectin, which is required for attachment to the ECM (50). This study along with other independent studies found that survivin is released from cancer cells in secretory vesicles (53, 67). In fact, survivin along with HSP90α and AHA1 were present in these secretory vesicles, suggesting that these proteins have extracellular functions in cell motility and metastasis. GRP94 modulates secretion of the HSP90α-AHA1 complex, along with survivin, toward the direction of cellular movement.
As presented in Fig. 9, this report highlights that GRP94 is required for the transport of integrin to the cell surface, F-actin toward the cellular cortex, and secretory vesicles containing survivin toward the direction of the migration. These defects in intracellular transport in GRP94 knockdown cells produced altered filopodia formation and attachment to the matrix, causing random movements instead of motility toward the direction of the wound, which in turn caused defects in cell migration and tumorigenesis.
Author Contributions
S. G. and B. S. J. B. conceived and coordinated the study and prepared the manuscript. S. G. and H. E. S. designed, performed, and analyzed the microscopic data. S. G., N. A. G., and R. T. D. designed, performed, and analyzed the proteomics data. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank the laboratory of Dr. Yair Argon (University of Pennsylvania, Philadelphia, PA) for kindly providing the canine GRP94-KDEL-GFP plasmid for this study.
This work was supported by National Institutes of Health Grants CA109265 (to B. S. J. B.) and DK095911 (to R. T. D.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table 1 and Videos 1–7.
- ECM
- extracellular matrix
- ER
- endoplasmic reticulum
- KD
- knockdown
- SILAC
- stable isotope labeling by amino acids in cell culture.
References
- 1. Etienne-Manneville S., and Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 2. Rodriguez O. C., Schaefer A. W., Mandato C. A., Forscher P., Bement W. M., and Waterman-Storer C. M. (2003) Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat. Cell Biol. 5, 599–609 [DOI] [PubMed] [Google Scholar]
- 3. Small J. V., Stradal T., Vignal E., and Rottner K. (2002) The lamellipodium: where motility begins. Trends Cell Biol. 12, 112–120 [DOI] [PubMed] [Google Scholar]
- 4. Machesky L. M. (2008) Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 582, 2102–2111 [DOI] [PubMed] [Google Scholar]
- 5. Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 [DOI] [PubMed] [Google Scholar]
- 6. Mattila P. K., and Lappalainen P. (2008) Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 9, 446–454 [DOI] [PubMed] [Google Scholar]
- 7. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., and Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 8. Woods A. J., White D. P., Caswell P. T., and Norman J. C. (2004) PKD1/PKCμ promotes αvβ3 integrin recycling and delivery to nascent focal adhesions. EMBO J. 23, 2531–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Proux-Gillardeaux V., Gavard J., Irinopoulou T., Mège R. M., and Galli T. (2005) Tetanus neurotoxin-mediated cleavage of cellubrevin impairs epithelial cell migration and integrin-dependent cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 102, 6362–6367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wanderling S., Simen B. B., Ostrovsky O., Ahmed N. T., Vogen S. M., Gidalevitz T., and Argon Y. (2007) GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol. Biol. Cell 18, 3764–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Randow F., and Seed B. (2001) Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat. Cell Biol. 3, 891–896 [DOI] [PubMed] [Google Scholar]
- 12. Liu B., and Li Z. (2008) Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood 112, 1223–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rachidi S., Sun S., and Li Z. (2015) Endoplasmic reticulum heat shock protein gp96/grp94 is a pro-oncogenic chaperone, not a tumor suppressor. Hepatology 61, 1766–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shelton S. N., Shawgo M. E., Matthews S. B., Lu Y., Donnelly A. C., Szabla K., Tanol M., Vielhauer G. A., Rajewski R. A., Matts R. L., Blagg B. S., and Robertson J. D. (2009) KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol. 76, 1314–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ostrovsky O., Makarewich C. A., Snapp E. L., and Argon Y. (2009) An essential role for ATP binding and hydrolysis in the chaperone activity of GRP94 in cells. Proc. Natl. Acad. Sci. U.S.A. 106, 11600–11605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peterson L. B., Eskew J. D., Vielhauer G. A., and Blagg B. S. (2012) The hERG channel is dependent upon the Hsp90α isoform for maturation and trafficking. Mol. Pharm. 9, 1841–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J., Golland P., and Sabatini D. M. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bolte S., and Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 19. Okado-Matsumoto A., and Fridovich I. (2001) Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 276, 38388–38393 [DOI] [PubMed] [Google Scholar]
- 20. Steen H., and Mann M. (2004) The ABC's (and XYZ's) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 5, 699–711 [DOI] [PubMed] [Google Scholar]
- 21. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 22. Zhang L., Zhao H., Blagg B. S., and Dobrowsky R. T. (2012) C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. J. Proteome Res. 11, 2581–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang L., Yu C., Vasquez F. E., Galeva N., Onyango I., Swerdlow R. H., and Dobrowsky R. T. (2010) Hyperglycemia alters the schwann cell mitochondrial proteome and decreases coupled respiration in the absence of superoxide production. J. Proteome Res. 9, 458–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 25. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 26. Sreedhar A. S., Kalmár E., Csermely P., and Shen Y. F. (2004) Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 562, 11–15 [DOI] [PubMed] [Google Scholar]
- 27. Chen Y. A., and Scheller R. H. (2001) SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2, 98–106 [DOI] [PubMed] [Google Scholar]
- 28. Meng J., and Wang J. (2015) Role of SNARE proteins in tumourigenesis and their potential as targets for novel anti-cancer therapeutics. Biochim. Biophys. Acta 1856, 1–12 [DOI] [PubMed] [Google Scholar]
- 29. Fehrenbacher N., Bastholm L., Kirkegaard-Sørensen T., Rafn B., Bøttzauw T., Nielsen C., Weber E., Shirasawa S., Kallunki T., and Jááttelá M. (2008) Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 68, 6623–6633 [DOI] [PubMed] [Google Scholar]
- 30. Luo M. L., Gong C., Chen C. H., Hu H., Huang P., Zheng M., Yao Y., Wei S., Wulf G., Lieberman J., Zhou X. Z., Song E., and Lu K. P. (2015) The Rab2A GTPase promotes breast cancer stem cells and tumorigenesis via Erk signaling activation. Cell Rep. 11, 111–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Culine S., Honoré N., Tavitian A., and Olofsson B. (1992) Overexpression of the ras-related rab2 gene product in peripheral blood mononuclear cells from patients with hematological and solid neoplasms. Cancer Res. 52, 3083–3088 [PubMed] [Google Scholar]
- 32. King S. J., Nowak K., Suryavanshi N., Holt I., Shanahan C. M., and Ridley A. J. (2014) Nesprin-1 and nesprin-2 regulate endothelial cell shape and migration. Cytoskeleton 71, 423–434 [DOI] [PubMed] [Google Scholar]
- 33. Cartwright S., and Karakesisoglou I. (2014) Nesprins in health and disease. Semin. Cell Dev. Biol. 29, 169–179 [DOI] [PubMed] [Google Scholar]
- 34. Shah S. P., Roth A., Goya R., Oloumi A., Ha G., Zhao Y., Turashvili G., Ding J., Tse K., Haffari G., Bashashati A., Prentice L. M., Khattra J., Burleigh A., Yap D., et al. (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chung C. Y., Lee S., Briscoe C., Ellsworth C., and Firtel R. A. (2000) Role of RAC in controlling the actin cytoskeleton and chemotaxis in motile cells. Proc. Natl. Acad. Sci. U.S.A. 97, 5225–5230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo F., Debidda M., Yang L., Williams D. A., and Zheng Y. (2006) Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J. Biol. Chem. 281, 18652–18659 [DOI] [PubMed] [Google Scholar]
- 37. Sokac A. M., Co C., Taunton J., and Bement W. (2003) Cdc42-dependent actin polymerization during compensatory endocytosis in Xenopus eggs. Nat. Cell Biol. 5, 727–732 [DOI] [PubMed] [Google Scholar]
- 38. Friederichs J. M., Ghosh S., Smoyer C. J., McCroskey S., Miller B. D., Weaver K. J., Delventhal K. M., Unruh J., Slaughter B. D., and Jaspersen S. L. (2011) The SUN protein Mps3 is required for spindle pole body insertion into the nuclear membrane and nuclear envelope homeostasis. PLoS Genet. 7, e1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghosh S., Gardner J. M., Smoyer C. J., Friederichs J. M., Unruh J. R., Slaughter B. D., Alexander R., Chisholm R. D., Lee K. K., Workman J. L., and Jaspersen S. L. (2012) Acetylation of the SUN protein Mps3 by Eco1 regulates its function in nuclear organization. Mol. Biol. Cell 23, 2546–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jaspersen S. L., and Ghosh S. (2012) Nuclear envelope insertion of spindle pole bodies and nuclear pore complexes. Nucleus 3, 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haque F., Mazzeo D., Patel J. T., Smallwood D. T., Ellis J. A., Shanahan C. M., and Shackleton S. (2010) Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J. Biol. Chem. 285, 3487–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dawe H. R., Adams M., Wheway G., Szymanska K., Logan C. V., Noegel A. A., Gull K., and Johnson C. A. (2009) Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J. Cell Sci. 122, 2716–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamashiro S., Yamakita Y., Ono S., and Matsumura F. (1998) Fascin, an actin-bundling protein, induces membrane protrusions and increases cell motility of epithelial cells. Mol. Biol. Cell 9, 993–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo W., and Giancotti F. G. (2004) Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 5, 816–826 [DOI] [PubMed] [Google Scholar]
- 45. Chan K. T., Cortesio C. L., and Huttenlocher A. (2007) Integrins in cell migration. Methods Enzymol. 426, 47–67 [DOI] [PubMed] [Google Scholar]
- 46. Caswell P. T., Vadrevu S., and Norman J. C. (2009) Integrins: masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 10, 843–853 [DOI] [PubMed] [Google Scholar]
- 47. Subramani D., and Alahari S. K. (2010) Integrin-mediated function of Rab GTPases in cancer progression. Mol. Cancer 9, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hasan N., and Hu C. (2010) Vesicle-associated membrane protein 2 mediates trafficking of α5β1 integrin to the plasma membrane. Exp. Cell Res. 316, 12–23 [DOI] [PubMed] [Google Scholar]
- 49. Ghosh S., Shinogle H. E., Garg G., Vielhauer G. A., Holzbeierlein J. M., Dobrowsky R. T., and Blagg B. S. (2015) Hsp90 C-terminal inhibitors exhibit antimigratory activity by disrupting the Hsp90α/Aha1 complex in PC3-MM2 cells. ACS Chem. Biol. 10, 577–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sung B. H., Ketova T., Hoshino D., Zijlstra A., and Weaver A. M. (2015) Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 6, 7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mehrotra S., Languino L. R., Raskett C. M., Mercurio A. M., Dohi T., and Altieri D. C. (2010) IAP regulation of metastasis. Cancer Cell 17, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Altieri D. C. (2015) Survivin: the inconvenient IAP. Semin. Cell Dev. Biol. 39, 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khan S., Bennit H. F., and Wall N. R. (2015) The emerging role of exosomes in survivin secretion. Histol. Histopathol. 30, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Samadi A. K., Zhang X., Mukerji R., Donnelly A. C., Blagg B. S., and Cohen M. S. (2011) A novel C-terminal HSP90 inhibitor KU135 induces apoptosis and cell cycle arrest in melanoma cells. Cancer Lett. 312, 158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Solier S., Kohn K. W., Scroggins B., Xu W., Trepel J., Neckers L., and Pommier Y. (2012) Heat shock protein 90α (HSP90α), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc. Natl. Acad. Sci. U.S.A. 109, 12866–12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng H. C., Takahashi H., Li X. H., Hara T., Masuda S., Guan Y. F., and Takano Y. (2008) Overexpression of GRP78 and GRP94 are markers for aggressive behavior and poor prognosis in gastric carcinomas. Hum. Pathol. 39, 1042–1049 [DOI] [PubMed] [Google Scholar]
- 57. Sanz-Pamplona R., Aragüés R., Driouch K., Martín B., Oliva B., Gil M., Boluda S., Fernández P. L., Martínez A., Moreno V., Acebes J. J., Lidereau R., Reyal F., Van de Vijver M. J., and Sierra A. (2011) Expression of endoplasmic reticulum stress proteins is a candidate marker of brain metastasis in both ErbB-2+ and ErbB-2− primary breast tumors. Am. J. Pathol. 179, 564–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nyga A., Cheema U., and Loizidou M. (2011) 3D tumour models: novel in vitro approaches to cancer studies. J. Cell Commun. Signal. 5, 239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Borkovich K. A., Farrelly F. W., Finkelstein D. B., Taulien J., and Lindquist S. (1989) hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol. Cell Biol. 9, 3919–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Trepel J., Mollapour M., Giaccone G., and Neckers L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McCormick P. J., and Babiarz B. (1984) Expression of a glucose-regulated cell surface protein in early mouse embryos. Dev. Biol. 105, 530–534 [DOI] [PubMed] [Google Scholar]
- 62. Kim S. K., Kim Y. K., and Lee A. S. (1990) Expression of the glucose-regulated proteins (GRP94 and GRP78) in differentiated and undifferentiated mouse embryonic cells and the use of the GRP78 promoter as an expression system in embryonic cells. Differentiation 42, 153–159 [DOI] [PubMed] [Google Scholar]
- 63. Barnes J. A., and Smoak I. W. (1997) Immunolocalization and heart levels of GRP94 in the mouse during post-implantation development. Anat. Embryol. 196, 335–341 [DOI] [PubMed] [Google Scholar]
- 64. Ostrovsky O., Ahmed N. T., and Argon Y. (2009) The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol. Biol. Cell 20, 1855–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Honda R., Körner R., and Nigg E. A. (2003) Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol. Biol. Cell 14, 3325–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tamm I., Wang Y., Sausville E., Scudiero D. A., Vigna N., Oltersdorf T., and Reed J. C. (1998) IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 58, 5315–5320 [PubMed] [Google Scholar]
- 67. Khan S., Jutzy J. M., Aspe J. R., McGregor D. W., Neidigh J. W., and Wall N. R. (2011) Survivin is released from cancer cells via exosomes. Apoptosis 16, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.