

Abstract
Multiple myeloma is an aggressive hematopoietic cancer of plasma cells. The recent emergence of three effective FDA-approved proteasome-inhibiting drugs, bortezomib (Velcade®), carfilzomib (Kyprolis®), and ixazomib (Ninlaro®), confirms that proteasome inhibitors are therapeutically useful against neoplastic disease, in particular refractory multiple myeloma and mantle cell lymphoma. This study describes the synthesis, computational affinity assessment, and preclinical evaluation of TIR-199, a natural product-derived syrbactin structural analog. Molecular modeling and simulation suggested that TIR-199 covalently binds each of the three catalytic subunits (β1, β2, and β5) and revealed key interaction sites. In vitro and cell culture-based proteasome activity measurements confirmed that TIR-199 inhibits the proteasome in a dose-dependent manner and induces tumor cell death in multiple myeloma and neuroblastoma cells as well as other cancer types in the NCI-60 cell panel. It is particularly effective against kidney tumor cell lines, with >250-fold higher anti-tumor activities than observed with the natural product syringolin A. In vivo studies in mice revealed a maximum tolerated dose of TIR-199 at 25 mg/kg. The anti-tumor activity of TIR-199 was confirmed in hollow fiber assays in mice. Adverse drug reaction screens in a kidney panel revealed no off-targets of concern. This is the first study to examine the efficacy of a syrbactin in animals. Taken together, the results suggest that TIR-199 is a potent new proteasome inhibitor with promise for further development into a clinical drug for the treatment of multiple myeloma and other forms of cancer.
Keywords: computer modeling, multiple myeloma, natural product, neuroblastoma, proteasome, TIR-199, anti-tumor activity, chemical synthesis, hollow fiber assays, syringolin A
Introduction
Multiple myeloma (MM)4 is an aggressive hematopoietic cancer of plasma cells that develops in about 6 per 100,000 people/year. MM is considered to be treatable but incurable, with a 5-year survival rate of 45%. After non-Hodgkin lymphoma, MM is the second most common hematological malignancy in the United States and constitutes 1% of all cancers (1). The current therapeutic options include six major drug classes: classic drugs like chemotherapeutic agents, corticosteroids, and interferon as well as immunomodulatory drugs (e.g. thalidomide), proteasome inhibitors, histone deacetylase inhibitors (e.g. panobinostat), and the first FDA-approved monoclonal antibody against MM, daratumumab (Darzalex®).
The recent FDA approval of the three proteasome inhibitors bortezomib (Velcade®), carfilzomib (Kyprolis®), and ixazomib (Ninlaro®) shows that the proteasome is a valuable therapeutic target against neoplastic disease (2, 3). Notably, it has been shown that actively dividing cancer cells are more sensitive to proteasome inhibition than quiescent or differentiated normal cells. For example, MM cells are significantly more sensitive to the pro-apoptotic effects of bortezomib (BTZ)-induced proteasome inhibition than are healthy bone marrow cells or peripheral blood mononuclear cells (4). The differential sensitivity of cells to proteasome inhibition may in part be due to the fact that cancer cells require a higher rate of protein turnover than normal cells and, therefore, may be more susceptible to losing proteasome function. Proteasome inhibition also enhances the sensitivity of cancer cells to traditional chemotherapies, providing a rationale for the development of combination therapies.
Despite the fact that proteasome inhibition is a validated strategy for therapy of MM (5–7) and mantle cell lymphoma (8, 9), this disease remains challenging because relapses are common and usually associated with increasing chemoresistance (10). Moreover, proteasome inhibitors like BTZ can induce peripheral neuropathy and other toxicities that may decrease efficacy by compromising the ability to deliver therapy at optimal doses. Thus, there are a number of shortcomings, and there is an urgent need to develop next generation proteasome inhibitors with improved safety profiles for therapeutic use.
We have discovered and developed a novel class of proteasome inhibitors referred to as syrbactins (11, 12). Our previous findings indicated that the natural product syringolin A (SylA) inhibits the proteasome (12–15) and induces cell death in a number of tumor cell types, including MM and neuroblastoma (NB) cells (11, 12, 14). SylA reacts irreversibly with the N-terminal Thr of the active site in the β5 pocket by a 1,4-addition of the hydroxyl group of the Thr to the α,β-unsaturated carboxamide moiety of SylA (12). SylA preferentially inhibits the proteasomal β5 subunit (chymotrypsin-like; CT-L) activity with a Ki value of 0.843 μm, weakly inhibits the β2 subunit (trypsin-like; T-L) activity with a Ki value of 6.7 μm, and has no effects on the β1 subunit (caspase-like; C-L) activity (12, 15). We previously synthesized and evaluated a number of SylA analogs (13–22), and three other groups have designed syringolin variants (23–26). In this study, we synthesized TIR-199, one of the most potent SylA-derived compounds to date. We demonstrate that TIR-199 inhibits the proteasome activity and impedes MM and other tumor cell growth, with a significantly higher potency than the natural product SylA, both in vitro and in vivo. Importantly, this represents the first study to examine the efficacy of a syrbactin in animals.
Experimental Procedures
Total Synthesis of Syrbactin Structural Analog TIR-199 and Reagents
The synthetic route to TIR-199 (MW 534) is provided in Scheme 1 and described in detail in the supplemental Methods. Most of the route follows our reported syringolin A synthesis (22), with the exception of the use of alaninol. From an (S)-vinylglycine starting material, the route entails 10 steps and proceeds in 10% overall yield. Stocks were prepared in DMSO (10 mm). The natural product SylA and BTZ served as controls throughout the study and were dissolved in sterile water and DMSO, respectively.
SCHEME 1.
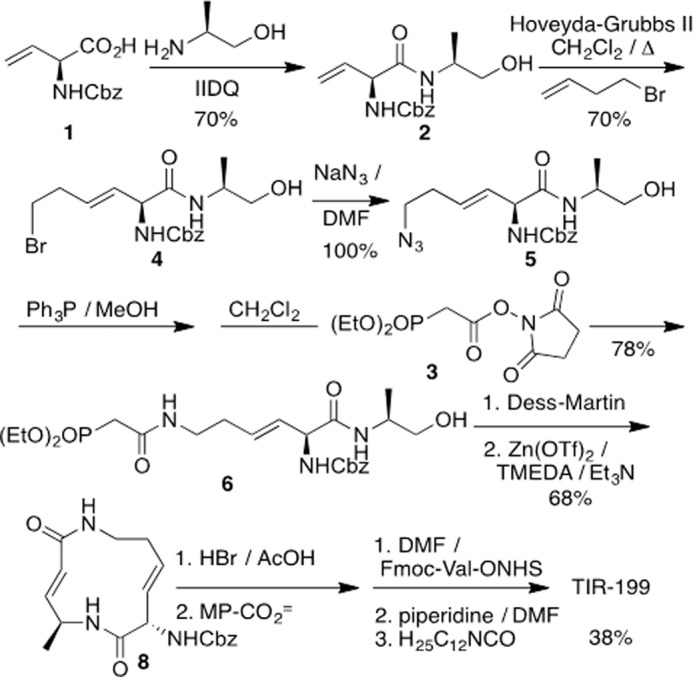
Total synthesis of TIR-199. Molecular weight of TIR-199 is 534.
Computational Assessment of Selectivity
We used computational modeling and energetic analysis of the covalently bound complexes between TIR-199 and each catalytic subunit of the human proteasome to understand the selectivity of TIR-199. Because no experimental structure is available for the human proteasome, we performed homology modeling to derive the human proteasome atomic structure from the x-ray crystallographic data of the Bos taurus proteasome (Protein Data Bank code 1IRU), which has a sequence similarity of 99.6% to that of Homo sapiens. Molecular recognition takes place within a distinct binding pocket at each catalytic site in the proteasome, which includes the active catalytic enzyme and the domain directly adjacent to the active site (Fig. 2A). Therefore, three binding pocket structures were constructed from the homology model for use in all subsequent calculations, simulations, and analyses: the C-L (catalytic β1 and neighboring β2), T-L (catalytic β2 and neighboring β3), and CT-L (catalytic β5 and neighboring β6) binding pockets. AutoDock version 4.2 was used to determine low energy conformational states of TIR-199 within each of the catalytic receptors when bound covalently to the catalytic Thr-1 active site (27). AMBER version 11 was subsequently used to perform molecular dynamics simulations starting with the bound state conformations obtained from docking (28). The AMBER GAFF parameters were applied to the novel ligand TIR-199, whereas the AMBER 99SB protein force field parameterized the protein structures. Covalent bonds were manually formed using the AMBER xleap utility, and parameters bridging the GAFF and 99SB systems were derived from the GAFF force field (29). Implicit solvent Langevin dynamics simulated all systems. Ten thousand equilibration steps were performed at 100, 200, and 298 K. Production simulations of each system were performed for a 10-ns simulation time at 298 K on the XSEDE national supercomputer system. For all simulations, a selection of protein atoms within 10 Å of the ligand's initial positions defined the unconstrained, mobile region of the simulation, whereas the rest of the protein structure was held fixed. Trajectories of the molecular dynamics simulations were subject to molecular mechanics/generalized Born/surface area energetic post-simulation analysis to calculate accurate ligand-receptor affinities over each recorded snapshot along the simulation trajectories (30, 31). The total molecular mechanics/generalized Born/surface area interaction energy was calculated for every frame in each trajectory. The energies were separated into polar interactions, combining Coulombic and generalized Born interaction energies, and non-polar interactions, combining van der Waals and non-polar solvation solvent-accessible surface area interaction energies.
FIGURE 2.
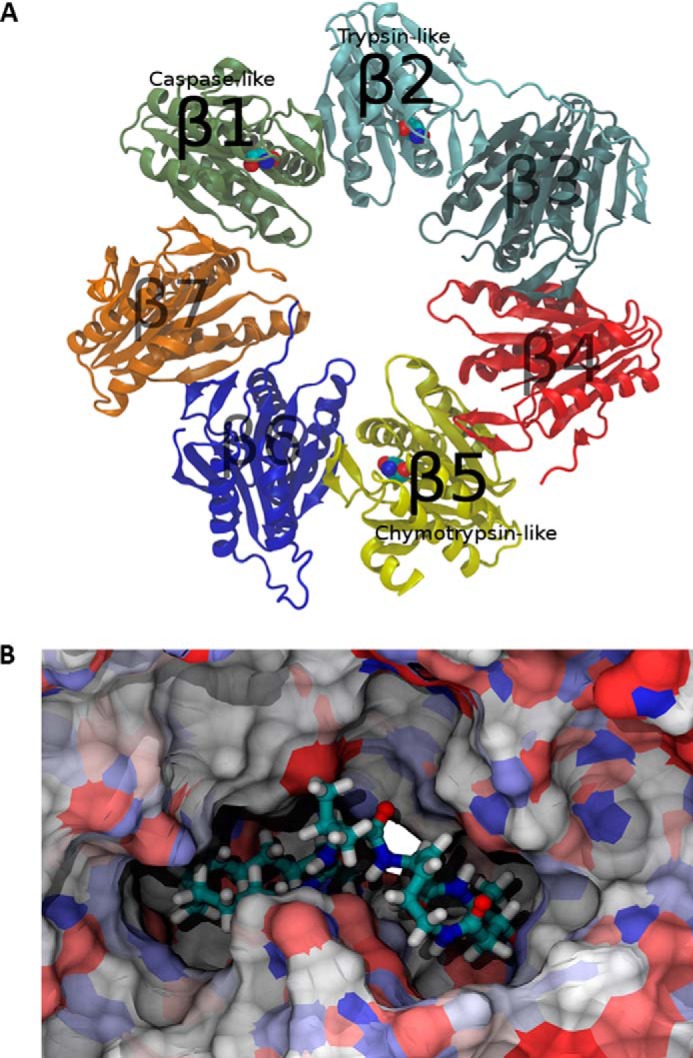
Human proteasome catalytic β ring homology model. A, protein domains are differentiated by color. Catalytic active site Thr-1 residues shown with space fill graphical representations. B, TIR-199 bound to the CT-L subunit of the human proteasome. A unique pocket exposed to the surface of the proteasome complex is visible behind TIR-199. Extension of the hydrophobic tail into this pocket leads to increased affinity and stability.
In Vitro Proteasome Activity Assay
To determine the anti-proteasome activity of TIR-199 in the in vitro environment, we measured the three catalytic activities (β1, β2, and β5) as described previously (12, 13, 15). This assay uses purified 20S constitutive proteasome from human erythrocytes or immunoproteasome from human peripheral blood mononuclear cells (Enzo Life Sciences) and luminogenic substrates (Z-LRR-GloTM,Z-nLPnLD-GloTM, and Suc-LLVY-GloTM), specific for the three β1, β2, and β5 catalytic subunit activities (also referred to T-L, C-L, and CT-L activities, respectively). The 20S proteasome and the specific luminogenic substrates were incubated individually with compounds at increasing concentrations (0–10 μm). Controls included the natural product SylA (20 μm) and BTZ (0.01 μm). Following cleavage by the 20S proteasome, the substrate for luciferase (aminoluciferin) is released, allowing the luciferase reaction to produce light. The luminescence was recorded as relative light units after a 30-min incubation on a 96-well microplate luminometer.
In Vivo Proteasome Activity Assay
To determine the anti-proteasome activity of TIR-199 in the culture environment, the proteasome-Glo inhibition assay was performed as described previously (13, 21). Solid white 96-well microtiter cell culture plates were seeded with cells, and proteasome inhibition was measured using the proteasome GloTM reagent according to the manufacturer's instructions (Promega). In brief, MM1.RL or MYCN2 cells were treated with TIR-199 at different concentrations (0–10 μm) as indicated and incubated for 2 h, followed by incubation for 10 min with 100 μl of proteasome Glo reagent, containing the bioluminescent substrates Suc-LLVY-aminoluciferin, Z-nLPnLD-aminoluciferin, and Z-LRR-aminoluciferin to measure the CT-L, C-L, and T-L activities, respectively. Luminescence was measured with a Multi-Mode SynergyTM MX microplate reader (BioTek, Inc.) and expressed as relative light units. SylA and BTZ were used as controls.
In Vivo Proteasome Sensor Assay
The proteasome sensor activity assay was performed in HEK-293 cells according to the manufacturer's instructions (Clontech). ZsProSensor-1 is a proteasome-sensitive fluorescent reporter, producing a fusion of green fluorescent protein (GFP) with a degradation domain that targets the protein for rapid degradation by the proteasome. As a consequence, green fluorescence accumulates in those cells in which the proteasome is inhibited. Unlike the previous cell culture-based assay, this test does not require the exogenous addition of bioluminescent substrates because it expresses an internal probe. Cells were transfected with the ZsProSensor-1 vector and tested in the presence of increasing concentrations (0–1 μm) of TIR-199 or BTZ for 24 h. GFP expression was quantified using an ELISA reader and pictures taken with an immunofluorescence microscope.
Mammalian Cell Cultures
The MM cell line MM1.RL is derived from the parent cell line MM1, established from peripheral blood of a multiple myeloma patient with acquired resistance to steroid-based (dexamethasone) therapy and was kindly provided by N. Krett (Northwestern University) (32). The human NB cell line MYCN2 was generated with cell line SH-EP, an S-type subclone of the parent cell line SK-N-SH. MYCN2 cells were kindly provided by Dr. J. Shohet (Texas Children's Hospital) (33). Human embryonic kidney 293 (HEK-293) cells were from the American Type Culture Collection (ATCC) (Manassas, VA). HepG2 cells were tested at Cerep, Inc. (Redmond, WA). Cell lines were maintained in RPMI 1640 medium (MM1.RL, MYCN2), DMEM medium (HEK-293) (Mediatech Inc., Manassas, VA) or minimum essential medium α growth medium (HepG2). RPMI 1640 and DMEM medium contained 10% (v/v) heat-inactivated fetal bovine serum (Invitrogen), supplemented with penicillin (100 units/ml) and streptomycin (100 μg/ml). Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2 and seeded 16–24 h before the start of the assay.
Cell Viability Assay
The CellTiter 96® aqueous non-radioactive cell proliferation assay is a homogeneous, colorimetric method for determining the number of viable cells in cell culture assays. The assay is composed of solutions of a novel tetrazolium compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfofenyl)-2H-tetrazolium, inner salt; MTS) and an electron coupling reagent (phenazine methosulfate) (Promega, San Luis Obispo, CA). MTS is bioreduced into soluble formazan product by dehydrogenase enzymes found in metabolically active cells. The viability of cancer cells was determined after 24 h of treatment with TIR-199 at the indicated concentrations (0–10 μm) by measuring the absorbance of the formazan product at 490 nm using a Multi-Mode SynergyTM MX microplate reader (BioTek, Inc., Winooski, VT) as described previously (14). Data were expressed in percentage of cell viability relative to untreated control cells. SylA and BTZ were used as positive controls.
NCI-60 Human Tumor Cell Line Screen
The NCI-60 cell line panel includes 60 human tumor cell lines. The effect of SylA and TIR-199 on tumor cell growth was tested at the National Cancer Institute, Developmental Therapeutics Program (NCI-DTP), according to their standard protocols and as published previously (34). For additional information, see the NCI-DTP Web site.
Adverse Drug Reaction (ADR) Profiling
The ADR profile was performed at Cerep, Inc. Many ADRs are linked to off-target activities at a great variety of cellular receptors and enzymes. The effect of TIR-199 (10 μm) against the kidney organ system was tested by measuring 17 molecular targets selected on the basis of known associations of individual targets with serious ADRs as well as statistical associations derived from Cerep's proprietary BioPrint®. The following receptors/targets were individually tested: 5-HT2A, 5-HT2C, α2B-adrenergic, β1-adrenergic, dopamine 2S, muscarinic acetylcholine (M2, M4, M5), neurokinin 2, urotensin, and parathyroid hormone 1 receptor. In addition, activation of adenylate cyclase C and inhibition of enzymes COX2, angiotensin-converting enzyme, dipeptidyl peptidase IV, phospholipase C, and acetylcholinesterase were measured.
In Vitro Cytotoxicity Assay
This high content cytotoxicity assay was performed at Cerep, Inc. Five end points were simultaneously measured in individual cells, avoiding the drawbacks of classic in vitro cytotoxicity assays, which measure nonspecific and late occurring cytotoxic events. The five end points are cellular parameters, such as mitochondrial membrane potential (tetramethylrhodamine methyl ester (TMRM)) and intracellular free calcium (Fluo-4), and more classic parameters, such as nuclear size (Hoechst), membrane permeability (TOTO-3), and cell number (Hoechst), in live HepG2 cells. In brief, HepG2 cells (passage 1–15) were plated in 96-well poly-d-lysine-coated plates at 3,000 cells/well in minimum essential medium α growth medium (HepG2) 16–24 h before the start of the assay. One hour before the addition of test compound, the cells were equilibrated with assay medium containing 1% FBS. Cells were treated with TIR-199 (100 nm) or BTZ (5 nm) for 72 h. At the end of the incubation period, the cells were loaded for 1 h with a dye mixture containing Hoechst TMRM, Fluo-4, and TOTO-3. Plates were scanned with an automated fluorescent microscope. Image analysis software was used to quantitate surface area (nuclear size) and fluorescence intensity in defined cellular areas. Data were normalized and expressed as a percentage of effect relative to the untreated controls. Data were also normalized using a reference compound (cerivastatin), whose maximum effect at any concentration was considered the 100% effect.
In Vivo Acute Toxicity Study
The maximum tolerated dose (MTD) study was performed at the NCI-DTP. To investigate acute toxicity of TIR-199 in vivo, the MTD was determined in female athymic nude mice. TIR-199 was given by intraperitoneal (IP) injection QD × 1 on day 0, at 12.5, 25.0, and 50.0 mg/kg/dose (groups 1–3; n = 1/group). Injection volume was 0.05, 0.1, or 0.2 ml/10 g of body weight (2.5 mg/ml, homogeneous smooth suspension in 10% DMSO in saline/Tween 80). The mice were held for 14 days post-dosing to monitor them for delayed toxicity (bone marrow suppression or irreversible liver/kidney and other organ damage). For additional information, see https://dtp.cancer.gov.
In Vivo Hollow Fiber Assay
The in vivo hollow fiber assay was performed at the NCI-DTP. A solid tumor efficacy mouse model based on cell growth inside biocompatible hollow fibers was used to provide quantitative indices of drug efficacy in heterogeneous tumors with minimal time and material expenditures (35). Small hollow fibers (1 mm in diameter, 2 cm long, molecular mass exclusion of 500,000 Da), made of polyvinylidene fluoride and containing cells from human tumors, were inserted underneath the skin and in the body cavity of the mouse. A standard panel of 12 tumor cell lines, including lung, breast, colon, melanoma, ovarian, and central nervous system, was used for the routine hollow fiber screening of in vitro activities. Each mouse received three tumor cell lines as intraperitoneal implants and three as subcutaneous implants. A total of 24 mice were used in 8 groups (n = 3/group, each group representing two dose levels with 4 experiments, 3 cell lines/experiment) and treated by intraperitoneal injection with TIR-199 starting on day 3 or 4 following fiber implantation and continuing daily for 4 days. TIR-199 was administered at two doses (9.4 or 6.3 mg/kg/dose). The selected high and low doses were based on the MTD determined in the in vivo acute toxicity study, using the formulas, high dose = (MTD × 1.5)/4 and low dose = 0.67 × high dose. The fibers were collected from the mice on the day following the fourth compound treatment and subjected to the stable end point 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. The optical density of each sample was determined by spectrophotometry at 540 nm, and the mean of each treatment group was calculated. The percent net growth for each cell line in each treatment group is calculated and compared with the percent net growth in the vehicle-treated controls. A 50% or greater reduction in percent net growth in the treated samples compared with the vehicle control samples is considered a positive result. Each positive result is given a score of 2, and all of the scores are totaled for a given compound. The maximum possible score for an agent is 96 (12 cell lines × 2 sites × 2 dose levels × 2). A compound is considered effective and of potential interest for further xenograft studies at NCI-DTP, if it has a combined intraperitoneal + subcutaneous score of ≥20. Appropriate controls were included (n = 12 for blank fibers, n = 24 for vehicle controls, and n = 24 for positive controls using paclitaxel). For additional information, see https://dtp.cancer.gov.
Statistical Analyses
All experiments were performed in three independent experiments (n = 3) unless otherwise stated. Error bars indicate the S.E. Data were prepared using Excel (Microsoft, Inc., Redmond, WA) and Prism version 6 (GraphPad Software, La Jolla, CA).
Results
Structure and Synthesis of TIR-199
The need to improve both the intrinsic potency and physicochemical properties of the syringolins led us to related natural products, the glidobactins, which were discovered based on their activity against tumor cell lines (36, 37). They share a 12-membered macrodilactam, but the glidobactins have a much less hydrophilic side chain and also differ in the amino acid (valine versus alanine) used biosynthetically to form the α,β-unsaturated amide (Fig. 1). Because that grouping is the pharmacophore of both the syringolins and glidobactins that reacts with the essential Thr-1 hydroxy group of proteasome β subunits, we assumed that a less sterically demanding group adjacent to the unsaturated amide could enhance activity. These considerations suggested a number of structural analogs, one of which, TIR-199, proved the most interesting (Fig. 1).
FIGURE 1.

The chemical structures of SylA, GlbA, and their structural analog TIR-199. SylA is a natural product from Pseudomonas syringae pv. syringae. GlbA is from Polyangium brachysporum. Together, this group of proteasome inhibitors is referred to as syrbactins. TIR-199 is one of several syrbactin-derived structural analogs with superior inhibitory activities. The molecular weight of TIR-199 is 534.
The synthetic route to TIR-199 is provided in Scheme 1 and described in detail in the supplemental Methods. Most of the route follows our reported syringolin A synthesis (22), with the exception of the use of alaninol. From an (S)-vinylglycine starting material, the route entails 10 steps and proceeds in 10% overall yield.
Computational Modeling and Docking of TIR-199 into the Proteasome
Structure-activity relationships of the dozens of natural and synthetic syrbactins were examined to assist in understanding the intrinsic activity of TIR-199 against the proteasome (Fig. 2A). Replacement of the syringolin R3 with a straight-chain alkane was first reported by Kaiser's group (15) and, while expected to significantly improve the cell-based activity by eliminating charge, also benefitted its intrinsic potency. Regarding the change of the larger R1 of syringolin A to a methyl group, the proteasome's catalytic Thr-1 hydroxyl approaches the unsaturated lactam adjacent to this position, making a larger grouping there a significant steric impediment (Fig. 2B). Syringolin A is more potent than its monounsaturated natural relative syringolin B (A-B = CH2CH2), and conformational analysis suggests that the effect of the second alkene is to introduce strain into the macrodilactam and make it more reactive with the proteasome.
The bound state affinities for TIR-199 to each active site within the human proteasome were calculated using molecular mechanics/generalized Born/surface area energetic post-simulation analysis of molecular dynamics trajectories. The most favorable interaction was found between TIR-199 and CT-L subunit due to unique non-polar stabilization. With decreasing non-polar affinity, the selectivity continues with the C-L subunit, followed by the T-L subunit. The average polar and non-polar potential energies are summarized in Table 1.
TABLE 1.
Bound state potential energy between TIR-199 and each catalytic subunit of the human proteasome
Values are calculated for and averaged over each simulation snapshot of the simulation trajectories (kcal/mol).
| Proteasome/TIR-199 affinity | Polar | Non-polar | Total |
|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | |
| C-L | −26.24 | −36.37 | −62.61 |
| T-L | −25.96 | −32.39 | −58.35 |
| CT-L | −26.66 | −41.6 | −68.26 |
TIR-199 Inhibits Catalytic Subunit Activities of the Proteasome
To measure the effect of TIR-199 on the proteasome, we measured the β1 (C-L), β2 (T-L), and β5 (CT-L) catalytic subunit activities of the proteasome. In vitro experiments with either purified 20S constitutive proteasome from human erythrocytes or 20S immunoproteasome from human peripheral blood mononuclear cells showed that TIR-199 strongly inhibited CT-L and T-L but not C-L activity of the proteasome in a dose-dependent manner (Fig. 3). BTZ was used as a control and inhibited all three constitutive proteasome activities indiscriminately but, interestingly, did not inhibit the immunoproteasome T-L activity. TIR-199 displayed significantly improved potencies on the constitutive proteasome CT-L activity compared with SylA (Ki of 0.018 and 0.843 μm (12), respectively) but was less potent than BTZ (Ki < 0.01 μm) (Fig. 3). Interestingly, TIR-199 was less effective against the immunoproteasome CT-L activity (Ki of ∼0.075 μm) (Fig. 3B). Table 2 compares the activity of TIR-199 against previously reported syrbactins (SylA, GlbA, and SylA-GlbA hybrid) and bortezomib (12, 13, 38). The activity of TIR-199 was comparable with the activity of the SylA-GlbA hybrid.
FIGURE 3.

TIR-199 inhibits the proteasome activity in vitro. The inhibitory effect of increasing concentrations of TIR-199 (0–5 μm) on the CT-L, C-L, and T-L catalytic subunit activities of the constitutive proteasome (A) and immunoproteasome (B) were measured, using luminogenic substrates as described under “Experimental Procedures.” BTZ was used as a positive control (0.01 μm). The relative light units for each catalytic subunit activity were expressed as Remaining Activity in %, where 100% activity represents the control (untreated proteasome). Data represent the average of three independent experiments (n = 3); bars, mean ± S.E. (error bars).
TABLE 2.
Inhibitory effects of syrbactins on proteasome activity in vitro
Three proteasomal activities were measured: CT-L (β5), T- L (β2), and C-L (β1). Values in this table are either derived from this study (Fig. 3A) or are historic values from the published literature. ND, not determined.
To verify the potency against the proteasome in actively growing cell cultures, we tested TIR-199 in a cell-based, in vivo proteasome activity assay. TIR-199 strongly inhibited the catalytic C-L, T-L, and CT-L subunit activities in MM1.RL cells in a dose-dependent manner (Fig. 4, A, C, and E). Similar data were obtained with MYCN2 NB cells, confirming the activity of proteasome inhibition in two distinct cancer cell types (Fig. 4, B, D, and F). Slightly higher doses of TIR-199 were required in NB cells to achieve the effects observed in MM cells. The T-L activity in NB cells was not significantly reduced.
FIGURE 4.

TIR-199 inhibits the proteasomal activity and cell viability of actively dividing cancer cells. TIR-199 inhibited the CT-L (A and B), C-L (C and D), and T-L (E and F) catalytic subunit activities of the dexamethasone-resistant MM cell line MM1.RL (A, C, and E) and the NB cell line MYCN2 (B, D, and F), respectively. Cells were treated for 2 h with TIR-199 at various concentrations (0–10 μm). SylA and BTZ were used as controls. The proteasomal activities were measured as outlined under “Experimental Procedures.” TIR-199 induced rapid death of MM1.RL (G) and MYCN2 cells (H). Cells were treated over a period of 24 h with TIR-199 at various concentrations (0–10 μm). SylA and BTZ were used as a control. The viability of cells is expressed as percentage of cell survival relative to untreated control cells and was determined by an MTS assay as outlined under “Experimental Procedures.” Data represent the average of three independent experiments (n = 3); bars, mean ± S.E. (error bars).
The effect of TIR-199 on the proteasome in actively growing cells was independently confirmed with the proteasome sensor activity assay in which the blockage of proteasome function is reflected by the accumulation of an internally overexpressed substrate (rather than externally added substrate, as shown in Fig. 4) fused to GFP. TIR-199-treated cells as well as BTZ-treated cells showed strong accumulation of GFP compared with untreated control cells (Fig. 5). This result shows that TIR-199 is able to penetrate the membrane of intact cells and inhibits the proteasome of actively dividing cells.
FIGURE 5.
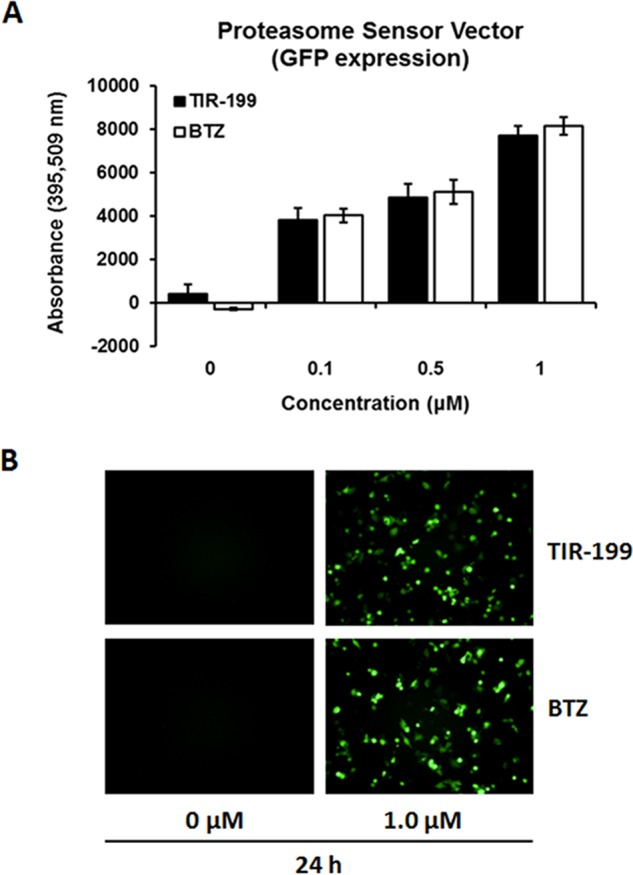
TIR-199 inhibited the proteasome activity in human embryonic kidney cells HEK-293 cells using a proteasome sensor cell transfection assay. A, HEK-293 cells transfected with the ZsProSensor-1 plasmid were treated with the indicated concentrations of TIR-199 and BTZ and quantified for GFP expression as a marker for proteasome inhibition. B, fluorescent microscope images of GFP-expressing cells treated with TIR-199, BTZ, or control, after 24 h of exposure. Error bars, S.E.
TIR-199 Induces Cell Death in Multiple Myeloma and Neuroblastoma Cells
To investigate whether TIR-199 induces cancer cell death, we tested increasing TIR-199 concentrations against MM and NB cells. TIR-199 killed the MM cell line MM1.RL very rapidly at the lowest concentration (0.05 μm) and inhibited NB cell viability in a dose-dependent manner between 0 and 10 μm (Fig. 4, G and H). The concentrations to induce 50% cell death (IC50) were estimated at <0.05 and ∼0.1 μm for MM and NB cells, respectively. These IC50 values reflect a >250-fold increase in potency compared with SylA that has an IC50 of 20–25 μm (11) and up to 39 μm in MM1.RL cells (14). The fact that TIR-199 is able to induce strong cell death in the MM1.RL cell line is particularly important and confirms that proteasome inhibition is a successful strategy to treat dexamethasone-resistant MM tumors. Table 3 compares the effect of TIR-199 on cancer cell death against previously reported syrbactins (SylA, GlbA, and SylA-GlbA hybrid) and bortezomib (11, 13, 14). The activity of TIR-199 was better than or comparable with that of GlbA and SylA-GlbA hybrid.
TABLE 3.
Effect of TIR-199 and other syrbactins on cancer cell death
BTZ was used as a control for comparison. MYCN2, neuroblastoma cell line SH-EP, which was previously transfected with a tetracycline on/off plasmid system to allow for controlled MYCN expression (33). SH-EP, S-type subclone of the cell line SK-N-SH. Data represent the mean values of three independent experiments, each performed in duplicate wells (n = 6). Values are either derived from this study (Fig. 4, G and H) or are historic values from the published literature. ND, not determined.
NCI-60 Human Tumor Cell Line Screen with TIR-199
To explore its utility in other cancer cell types, TIR-199 was tested at the NCI-DTP. The results confirmed that TIR-199 induces dose-dependent cell death in a wide range of tumor cell types, including breast, CNS, colon, kidney, lung, ovarian, skin, and prostate cancer and leukemia. Of note, TIR-199 was active in a panel of kidney tumor cell lines, with exceptionally high activities in four cell lines (RXF 393, TK-10, A498, and SN12C) (Fig. 6). Compared with other groups of the NCI-60 cell panel, the renal cancer panel often does not respond well in drug screens, and, therefore, the results with TIR-199 are particularly encouraging.5 TIR-199 also exhibited high activity in other cell lines, including RPMI-8226 (leukemia), NCI-H522 (non-small cell lung cancer), KM12 (colon cancer), SNB-75 (CNS cancer), LOX IMVI (melanoma), OVCAR-3 (ovarian cancer), PC-3 (prostate cancer), and BT-549 (breast cancer). Overall, our data suggest that TIR-199 is a promising anticancer agent with broad application potential against various tumor groups and selective activities in certain cell lines within each tumor group. To further demonstrate the improved anti-tumor cell activity, the initial one-dose mean graph of TIR-199 was compared side-by-side with that for SylA, clearly showing that TIR-199 has significantly higher activity potential than the natural product SylA (Fig. 7).
FIGURE 6.

Antiproliferative effect of proteasome inhibitor TIR-199 in the NCI-60 human tumor cell line panel. Dose-response curves for TIR-199 (NSC:761526). A, a panel of 60 human tumor cell lines representing nine different cancerous tissues of origin (leukemia, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, and breast cancer) was tested at the NCI-DTP in the presence of TIR-199 at five concentrations. TIR-199 was tested over a 10,000-fold concentration range in a 2-day assay and exhibited dose-dependent growth inhibition to various degrees, in all tested tumor cell lines. B, dose-response data of A were used to calculate three end points for each cell line: GI50 (the log10 of the concentration that caused 50% growth inhibition), TGI (the log10 of the concentration that caused total growth inhibition), and LC50 (the log10 of the concentration that caused 50% lethality). For each end point, the mean across all the cell lines was calculated. The GI50 data are graphed as the difference of the GI50 for a particular cell line from the mean GI50. Cell lines that are more sensitive are represented as bars deflecting to the right of the mean, and less sensitive cell lines project to the left of the mean. TGI and LC50 mean graphs are generated in a similar fashion. One representative data set of three independent experiments is shown (n = 3), except for the leukemia cell panel (n = 2). For additional information about the NCI-60 cell line panel, see Ref. 34 and “Experimental Procedures.”
FIGURE 7.

Mean graph display of NCI-60 cell line screening data for TIR-199. Shown is a one-dose mean graph display of NCI-60 cell line screening data for TIR-199 (NSC:761526) (A) and SylA (NSC:749671) (B) at 10 μm. Bars deflecting to the right of the mean indicate high sensitivity, and bars deflecting to the left of the mean indicate low sensitivity to TIR-199 (A) and SylA (B). For additional information about the NCI-60 human cancer cell line screen, see Ref. 34 and “Experimental Procedures.” Experiments were performed at the NCI-DTP.
Preclinical in Vitro and in Vivo Evaluation of TIR-199
Drug-induced toxicity is one of the major causes of failure during drug development and the major reason for removal of approved drugs from the market. To assess the potential of TIR-199 for preclinical development, a broad range of biological screening tests were performed. Because one potential therapeutic area identified by the NCI-DTP for TIR-199 is renal cancer (Fig. 6), we were interested in an ADR assessment for kidney targets. The Cerep ADR kidney panel included 17 assays, which determine the binding of TIR-199 (10 μm) to the following targets/receptors: 5-HT2A, 5-HT2C, α2B-adrenergic, β1-adrenergic, dopamine 2S, muscarinic acetylcholine (M2, M4, M5), neurokinin 2, urotensin, and parathyroid hormone 1 receptor. In addition, activation of adenylate cyclase C and inhibition of enzymes COX2, angiotensin-converting enzyme, dipeptidyl peptidase IV, phospholipase C, and acetylcholinesterase were measured. For the radioligand binding assays to the receptors, the strongest antagonism observed was 8% at the test concentration. Functional assays of parathyroid hormone 1 receptor and adenylate cyclase activation showed no effect of TIR-199. The enzymatic activity most affected by it was dipeptidyl peptidase IV, which was inhibited 26% at the test concentration. Because an effective concentration for proteasome inhibition by TIR-199 is 1,000-fold lower than these test concentrations, none of these off-target effects is considered significant (data not shown).
Additional assays were performed to determine the cytotoxicity of TIR-199 in HepG2 cells. Readouts were based on five end points that are measured simultaneously in individual cells: cell number (decreased numbers indicate cell death and/or decreased proliferation), free calcium release (uncontrolled increase in cytoplasmic calcium indicates cellular toxicity), membrane permeability (disruption of the cytoplasmic membrane indicates late cellular toxicity), mitochondrial membrane potential (indicator of respiratory capacity and cellular energetics), and nuclear size (nuclear shrinkage by chromatin condensation indicates apoptotic cell death). TIR-199 (100 nm) was tested and compared in parallel with BTZ (5 nm). The results are summarized in Table 4. Other than in direct cell killing, TIR-199 and BTZ had similar effects on intracellular calcium, nuclear size, membrane permeability, and mitochondrial membrane potential.
TABLE 4.
Percentage of effect (percentage of cytotoxicity) relative to untreated controls
Values include the following: cell number, percentage reduction; intracellular free calcium, percentage increase; nuclear size, percentage reduction; membrane permeability, percentage increase; mitochondrial membrane potential, percentage reduction. Data represent the mean of triplicate wells (n = 3).
| Test | TIR-199 (100 nm) | BTZ (5 nm) |
|---|---|---|
| % | % | |
| Cell number | 62 | 98 |
| Intracellular free Ca2+ | 27 | 29 |
| Nuclear size | 84 | 92 |
| Membrane permeability | 93 | 99 |
| Mitomembrane potential | 98 | 100 |
Due to the encouraging results of the NCI-60 cell line panel (Fig. 6), the NCI-DTP performed in vivo studies in mice to determine the MTD and moved TIR-199 forward into in vivo hollow fiber assays. Non-tumored athymic nude female mice were injected (intraperitoneally) with a single dose of TIR-199 at 12.5, 25, and 50 mg/kg/dose (groups 1–3; n = 1/group). At 25 mg/kg, treated mice were alive at day 19, whereas at 50 mg/kg, the mice died on day 2, suggesting an MTD for TIR-199 of 25 mg/kg (Table 5). For comparison, the classical anticancer drug paclitaxel (Taxol®) is highly active at 15 mg/kg and lethal at a 30 mg/kg.5 Importantly, TIR-199 at 9.4 mg/kg/dose and 6.3 mg/kg/dose (total of four doses, intraperitoneally) was effective in hollow fiber studies in mice using 12 tumor cell lines (hollow fibers implanted either intraperitoneally or subcutaneously) and inhibited the cell proliferation to various degrees in representing tumors of the breast, CNS, colon, lung, ovary, and skin (Table 6). Most notably, TIR-199 inhibited colon, melanoma, and ovarian tumor cells. Overall, tumor cells in intraperitoneally implanted hollow fibers responded more readily to intraperitoneally administered TIR-199, with the exception of melanoma tumors, which were also significantly inhibited in subcutaneously implanted hollow fibers. Most but not all tumor cells that were inhibited by TIR-199 in the in vivo hollow fiber assay were also highly responsive in the NCI-60 cell line screen (Fig. 6).
TABLE 5.
In vivo acute toxicity study
The MTD of TIR-199 was determined in athymic nude female mice as described under “Experimental Procedures.” TIR-199 was tested at three doses (intraperitoneal single injection), and an MTD of 25 mg/kg was determined. Groups 1–3; n = 1/group. Experiments were performed at the NCI-DTP. IP, intraperitoneal.
| Group | Dose (IP) | Schedule | Death days | Survival/Total (day 19) |
|---|---|---|---|---|
| 1 | 12.5 mg/kg/dose | Daily × 1, day 0 | 1/1 | |
| 2 | 25.0 mg/kg/dose | Daily × 1, day 0 | 1/1 | |
| 3 | 50.0 mg/kg/dose | Daily × 1, day 0 | 2 | 0/1 |
TABLE 6.
In vivo hollow fiber assays to assess anti-tumor efficacy of TIR-199 (NSC:761526) in mice
Twelve cancer cell lines were tested in mice, representing the following tumor groups: colon (COLO 205, SW-620), skin/melanoma (LOX IMVI, MDA-MB-435, UACC-62), breast (MDA-MB-231), non-small cell lung (NCI-H23, NCI-H522), ovary (OVCAR-3, OVCAR-5), and CNS (SF-295, U251). These 12 cell lines represent a standard panel selected by the NCI-DTP based on good tumor growth performance in the hollow fiber assay. Cell lines were implanted (intraperitoneally or subcutaneously), and mice were treated (intraperitoneally) at 6.4 or 9.3 mg/kg/dose daily for 4 days, as described under “Experimental Procedures.” Values represent the percentage of cell growth. Underlined values indicate cell lines with ≥50% growth inhibition, which is considered a positive result to determine the total score. The total score for TIR-199 was 20 (intraperitoneal 2 × 8 cell lines = 16 and subcutaneous 2 × 2 cell lines = 4). Experiments were performed at the NCI-DTP. HF, hollow fiber; IP, intraperitoneal; SC, subcutaneous.
| Cell line | Percentage of cell growth |
||||
|---|---|---|---|---|---|
| HF implant (IP) 6.30 mg/kg (IP) | HF implant (IP) 9.40 mg/kg (IP) | HF implant (SC) 6.30 mg/kg (IP) | HF implant (SC) 9.40 mg/kg (IP) | Tumor group | |
| COLO 205 | 78 | 25 | 100 | 60 | Colon |
| SW-620 | 109 | 91 | 106 | 73 | Colon |
| LOX IMVI | 162 | 53 | 107 | 28 | Skin/melanoma |
| MDA-MB-435 | 44 | 24 | 80 | 48 | Skin/melanoma |
| UACC-62 | 115 | 88 | 114 | 106 | Skin/melanoma |
| MDA-MB-231 | 92 | 66 | 87 | 97 | Breast |
| NCI-H23 | 85 | 75 | 101 | 108 | Non-small cell lung |
| NCI-522 | 95 | 49 | 124 | 96 | Non-small cell lung |
| OVCAR-3 | 97 | 45 | 93 | 65 | Ovary |
| OVCAR-5 | 44 | 22 | 95 | 75 | Ovary |
| SF-295 | 72 | 48 | 95 | 77 | CNS |
| U251 | 97 | 54 | 115 | 97 | CNS |
Discussion
Natural products continue to offer attractive lead compounds for novel anticancer therapeutics (39), with the syrbactins as just one recent example. It is notable that the Food and Drug Administration-approved proteasome inhibitor carfilzomib closely resembles the peptide natural product epoxomicin. Likewise, TIR-199 addresses deficits of the original lead, SylA, through reduction of hydrophilicity and enhancement of potency. Its ligand efficiency, a broadly used measure of the potential of a compound for pharmaceutical lead development (40), based on its potency at the proteasome CT-L site, is 0.28, within the desirable range.
Our understanding of TIR-199 activity was enhanced through simulation of its interaction with the chemical environment of each subunit when bound to the three catalytic sites of the proteasome. This approach revealed an interaction of the TIR-199 side chain with a proteasome pocket that is absent in any previous drug-proteasome structure (41). A unique pocket behind the binding site of the CT-L subunit led to exceptionally high non-polar affinity and conformational stability of TIR-199 in the binding site (Fig. 2B). In addition, a unique rotation after the peptide bond was required for the ligand to extend into the deep pocket, lowering the internal energy of the ligand by 2.5–3 kcal/mol relative to the other starting conformations.
In the in vitro experiments, TIR-199 inhibited the β2 and β5 catalytic subunit activities in a dose-dependent fashion, with the highest affinity toward the β5 subunit and only minimal activity toward the β1 subunit. This is similar to what was observed with SylA (12, 15). Interestingly, in the cell-based proteasome activity assay, all three activities were inhibited in a similar fashion in MM but not in NB cells. The reason for these differences is not clear but may in part be due to cell type-specific differences and/or differences in assay conditions (in vitro proteasome activity assay versus a cell-based in vivo proteasome activity assay) and the different sources of luminogenic substrates used in each assay. TIR-199 strongly induced tumor cell death in MM and NB cells but also in a wide range of other tumor cell lines, as shown in the NCI-60 cell panel screening results (Fig. 6). Importantly, four of eight cell lines in the renal cancer panel that traditionally responds poorly to compounds tested in the NCI-DTP drug pipeline were exceptionally sensitive to TIR-199. In the hollow fiber mouse tumor model, TIR-199 was most notably active in colon, melanoma, and ovarian tumors (Table 6). Of note, several leading drugs like bortezomib, paclitaxel, romidepsin, eribulin, sipuleucel-T, and dinutuximab (Ch14.18) were initially studied at the NCI-DTP using hollow fiber assays in mice to obtain quantitative indices of in vivo drug efficacy.
Proteasome inhibitors might be explored for use in diseases unrelated to cancer, in which aberrant regulation of the proteasome or immunoproteasome has been observed. Such diseases include Huntington disease (42), Alzheimer disease (43), macular degeneration (44), inflammatory bowel disease (Crohn disease, ulcerative colitis) (45, 46), and rheumatoid arthritis (Sjogren syndrome) (47). In some instances, the use of proteasome-specific inhibitors that directly target the catalytic core may be effective. For example, the proteasomal CT-L activity is increased in neurosensory retina with disease progression in age-related macular degeneration (44). In Crohn disease and ulcerative colitis, proteasome inhibitors may be effective in blocking the proteasome-mediated activation of the NF-κB pathway in inflammatory bowel disease patients (45, 46). In other instances, inhibitors that alter the expression of immunoproteasome-specific subunits might lead to novel treatment options. For example, the immunoproteasome-specific subunits LMP2 or LMP7 are increased in Huntington disease neurodegeneration (42), and tissue-specific up-regulation of LMP7 is characteristic in patients with Sjogren syndrome (47).
In conclusion, this is the first study that tests a syrbactin-related proteasome inhibitor in animals. We demonstrate that TIR-199 has broad range anti-tumor activity in vivo. Therefore, TIR-199 and novel TIR-199-derived analogs bear significant potential for further preclinical development into a clinical drug to treat MM, kidney, and other cancer forms.
Author Contributions
J. O.-A. carried out all proteasome activity and cell viability experiments. T. R. I.-R. and S. A. synthesized TIR-199. C. C. R. and C. A. C. made the structural models. L. P. Y. performed the statistical analyses of proteasome and cell viability assays. The NCI-DTP screened the NCI 60-cell panel and performed the in vivo MTD and hollow fiber studies. A. S. B. and M. C. P. designed the study. A. S. B., M. C. P., C. A. C, and C. C. R. wrote the paper with comments from all other authors.
Supplementary Material
Acknowledgments
We thank the National Cancer Institute, Developmental Therapeutics Program (NCI-DTP) for performing the NCI-60 human tumor cell line screen, in vivo acute toxicity study, and the in vivo hollow fiber assays with TIR-199. We thank Dr. Paul Grothaus (NCI-DTP) for support and for insightful discussion of results. We are grateful to Nancy L. Krett (Northwestern University, Chicago, IL) and Jason Shohet (Texas Children's Hospital, Houston, TX) for providing multiple myeloma cell line MM1.RL and neuroblastoma cell line MYCN2, respectively. We are indebted to Dr. Dirk Geerts (Rotterdam, The Netherlands) for excellent scientific discussion and for editing the manuscript. Cerep, Inc. performed the ADR profiling and HepG2 in vitro cytotoxicity studies.
This work was supported in part by funding from the Robert C. Perry Fund of the Hawaii Community Foundation (HCF 09ADVC-45411) (to A. S. B.) and internal funds from the Daniel K. Inouye College of Pharmacy (to A. S. B.). Further support was provided by the University of California Cancer Research Coordinating Committee (to M. C. P.) and postdoctoral fellowships from UC-MEXUS and CONACYT (to T. R. I.-R.). This work was also supported by contributions via experiment.com from Francois Souchay, Yong Rok Lee, Bruce Liu, Ann Adenbaum, Alan Schramm, and Robert Patton. A. S. B. is a named inventor of a United States patent (US 8,597,904, December 3, 2013) that relates to pharmaceutical compositions for the treatment of conditions responsive to proteasome inhibition. This patent is assigned to PONO Pharma Inc. (Honolulu, HI), for which A. S. B. currently serves as an independent agent. M. C. P. is a named inventor on a United States patent application (US 2015-0141392 A1) assigned to the University of California concerning synthetic macrocyclic compounds having proteasome inhibitory activity.

This article contains supplemental Methods.
P. Grothaus, personal communication.
- MM
- multiple myeloma
- BTZ
- bortezomib
- SylA
- syringolin A
- MTD
- maximum tolerated dose
- CT-L
- chymotrypsin-like
- C-L
- caspase-like
- T-L
- trypsin-like
- GlbA
- glidobactin A
- NB
- neuroblastoma
- Z
- benzyloxycarbonyl
- HEK
- human embryonic kidney
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfofenyl)-2H-tetrazolium, inner salt
- NCI-DTP
- National Cancer Institute, Developmental Therapeutics Program
- ADR
- adverse drug reaction.
References
- 1. Raab M. S., Podar K., Breitkreutz I., Richardson P. G., and Anderson K. C. (2009) Multiple myeloma. Lancet 374, 324–339 [DOI] [PubMed] [Google Scholar]
- 2. Bross P. F., Kane R., Farrell A. T., Abraham S., Benson K., Brower M. E., Bradley S., Gobburu J. V., Goheer A., Lee S. L., Leighton J., Liang C. Y., Lostritto R. T., McGuinn W. D., Morse D. E., Rahman A., Rosario L. A., Verbois S. L., Williams G., Wang Y. C., and Pazdur R. (2004) Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 10, 3954–3964 [DOI] [PubMed] [Google Scholar]
- 3. Herndon T. M., Deisseroth A., Kaminskas E., Kane R. C., Koti K. M., Rothmann M. D., Habtemariam B., Bullock J., Bray J. D., Hawes J., Palmby T. R., Jee J., Adams W., Mahayni H., Brown J., Dorantes A., Sridhara R., Farrell A. T., and Pazdur R. (2013) U.S. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 19, 4559–4563 [DOI] [PubMed] [Google Scholar]
- 4. Adams J. (2004) The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 4, 349–360 [DOI] [PubMed] [Google Scholar]
- 5. Richardson P. G., Barlogie B., Berenson J., Singhal S., Jagannath S., Irwin D., Rajkumar S. V., Srkalovic G., Alsina M., Alexanian R., Siegel D., Orlowski R. Z., Kuter D., Limentani S. A., Lee S., Hideshima T., Esseltine D. L., Kauffman M., Adams J., Schenkein D. P., and Anderson K. C. (2003) A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 348, 2609–2617 [DOI] [PubMed] [Google Scholar]
- 6. Richardson P. G., Sonneveld P., Schuster M., Irwin D., Stadtmauer E., Facon T., Harousseau J. L., Ben-Yehuda D., Lonial S., Goldschmidt H., Reece D., Miguel J. S., Bladé J., Boccadoro M., Cavenagh J., et al. (2007) Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final time-to-event results of the APEX trial. Blood 110, 3557–3560 [DOI] [PubMed] [Google Scholar]
- 7. Richardson P. G., Sonneveld P., Schuster M. W., Irwin D., Stadtmauer E. A., Facon T., Harousseau J. L., Ben-Yehuda D., Lonial S., Goldschmidt H., Reece D., San-Miguel J. F., Bladé J., Boccadoro M., Cavenagh J., et al. (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 352, 2487–2498 [DOI] [PubMed] [Google Scholar]
- 8. Fisher R. I., Bernstein S. H., Kahl B. S., Djulbegovic B., Robertson M. J., de Vos S., Epner E., Krishnan A., Leonard J. P., Lonial S., Stadtmauer E. A., O'Connor O. A., Shi H., Boral A. L., and Goy A. (2006) Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 24, 4867–4874 [DOI] [PubMed] [Google Scholar]
- 9. Strauss S. J., Maharaj L., Hoare S., Johnson P. W., Radford J. A., Vinnecombe S., Millard L., Rohatiner A., Boral A., Trehu E., Schenkein D., Balkwill F., Joel S. P., and Lister T. A. (2006) Bortezomib therapy in patients with relapsed or refractory lymphoma: potential correlation of in vitro sensitivity and tumor necrosis factor α response with clinical activity. J. Clin. Oncol. 24, 2105–2112 [DOI] [PubMed] [Google Scholar]
- 10. Lü S., and Wang J. (2013) The resistance mechanisms of proteasome inhibitor bortezomib. Biomark. Res. 1, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coleman C. S., Rocetes J. P., Park D. J., Wallick C. J., Warn-Cramer B. J., Michel K., Dudler R., and Bachmann A. S. (2006) Syringolin A, a new plant elicitor from the phytopathogenic bacterium Pseudomonas syringae pv. syringae, inhibits the proliferation of neuroblastoma and ovarian cancer cells and induces apoptosis. Cell Prolif. 39, 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Groll M., Schellenberg B., Bachmann A. S., Archer C. R., Huber R., Powell T. K., Lindow S., Kaiser M., and Dudler R. (2008) A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 452, 755–758 [DOI] [PubMed] [Google Scholar]
- 13. Archer C. R., Groll M., Stein M. L., Schellenberg B., Clerc J., Kaiser M., Kondratyuk T. P., Pezzuto J. M., Dudler R., and Bachmann A. S. (2012) Activity enhancement of the synthetic syrbactin proteasome inhibitor hybrid and biological evaluation in tumor cells. Biochemistry 51, 6880–6888 [DOI] [PubMed] [Google Scholar]
- 14. Archer C. R., Koomoa D. L., Mitsunaga E. M., Clerc J., Shimizu M., Kaiser M., Schellenberg B., Dudler R., and Bachmann A. S. (2010) Syrbactin class proteasome inhibitor-induced apoptosis and autophagy occurs in association with p53 accumulation and Akt/PKB activation in neuroblastoma. Biochem. Pharmacol. 80, 170–178 [DOI] [PubMed] [Google Scholar]
- 15. Clerc J., Groll M., Illich D. J., Bachmann A. S., Huber R., Schellenberg B., Dudler R., and Kaiser M. (2009) Synthetic and structural studies on syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proc. Natl. Acad. Sci. U.S.A. 106, 6507–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anshu A., Thomas S., Agarwal P., Ibarra-Rivera T. R., Pirrung M. C., and Schönthal A. H. (2011) Novel proteasome-inhibitory syrbactin analogs inducing endoplasmic reticulum stress and apoptosis in hematological tumor cell lines. Biochem. Pharmacol. 82, 600–609 [DOI] [PubMed] [Google Scholar]
- 17. Clerc J., Florea B. I., Kraus M., Groll M., Huber R., Bachmann A. S., Dudler R., Driessen C., Overkleeft H. S., and Kaiser M. (2009) Syringolin A selectively labels the 20 S proteasome in murine EL4 and wild-type and bortezomib-adapted leukaemic cell lines. Chembiochem 10, 2638–2643 [DOI] [PubMed] [Google Scholar]
- 18. Clerc J., Li N., Krahn D., Groll M., Bachmann A. S., Florea B. I., Overkleeft H. S., and Kaiser M. (2011) The natural product hybrid of Syringolin A and Glidobactin A synergizes proteasome inhibition potency with subsite selectivity. Chem. Commun. (Camb.) 47, 385–387 [DOI] [PubMed] [Google Scholar]
- 19. Clerc J., Schellenberg B., Groll M., Bachmann A. S., Huber R., Dudler R., and Kaiser M. (2010) Convergent synthesis and biological evaluation of Syringolin A and derivatives as eukaryotic 20S proteasome inhibitors. Eur. J. Org. Chem. 2010, 3991–4003 [Google Scholar]
- 20. Ibarra-Rivera T. R., Opoku-Ansah J., Ambadi S., Bachmann A. S., and Pirrung M. C. (2011) Syntheses and cytotoxicity of syringolin B-based proteasome inhibitors. Tetrahedron 67, 9950–9956 [Google Scholar]
- 21. Opoku-Ansah J., Ibarra-Rivera T. R., Pirrung M. C., and Bachmann A. S. (2012) Syringolin B-inspired proteasome inhibitor analogue TIR-203 exhibits enhanced biological activity in multiple myeloma and neuroblastoma. Pharm. Biol. 50, 25–29 [DOI] [PubMed] [Google Scholar]
- 22. Pirrung M. C., Biswas G., and Ibarra-Rivera T. R. (2010) Total synthesis of syringolin A and B. Org. Lett. 12, 2402–2405 [DOI] [PubMed] [Google Scholar]
- 23. Chiba T., Hosono H., Nakagawa K., Asaka M., Takeda H., Matsuda A., and Ichikawa S. (2014) Total synthesis of syringolin A and improvement of its biological activity. Angew. Chem. Int. Ed. Engl. 53, 4836–4839 [DOI] [PubMed] [Google Scholar]
- 24. Chiba T., Matsuda A., and Ichikawa S. (2015) Structure-activity relationship study of syringolin A as a potential anticancer agent. Bioorg Med. Chem. Lett. 25, 4872–4877 [DOI] [PubMed] [Google Scholar]
- 25. Totaro K. A., Barthelme D., Simpson P. T., Sauer R. T., and Sello J. K. (2015) Substrate-guided optimization of the syringolins yields potent proteasome inhibitors with activity against leukemia cell lines. Bioorg. Med. Chem. 23, 6218–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trivella D. B., Pereira A. R., Stein M. L., Kasai Y., Byrum T., Valeriote F. A., Tantillo D. J., Groll M., Gerwick W. H., and Moore B. S. (2014) Enzyme inhibition by hydroamination: design and mechanism of a hybrid carmaphycin-syringolin enone proteasome inhibitor. Chem. Biol. 21, 782–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 30. Massova I., and Kollman P. A. (2000) Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 18, 113–135 [Google Scholar]
- 31. Tsui V., and Case D. A. (2000) Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 56, 275–291 [DOI] [PubMed] [Google Scholar]
- 32. Greenstein S., Krett N. L., Kurosawa Y., Ma C., Chauhan D., Hideshima T., Anderson K. C., and Rosen S. T. (2003) Characterization of the MM.1 human multiple myeloma (MM) cell lines: a model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp. Hematol. 31, 271–282 [DOI] [PubMed] [Google Scholar]
- 33. Slack A., Chen Z., Tonelli R., Pule M., Hunt L., Pession A., and Shohet J. M. (2005) The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl. Acad. Sci. U.S.A. 102, 731–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shoemaker R. H. (2006) The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 6, 813–823 [DOI] [PubMed] [Google Scholar]
- 35. Hollingshead M. G., Alley M. C., Camalier R. F., Abbott B. J., Mayo J. G., Malspeis L., and Grever M. R. (1995) In vivo cultivation of tumor cells in hollow fibers. Life Sci. 57, 131–141 [DOI] [PubMed] [Google Scholar]
- 36. Oka M., Nishiyama Y., Ohta S., Kamei H., Konishi M., Miyaki T., Oki T., and Kawaguchi H. (1988) Glidobactins A, B and C, new antitumor antibiotics. I. Production, isolation, chemical properties and biological activity. J. Antibiot. 41, 1331–1337 [DOI] [PubMed] [Google Scholar]
- 37. Oka M., Numata K., Nishiyama Y., Kamei H., Konishi M., Oki T., and Kawaguchi H. (1988) Chemical modification of the antitumor antibiotic glidobactin. J. Antibiot. 41, 1812–1822 [DOI] [PubMed] [Google Scholar]
- 38. Adams J., Behnke M., Chen S., Cruickshank A. A., Dick L. R., Grenier L., Klunder J. M., Ma Y. T., Plamondon L., and Stein R. L. (1998) Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 8, 333–338 [DOI] [PubMed] [Google Scholar]
- 39. Cragg G. M., Grothaus P. G., and Newman D. J. (2009) Impact of natural products on developing new anti-cancer agents. Chem. Rev. 109, 3012–3043 [DOI] [PubMed] [Google Scholar]
- 40. Hopkins A. L., Keserü G. M., Leeson P. D., Rees D. C., and Reynolds C. H. (2014) The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 13, 105–121 [DOI] [PubMed] [Google Scholar]
- 41. Borissenko L., and Groll M. (2007) 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev. 107, 687–717 [DOI] [PubMed] [Google Scholar]
- 42. Díaz-Hernández M., Hernández F., Martín-Aparicio E., Gómez-Ramos P., Morán M. A., Castaño J. G., Ferrer I., Avila J., and Lucas J. J. (2003) Neuronal induction of the immunoproteasome in Huntington's disease. J. Neurosci. 23, 11653–11661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mishto M., Bellavista E., Santoro A., Stolzing A., Ligorio C., Nacmias B., Spazzafumo L., Chiappelli M., Licastro F., Sorbi S., Pession A., Ohm T., Grune T., and Franceschi C. (2006) Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains. Neurobiol. Aging 27, 54–66 [DOI] [PubMed] [Google Scholar]
- 44. Ethen C. M., Hussong S. A., Reilly C., Feng X., Olsen T. W., and Ferrington D. A. (2007) Transformation of the proteasome with age-related macular degeneration. FEBS Lett. 581, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fitzpatrick L. R., Small J. S., Poritz L. S., McKenna K. J., and Koltun W. A. (2007) Enhanced intestinal expression of the proteasome subunit low molecular mass polypeptide 2 in patients with inflammatory bowel disease. Dis. Colon Rectum 50, 337–348; discussion 348–350 [DOI] [PubMed] [Google Scholar]
- 46. Visekruna A., Joeris T., Seidel D., Kroesen A., Loddenkemper C., Zeitz M., Kaufmann S. H., Schmidt-Ullrich R., and Steinhoff U. (2006) Proteasome-mediated degradation of IκBα and processing of p105 in Crohn disease and ulcerative colitis. J. Clin. Invest. 116, 3195–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Egerer T., Martinez-Gamboa L., Dankof A., Stuhlmüller B., Dörner T., Krenn V., Egerer K., Rudolph P. E., Burmester G. R., and Feist E. (2006) Tissue-specific up-regulation of the proteasome subunit beta5i (LMP7) in Sjogren's syndrome. Arthritis Rheum. 54, 1501–1508 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.