

Abstract
The Hermansky Pudlak syndromes (HPS) constitute a family of disorders characterized by oculocutaneous albinism and bleeding diathesis, often associated with lethal lung fibrosis. HPS results from mutations in genes of membrane trafficking complexes that facilitate delivery of cargo to lysosome-related organelles. Among the affected lysosome-related organelles are lamellar bodies (LB) within alveolar type 2 cells (AT2) in which surfactant components are assembled, modified, and stored. AT2 from HPS patients and mouse models of HPS exhibit enlarged LB with increased phospholipid content, but the mechanism underlying these defects is unknown. We now show that AT2 in the pearl mouse model of HPS type 2 lacking the adaptor protein 3 complex (AP-3) fails to accumulate the soluble enzyme peroxiredoxin 6 (PRDX6) in LB. This defect reflects impaired AP-3-dependent trafficking of PRDX6 to LB, because pearl mouse AT2 cells harbor a normal total PRDX6 content. AP-3-dependent targeting of PRDX6 to LB requires the transmembrane protein LIMP-2/SCARB2, a known AP-3-dependent cargo protein that functions as a carrier for lysosomal proteins in other cell types. Depletion of LB PRDX6 in AP-3- or LIMP-2/SCARB2-deficient mice correlates with phospholipid accumulation in lamellar bodies and with defective intraluminal degradation of LB disaturated phosphatidylcholine. Furthermore, AP-3-dependent LB targeting is facilitated by protein/protein interaction between LIMP-2/SCARB2 and PRDX6 in vitro and in vivo. Our data provide the first evidence for an AP-3-dependent cargo protein required for the maturation of LB in AT2 and suggest that the loss of PRDX6 activity contributes to the pathogenic changes in LB phospholipid homeostasis found HPS2 patients.
Keywords: adaptor protein, lung, peroxiredoxin, phospholipid metabolism, trafficking, AP-3, Hermansky Pudlak syndrome, alveolar epithelial type 2 cell, lamellar body, peroxiredoxin 6
Introduction
Lamellar bodies are lysosome-related organelles of alveolar type 2 (AT2) cells within which pulmonary surfactant is synthesized, assembled, and stored and from which surfactant is secreted. Roughly 80% of surfactant by weight consists of phospholipids, most notably disaturated phosphatidylcholine (DSPC),3 that act in concert with surfactant-specific proteins (surfactant proteins A, B, and C) to lower surface tension at the air-liquid interface in the alveoli and thus stabilize alveolar inflation (1). There is a growing appreciation for an expanding repertoire of proteins that support the function of lamellar bodies, best illustrated by the published rat lamellar body proteome in which a variety of transmembrane, membrane-associated, and luminal proteins were identified to reside within lamellar bodies (2). However, little is known about the pathways that promote preferential targeting of cargo to lamellar bodies rather than other endosomal-lysosomal compartments.
The Hermansky Pudlak syndromes (HPS) constitute a family of genetic diseases with a common phenotype consisting of oculocutaneous albinism and bleeding diathesis; the most prevalent forms of HPS are also associated with a progressive lung fibrosis that is usually lethal within the 5th decade of life (3). The genes that are defective in HPS encode subunits of several multisubunit complexes, adaptor protein 3 (AP-3) and biogenesis of lysosome-related organelles complexes (BLOC)-1, -2, and -3, that contribute to membrane and protein trafficking to lysosome-related organelles. Although ubiquitously expressed, AP-3 and BLOC1, -2, and -3 play an important but incompletely understood role in regulating the trafficking of cell-specific cargoes to lysosome-related organelles, including melanosomes in pigment cells and likely dense granules in platelets (4–7). It has been unclear whether these complexes also participate in membrane trafficking to lamellar bodies and if so which components of the lamellar body proteome utilize them.
Peroxiredoxin 6 (PRDX6) is a well described luminal constituent of the lamellar body (2, 8). This bifunctional enzyme is best known for its antioxidant lipid peroxidase activity in the cytosol, but it also exhibits phospholipase A2 (PLA2) activity at a pH profile that matches the acidic environment of the lamellar body and plays a key role in lamellar body phospholipid homeostasis (9). PRDX6 facilitates the remodeling of unsaturated phosphatidylcholine (PC) in the lamellar body lumen by catalyzing deacylation of luminal PC, and it is thus thought to play a role in preferentially enriching DSPC in pulmonary surfactant (10). However, recent studies indicate that PRDX6 also plays a role in intraluminal degradation of lamellar body phospholipids, with Prdx6−/− mice exhibiting increased lamellar body phospholipid content and mice that overexpress PRDX6 exhibiting reduced lamellar body phospholipid content (11, 12). Although PRDX6 is primarily expressed in the cytosol, growing evidence indicates that a select fraction of PRDX6 enters the secretory pathway of AT2 cells where it is preferentially targeted to lamellar bodies (13, 14). PRDX6 does not share a long N-terminal hydrophobic domain used by peroxiredoxin-4 for entry into the secretory pathway (15), nor does PRDX6 share C-terminal domain homology that fosters endoplasmic reticulum localization of peroxiredoxin-4 (16). PRDX6 associates with 14-3-3ϵ in a MAPK-dependent fashion that is necessary for lysosomal targeting in cell lines (14), but the mechanism by which PRDX6 is targeted to lamellar bodies remains unclear.
Lamellar body enlargement with increased lamellar body surfactant phospholipid content is a common feature of HPS patients (17–20) and its mouse models (21–25). It is especially apparent in human HPS2 and the pearl mouse model in which AP-3 is essentially absent from AT2 cells (25) due to an inactivating mutation in the gene encoding the β3A subunit (26). The striking parallels in the increased phospholipid content observed in both the pearl and Prdx6−/− mouse models led us to investigate whether they might be linked. Our data demonstrate that PRDX6 trafficking to lamellar bodies requires AP-3 and that AP-3 is linked to PRDX6 via the transmembrane protein LIMP-2/SCARB2. Moreover, we show that loss of PRDX6 PLA2 activity contributes to surfactant phospholipid accumulation in AP-3-deficient pearl mice.
Experimental Procedures
Animals
Wild type (WT) C57BL/6J, mutant Ap3b1pe/Ap3b1pe (pearl), and transgenic pearl mice in which the Ap3b1 gene product was expressed in alveolar epithelial cells under the direction of the human SP-C promoter (3.7 kb; line HPS2mt Tg A) (25) were bred and maintained at the Laboratory Animal Facility at the Children's Hospital of Philadelphia or at Vanderbilt University as described previously (24). The pearl mouse expresses no detectable AP-3 in AT2 cells as indicated by expression of the μ subunit, whereas HPS2mt Tg A or Tg B expressed near normal levels of μ subunit as a marker of AP-3 complexes in AT2 cells (25). Drs. Michael Schwake and Paul Saftig supplied Limp-2−/− breeding pairs. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committees of The Children's Hospital Research Institute, the University of Pennsylvania School of Medicine, and the Vanderbilt University School of Medicine, and adhered to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Unless specified, both male and female mice were used between 8 and 10 weeks of age.
Isolation of Lung Fractions
Bronchoalveolar lavage was performed as described previously (24). Mouse lungs were lavaged with 1-ml aliquots of sterile Ca2+- and Mg2+-free 0.9% saline to a total of 5 ml instilled. Samples were centrifuged at 400 × g for 10 min at 4 °C, and cell-free supernatant was used for subsequent analysis. Enriched lamellar body fractions were obtained from post-lavage, freshly prepared lung homogenates via upward flotation through a sucrose step-gradient as described previously (27). Additional lung homogenates used for biochemical analyses were generated from flash-frozen lung tissue after lavage as described previously (24). Protein content of fractions was measured using the Coomassie Blue binding assay (Bio-Rad) with bovine IgG as the standard.
Alveolar Type 2 Cell Isolation
Alveolar type 2 cells were isolated from mouse lungs after bronchoalveolar lavage as described previously (24).
Immunofluorescence Microscopy
Primary cultured AT2 cells cultured on glass coverslips were rinsed with ice-cold PBS that was subsequently thoroughly removed. Digitonin solution (1 ml; 50 μg/ml digitonin in PBS with 100 mm KCl) was added and incubated for 10 min on ice with gentle shaking (at 100 rpm) on an orbital shaker. The cells were then washed twice with 2 ml of cold PBS to remove the remaining cellular material and were then fixed in paraformaldehyde (4% in PBS) for 10 min at room temperature. Following fixation, cells on coverslips were washed twice with PBS, and then blocked (1% BSA, 50 μg/ml digitonin in PBS) for 1 h at room temperature. Cells were immunolabeled with primary antibodies diluted in blocking solution for 1 h at room temperature. Primary antibodies included the following: monoclonal (mouse) antibody to PRDX6 (4A3, Abcam, Cambridge, MA; 1:100) and polyclonal (rabbit) antibody to LIMP2 (LS-B305, LifeSpan BioSciences, Inc. Seattle, WA; 1:250). Rat monoclonal antibody to Lamp1 (1D4B, DSHB, Iowa city, IA; 1:100) was used as a marker of lamellar bodies. The Lamp1 monoclonal antibody (1D4B), developed by J. T. August, was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD, National Institutes of Health, and maintained by the Department of Biology, University of Iowa (Iowa City, IA). After washing with PBS, secondary IgG antibodies were applied at 1:200 dilution in blocking solution for 1 h at room temperature: Alexa Fluor-488-conjugated goat anti-rat (green); Alexa Fluor-594-conjugated goat anti-mouse (red); and Alexa Fluor-674-conjuaged goat anti-rabbit (far red) (Molecular Probes, Eugene, OR). After a final washing (three times for 5 min each with PBS), the cells were mounted with ProLong Gold antifade reagent (Molecular probes, Eugene, OR). Immunofluorescence was imaged with an Olympus IX81 inverted microscope equipped with a ×60 lens and Slidebook software.
Phospholipid Analysis
Total phospholipid was determined from total phosphorus using the method of Marinetti (28). Total phosphatidylcholine was determined by application of sample to thin layer chromatography (27, 29). Spots were visualized with I2 vapor, and phosphatidylcholine was eluted with chloroform/methanol/H2O/acetic acid (60:35:4.5:0.5, v/v). The percent DSPC was determined using a neutral alumina column after treatment of lipids with OsO4 (27).
Immunoblotting
Western immunoblotting was performed as described previously (24). Primary antisera used included the following : Prdx6 (30, 31); surfactant protein B (SP-B; Abcam, catalog no. 40876); GAPDH (MAB374, Chemicon/Millipore); LAMP1 (1D4B, DSHB); Limp2 (L2T2, from Dr. Paul Saftig); and surfactant protein A (SP-A; Abcam, ab180865). Species-specific secondary antisera were all conjugated to IR dyes of either 680 or 800 nm wavelengths (Rockland). Visualization and densitometry of positive immunoblotting, where indicated, was accomplished using the Odyssey Imaging System (LiCOR Biosciences, Lincoln, NB), and band densities were quantified in the linear dynamic range. Densitometry of immunoblots was normalized to GAPDH for lung tissue and cell lysates and to SP-B or SP-A for lamellar body samples as neither of these surfactant proteins has demonstrated significant variation in these mouse models when equal LB total protein was loaded (24, 33).
Phospholipase A2 Activity Assay
Acidic PLA2 activity was measured as described previously (11, 32). PLA2 activity was measured at pH 4.0 in 40 mm sodium acetate and 5 mm EDTA buffer in lung homogenates and lamellar body fractions using 1-palmitoyl,2-[9,10-3H]palmitoyl-sn-3-glycoso-phosphorylcholine ([3H]DPPC) (11, 32).
Uptake and Degradation of DPPC
Uptake of [3H]choline-labeled DPPC in liposomes by the isolated lung was studied using a well established protocol (27). Briefly, unilamellar liposomes containing [3H]choline-labeled DPPC were instilled intratracheally into mice, and the lungs were then removed and either immediately homogenized or perfused for 2 h before homogenization. The specific activity of alveolar DPPC was calculated from the injected dose and the measured alveolar DPPC pool size. Uptake of DPPC was calculated from total lung (plus perfusate) disintegrations/min and the alveolar DPPC-specific activity. Samples of lung homogenate were analyzed for disintegrations/min in aqueous and lipid fractions (DPPC, lysophosphatidylcholine, and unsaturated phosphatidylcholine). The radiolabel in the aqueous fraction consists of choline, choline phosphate, CDP-choline, and possibly other metabolites (29). Total disintegrations/min were calculated from disintegrations/min recovered from the sum of aqueous, lysophosphatidylcholine, and unsaturated phosphatidylcholine fractions but cannot account for any DPPC that might have been resynthesized from [3H]lysophosphatidylcholine.
DSPC Synthesis
Synthesis of DSPC was measured as described previously (27). Mice were injected with a mixture of 10 μCi of [3H]methyl choline and 10 μCi of [1-14C]palmitate via tail vein followed by recovery of DSPC from total lung tissue homogenate and from an enriched lamellar body fraction as described above. Incorporation of each label was expressed as disintegrations/min of 14C or 3H per nmol of DSPC.
Surfactant Secretion
Secretion of radiolabeled surfactant from isolated AT2 cells was assessed as described previously (33). In brief, isolated AT2 cells were cultured in media supplemented with 0.5 μCi/dish of (methyl-3H)-choline (Amersham Biosciences) for 24 h. The medium was changed to Eagle's medium, and after 30 min a set of cells and media were harvested. The remaining cells were incubated with or without 1 mm ATP (Sigma) for 2 h, after which media and cells were harvested. Media samples were centrifuged to remove detached cells and debris, and cells were scraped from the dish into methanol. Phosphatidylcholine was extracted from all samples, and phospholipid secretion was calculated as disintegrations/min in phosphatidylcholine fractions from media divided by total disintegrations/min in phosphatidylcholine (cells and media combined).
Co-immunoprecipitation of Recombinant LIMP2 and PRDX6 Proteins
Cloning
The N-terminal His6-tagged luminal domain of hLIMP-2 (amino acids 35–430) and the full-length hPRDX6 were cloned from cDNA templates (ViGene, catalog no. CH840240) into PB-T-PAF, after the protein A tag (protein A-His6-hLIMP-2) and pet28MHL (GenBankTM accession number EF456735), respectively, using the In-Fusion CF Dry-Down PCR cloning kit (Clontech catalog no. 639605).
Cell Culture
293T cells were grown in DMEM (Basal Media, L110), supplemented with 10% FBS (Gemini, 900–108) at 37 °C in a 5% CO2 incubator. The protein A-His6-hLIMP-2 plasmid was transfected using ExFect transfection reagent (Vazyme, catalog no. 913051) using the manufacturer's protocols. A doxycycline-inducible stably transfected 293T cell line was selected according to the method described by Li et al. (34), using 10 μg/ml puromycin and 5 μg/ml blasticidin S.
His6.hPRDX6 Purification
Bacterial pellets were obtained from 1 liter of LB media initially cultured at 37 °C to an A600 = 1.0, after which cells were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 16 h at 15 °C. Then the bacterial pellets were sonicated, and the cleared cell lysate in Lysis Buffer (50 mm Hepes, pH 7.5, 500 mm NaCl, 2.5 mm imidazole) was loaded onto a 1-ml Ni2+-nitrilotriacetic acid metal-affinity resin column equilibrated in Wash Buffer (50 mm Hepes, pH 7.5, 500 mm NaCl, 15 mm imidazole) at 4 °C temperature. The column was washed with 50 ml of Wash Buffer, and the protein was eluted with 10 ml of Elution Buffer (50 mm Hepes, pH 7.5, 500 mm NaCl, 250 mm imidazole). The protein was further purified by gel filtration on a HighLoad Superdex200 column equilibrated with Gel Filtration Buffer (25 mm Hepes, pH 7, and 150 mm NaCl). Fractions containing protein were pooled and concentrated using Amicon ultracentrifugal filter with a 3-kDa cutoff membrane to a final concentration of 15 mg/ml. Protein yield was 30 mg/liter of bacterial culture.
Co-immunoprecipitation and Western Blot Analysis
After 48 h of induction with 1 μg/ml doxycycline, the culture medium (20 ml) of the 293T stable cell line expressing protein A-His6-hLIMP-2 fusion protein was harvested from two confluent 10-cm2 tissue culture dishes, centrifuged at 4000 rpm for 60 min, and the supernatant transferred into a 15-ml tube. A volume of 0.25 ml of 50% IgG beads 6FF (Solarbio, catalog no. 18600), pre-equilibrated in PBS buffer, was added, and batch binding was done for 120 min at 50 rpm and 4 °C in a shaker. The beads were separated in one open column and washed with 5 ml of PBS. Afterward, beads were incubated with purified His6-hPRDX6 (180 μg) in 0.1 ml of buffer at pH 7 (PBS) or at pH 5 (25 mm NaCH3COO, 300 mm NaCl). Subsequently, input controls and immunoprecipitate samples were analyzed by Western blot using anti-His6 peroxidase (Proteintech, 66005-1-lg) antibody.
Proximity Ligation Assay
Molecular interaction in vivo between Prdx6 and Limp2 was evaluated using the Duolink in situ proximity ligation assay (PLA) according to the manufacturer's instructions (Olink Bioscience; Sigma). Briefly, primary AT2 cells from WT, pearl, or Limp-2−/− mice were cultured on glass coverslips and fixed with cold ethanol/acetone mixture (1:1 in volume) for 5 min on ice. After fixation, cells were washed twice with cold PBS, followed by a 10-min incubation with 1% Triton X-100 solution in PBS and 40 min blocking in a 3% solution of bovine serum albumin in PBS containing 0.2% Triton X-100. Samples were incubated with primary antibodies against PRDX6 and LIMP2 (1:100 dilution in 0.2% Triton X-100 solution in PBS; PRDX6 MAB3478 mouse monoclonal antibody, Millipore; LIMP2 LS-B3225 rabbit polyclonal antibody, LifeSpan BioScience) for 1 h at room temperature. After three washes with PBS containing 0.2% Triton X-100, Duolink PLA probes were applied and incubated for 1 h in a humidity chamber at 37 °C. Unbound Duolink PLA probes were removed with wash buffer, and the samples were incubated in the ligation solution for 1 h. Amplification was accomplished by applying diluted polymerase to the sample for 100 min, and detection of the amplified probe was accomplished by adding diluted Duolink detection stock. Coverslips were then washed and mounted with Duolink mounting medium. Images were taken by a Zeiss LSM 710 META inverted confocal microscopy equipped with a ×63 lens (Vanderbilt University Medical Center Cell Imaging Shared Resource). Each puncta was considered a positive protein/protein interaction. AT2 cells were detected by their large vacuolar appearance clustered around cell nuclei in phase contrast images. The number of fluorescent puncta per AT2 cell was determined using ZEN imaging software (Zeiss).
Statistics
Student's t tests were performed with Prism 5.0 software for the Macintosh (GraphPad Software, San Diego). Results are expressed as means ± S.E. unless otherwise noted, and a p value of <0.05 was considered significant.
Results
Peroxiredoxin 6 Is Significantly Reduced in the Lamellar Bodies of pearl Mice
The pearl mouse lacks expression of AP-3 in most cell types due to inactivation of the Ap3b1 gene encoding the ubiquitously expressed β3A subunit (35). pearl mice display a pulmonary phenotype in which total phospholipid content in lung tissue is increased and lamellar bodies in AT2 cells are enlarged, suggestive of intracellular pulmonary surfactant retention (22–24). We performed a detailed analysis of surfactant pools in lung tissue, bronchoalveolar lavage, and a subcellular fraction enriched in lamellar bodies from pearl mice compared with C57BL6 (WT) mice (Fig. 1). Consistent with our previous report on compound homozygous HPS mice carrying the pearl and pale ear mutations (24), total lung tissue phospholipid was increased in pearl mice (1.9-fold over WT; Fig. 1A). The increase in total lung phospholipid of pearl mice was paralleled by specific increases in total PC (1.6-fold over WT) and DSPC (2.2-fold over WT). Similarly, the total phospholipid, PC, and DSPC content of cell-free bronchoalveolar lavage fluid containing secreted surfactant from pearl mice was increased compared with WT (Fig. 1B), suggesting that the enhanced tissue phospholipid was due to increased lamellar body stores that were subsequently secreted into the alveolar space. Examination of subcellular fractions of lung tissue enriched for lamellar bodies demonstrated a 1.6-fold increase in total phospholipid in lamellar body fractions from pearl mice compared with WT mice (Fig. 1C). The increased phospholipid content in both the cellular and cell-free compartments suggests that phospholipid metabolism within lamellar bodies might be compromised in AP-3-deficient pearl mice.
FIGURE 1.

Disrupted lamellar body phospholipid content in pearl mice is associated with loss of lamellar body PRDX6. A, total phospholipid, total PC, andtotal DSPC measured in lung tissue homogenate (n = 6 mice per genotype, mean ± S.E.). B, total phospholipid, total PC, and total DSPC measured in bronchoalveolar lavage fluid from wild type (WT) and pearl mice (n = 4 mice per genotype, mean ± S.E.). C, total phospholipid content measured in lamellar body fractions from WT and pearl mice (n = 3 samples per genotype, each prepared from three mice, mean ± S.E.). D, phospholipase A2 activity measured in lamellar body fractions isolated from WT and pearl mice (n = 4 samples from 2 to 3 mice of each strain, mean ± S.E.). E, representative immunoblots of PRDX6, SP-B, and GAPDH using lamellar body fractions (LB; 10 μg of protein) and alveolar type 2 cell lysate (AT2; 30 μg of protein) from wild type (WT) and pearl (pe) mice. F, immunolocalization of PRDX6 in WT and pearl isolated alveolar type 2 cells (mAT2) after depletion of cytosolic constituents, using LAMP1 immunostaining to identify lamellar bodies (representative of two experiments; bars, 5 μm).
Because the increase in total phospholipid pools in pearl mice was reminiscent of that of Prdx6−/− mice (11), we next examined acidic PLA2 activity and PRDX6 content in the lamellar body-enriched subcellular fraction from pearl and WT mice. The striking reduction of PLA2 activity in lamellar body fractions from pearl mice (Fig. 1D) was accompanied by significant reduction of PRDX6 protein content compared with WT lamellar body fractions (Fig. 1E). Consistent with prior observations that the majority of total cellular PRDX6 in AT2 cells is cytosolic, we confirmed that the bulk of total cellular PRDX6 content in isolated AT2 cells from both WT and pearl mice resided in a non-lamellar body cytosolic pool (Fig. 1E) suggesting a loss of selective trafficking of PRDX6 to lamellar bodies in these mice (13). By comparison, the content of another lamellar body cargo, surfactant protein B (SP-B), was similar in both WT and pearl lamellar body fractions and isolated AT2 cells indicating that SP-B does not require AP-3 for trafficking to lamellar bodies. Furthermore, immunofluorescence microscopy analysis of isolated AT2 cells after permeabilization to deplete cytosolic PRDX6 demonstrated that luminal PRDX6 could be detected in a fraction of LAMP1-positive lamellar bodies in WT AT2 but not in pearl AT2 (Fig. 1F). The variability of PRDX content between LAMP1-positive lamellar bodies may reflect the need for a permeabilization and cytosol depletion step prior to immunostaining due to the large amount of cytosolic PRDX6 in AT2 cells. Although this could result in permeabilization of some lamellar bodies as well, we expect that the effect would be similar for WT and pearl LB and AT2 cells. In addition, LAMP1 labels other lysosomal structures like lysosomes that may not contain PRDX6 normally; again this would be expected to be similar between WT and pearl LB. Finally, the process of lamellar body maturation is poorly understood, and variability in PRDX6 in LAMP1-positive lamellar bodies in WT cells may simply reflect lamellar body maturation and targeting of PRDX6 to more mature lamellar bodies. Regardless, there is a clear and consistent difference showing a striking depletion of PRDX6 from LAMP1-positive organelles in pearl AT2. Together, these data indicate that disruption of AP-3 trafficking in pearl AT2 cells results in loss of PRDX6 from lamellar bodies without altering total PRDX6 content in AT2 cells.
Depletion of PRDX6 Is Associated with Altered Intraluminal DPPC Metabolism in Lamellar Bodies of Pearl Mice
The lamellar body is a dynamic storage organelle in AT2 cells, receiving surfactant constituents from the secretory pathway in AT2 cells, modifying phospholipids to enrich DPPC content, secreting surfactant into the alveolar space, and accepting recycled alveolar surfactant via the endocytic pathway (36). The most significant biochemical alteration in the life cycle of surfactant phospholipids identified in the Prdx6−/− mouse model relative to WT littermates was reduced intraluminal degradation of DPPC (11). Within the acidic luminal environment of the lamellar body, the PLA2 activity of PRDX6 catalyzes the deacylation of PC in the sn-2 position to produce lysophosphatidylcholine (lyso-PC), a toxic metabolite that is rapidly reacylated or degraded (37). To test whether DPPC metabolism was impacted by loss of AP-3, we examined the fate of [3H]DPPC that was endocytosed by AT2 from the alveolar space of the lung by tracking labeled metabolites in total lung homogenates. Specifically, we quantified 3H-labeled lyso-PC and unsaturated PC after thin layer chromatography of the organic extracts of lung homogenates and the content of free [3H]choline and metabolites ([3H]choline phosphate and [3H]CDP-choline) within the aqueous phase. These compounds represent the products of phospholipid degradation ([3H]choline-labeled lyso-PC, choline, and metabolites) and reacylation of [3H]choline-labeled lyso-PC to DPPC (29). Compared with WT lungs, the metabolism of endocytosed DPPC was impaired in pearl lungs, with only 14% of starting label traceable in aggregate into lyso-PC, unsaturated PC, and the aqueous compartment, compared with ∼33% in WT lungs (Fig. 2A). The recovery of label in each individual compartment was also reduced, suggesting a proximal block in pearl mice at the deacylation step of the remodeling pathway. This block is consistent with the absence of PRDX6. These data demonstrate that the defect in DPPC metabolism in pearl mice phenocopies the primary biochemical finding of the Prdx6−/− mouse model (11).
FIGURE 2.
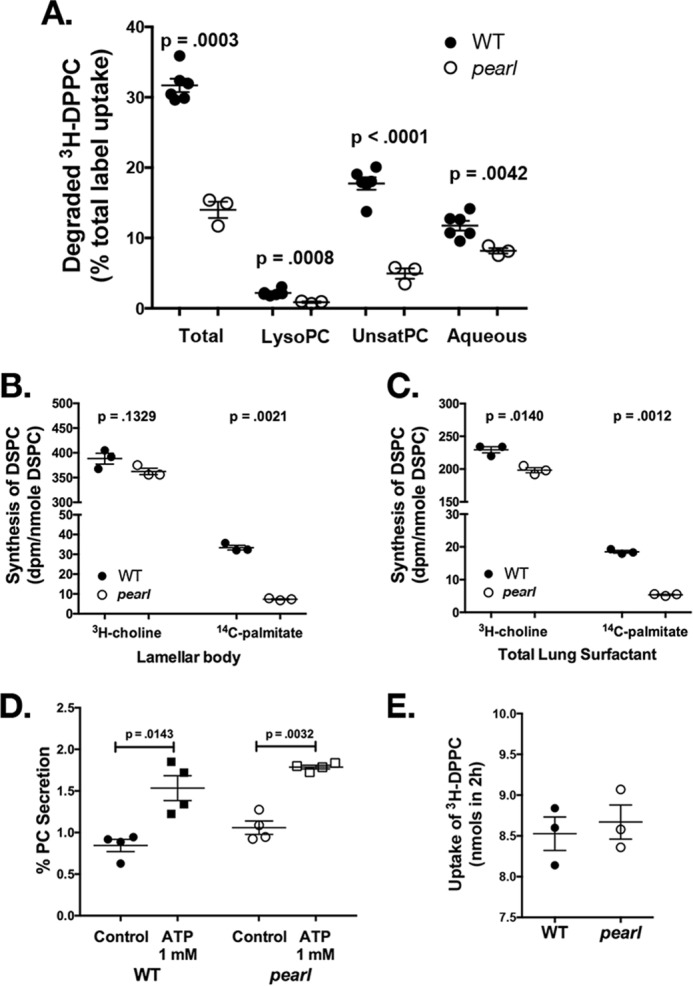
Disrupted phospholipid homeostasis in pearl mice is associated with reduced DPPC degradation and DPPC synthesis. A, degradation of intratracheally instilled [3H]DPPC and recovery of metabolites at 2 h by isolated perfused lungs of WT and pearl mice. The result for each fraction (lyso-PC, unsaturated phosphatidylcholine, aqueous) is expressed as a percentage of total recovered radioactivity, which is the sum of the recoveries for these three fractions (n = 6 WT and 3 pearl mice; mean ± S.E.). B and C, incorporation of radiolabeled [3H]choline and [14C]palmitate into DSPC of lamellar bodies (B) and total lung surfactant (C) isolated from WT and pearl mouse lungs. Substrates were administered via tail vein injection 24 h prior to isolation of DSPC from lamellar body fractions and total lung homogenate. DSPC isolated from these samples was analyzed for radioactivity and total phosphorus. Synthesis was expressed as disintegrations/min per nmol of DSPC (n = 3; mean ± S.E.). D, secretion of (methyl-3H)-choline-labeled phospholipid from AT2 cells isolated from WT and pearl mice, calculated as a percentage of recovered disintegrations/min associated with phospholipid in the culture medium after 2 h of secretagogues divided by total disintegrations/min in the cells plus culture medium (n = 4 experiments, mean ± S.E.). E, uptake of intratracheally instilled [3H]DPPC by isolated perfused lungs of WT and pearl mice between 5 and 120 min of lung perfusion (n = 3; mean ± S.E.).
Loss of lamellar body PRDX6 should also result in reduced synthesis of DPPC from palmitate via the remodeling pathway (Lands' cycle, or the deacylation-reacylation pathway). Although the remodeling pathway contributes less to AT2 cell lipogenesis than de novo lipogenesis from choline (via the Kennedy pathway, which is not catalyzed by PRDX6), it is believed to be important for the enrichment of surfactant DSPC by enabling the reacylation of unsaturated PC with palmitate incorporation at the sn-2 position (8). To test whether the remodeling pathway is affected in the pearl mouse, we injected 14C-labeled palmitate or [3H]choline intravenously and measured incorporation into DSPC by organic extraction and thin layer chromatography of surfactant isolated from LB preparations or of total lung homogenates. Incorporation of 14C-labeled palmitate was dramatically impaired in the pearl mouse model relative to WT mice whether we measured the incorporation of label into surfactant from lamellar body fractions (by 78%; p = 0.0021; Fig. 2B) or total lung homogenates (by 70%; p = 0.0012; Fig. 2C). By comparison, incorporation of [3H]choline into DSPC was not significantly reduced in lamellar bodies (by 7%; p = 0.1329; Fig. 2B) from pearl mice compared with WT littermates despite a small but significant reduction in total lung surfactant (by 14%; p = 0.014; Fig. 2C), indicating that de novo lipogenesis was only very modestly affected by the loss of AP-3 compared with the remodeling pathway. These data support a functional loss of lamellar body PRDX6 activity in pearl lungs.
To complete the characterization of the surfactant life cycle in pearl mice, we assessed basal and ATP-stimulated secretion of surfactant from isolated AT2 cells from WT and pearl mice. Surfactant was labeled overnight with [3H]choline added to the culture media, and recovery of [3H]PC from the medium before and after a 2-h exposure to ATP was measured as a percentage of total PC (total cell PC + total media PC). By this measure, the degree of both basal and ATP-stimulated secretion of PC was similar from pearl and WT AT2 cells (Fig. 2D). We also tested whether the defect in AP-3 in pearl mice affected surfactant endocytosis by measuring the incorporation of [3H]DPPC into lung tissue following intratracheal instillation into the lungs of WT and pearl mice for 2 h. Using this method, the rate of endocytosis of DPPC is typically ∼4 nmol/h in WT mice (11). After accounting for the increased endogenous bronchoalveolar phospholipid pool size of the pearl mice that would dilute the instilled [3H]DPPC, there was no significant difference in the rate of endocytosis of [3H]DPPC in pearl mice compared with WT mice (Fig. 2E). The lack of effect of the loss of AP-3 expression in pearl mice on surfactant secretion or DPPC endocytosis is consistent with the lack of a requirement for PRDX6 in these processes.
Together, our data demonstrate that the impairment of intraluminal DSPC metabolism within lamellar bodies of pearl AT2 cells contributes to the accumulation of DSPC and is consistent with prior observations in the Prdx6−/− mouse model. These data suggest that the surfactant defects in AP-3-deficient mice are at least partially due to defects in PRDX6 accumulation in lamellar bodies.
Reconstitution of the AP-3 Complex in Alveolar Type 2 Cells of Pearl Mice Corrects PRDX6 Trafficking and Restores Phospholipid Pool Sizes
To ensure that the loss of lamellar body PRDX6 and the surfactant lipid defects observed in pearl mice indeed reflected the loss of AP-3 expression in AT2 cells, we assessed whether the re-expression of the missing Ap3b1 gene would reverse the phenotypes. Transgenic expression of wild type Ap3b1 in alveolar epithelial cells in pearl mice, using the epithelial cell-specific mouse SP-C promoter to drive transgene expression, demonstrated a reduction in lamellar body size in transgene-positive (TG+) mice compared with transgene-negative littermates (TG−) and to parental pearl mice (25). Immunoblotting of isolated AT2 cells and enriched lamellar body fractions from lung tissue of TG+ and TG− mice revealed that lamellar body PRDX6 content of TG+ “rescued” pearl mice was significantly increased compared with TG− littermates and parental pearl mice, achieving levels comparable with WT mice (Fig. 3A). Isolated TG+ pearl AT2 cells also exhibited restoration of intraluminal PRDX6 in LAMP1-positive lamellar bodies when compared with TG− pearl AT2 cells (Fig. 3B). Restoration of lamellar body PRDX6 was associated with increased PLA2 activity (Fig. 3C) and with reductions in the total phospholipid content (Fig. 3D) in lamellar body fractions from TG+ mice relative to TG− mice. The net effect of transgenic rescue of Ap3b1 gene expression was restoration of total lung phospholipid levels (Fig. 3E). Although total phospholipid in bronchoalveolar lavage fluid of TG+ mice was reduced only modestly, PC and DSPC levels in total lung and in lavage fluid were significantly reduced toward WT levels (Fig. 3F). Thus, restoration of AP-3-mediated trafficking within AT2 cells of pearl mice resulted in correction of PRDX6 content and function within lamellar bodies, and this was reflected in improvement of phospholipid content in lamellar bodies, lung tissue, and BAL fluid.
FIGURE 3.

Reconstitution of AP-3 function in AT2 cells restores lamellar body PRDX6 and normalizes lung phospholipid homeostasis in pearl mice. pearl mice expressing the Ap3b1 transgene from an SP-C promoter (TG+) and transgene negative (TG−) littermates were compared with WT and genetically unmanipulated pearl mice (8–10 weeks old). A, representative immunoblots for PRDX6, SP-B, and GAPDH using AT2 lysates (20 μg each) and lamellar body fractions (25 μg each) from WT, pearl, TG+, and TG− mice. B, immunolocalization of PRDX6 in TG+ and TG− isolated alveolar type 2 cells after depletion of cytosolic constituents, using LAMP1 immunostaining to identify lamellar bodies (representative of two experiments; lamellar body noted by white arrow). C, phospholipase A2 activity measured in lamellar bodies from TG− and TG+ mice (n = 3, mean ± S.E.). D, total phospholipid measured in lamellar bodies from TG− and TG+ mice (n = 3; mean ± S.E.). E and F, total phospholipid (PL), total PC, and total DSPC were measured in total lung tissue (E) and bronchoalveolar lavage fluid (F) from TG− and TG+ mice (n = 3 mice of each genotype, mean ± S.E.).
LIMP-2/SCARB2 Is a Lamellar Body Transmembrane Protein Mistrafficked in AT2 from Pearl Mice
AP-3 is a cytosolic/membrane-associated protein, and typically facilitates the trafficking of transmembrane proteins via the recognition of cytoplasmically exposed sorting signals (38). Thus, it cannot directly engage a soluble luminal protein such as PRDX6. However, AP-3 can potentially foster the targeting of luminal proteins via their association with transmembrane proteins that contain AP-3-binding sorting signals. One such transmembrane protein is LIMP-2/SCARB2, a ubiquitously expressed lysosomal protein with an acidic dileucine motif (ERAPLI) that has been previously shown to bind directly to a site composed of δ and σ3A or σ3B subunits of AP-3 (39, 40). Moreover, LIMP-2/SCARB2 mediates the delivery of newly synthesized β-glucocerebrosidase, a luminal protein, to lysosomes in a pH-dependent manner (41). Because LIMP-2/SCARB2 has previously been identified in the lamellar body proteome (2), we considered that LIMP-2/SCARB2 might be delivered to lamellar bodies in an AP-3-dependent manner in AT2 cells. We therefore examined the content of LIMP-2/SCARB2 in enriched lamellar body fractions from WT and pearl mice by immunoblotting (Fig. 4A) and densitometry (Fig. 4B). As predicted, LIMP-2/SCARB2 was enriched in lamellar body fractions of WT AT2 cells but, like Prdx6, was dramatically reduced in lamellar body fractions from pearl mice despite comparable levels of surfactant proteins A and B, representative of luminal and membrane-associated lamellar body proteins. These observations were confirmed by immunofluorescence microscopy of WT and pearl AT2, which demonstrated a marked reduction in co-localization of LIMP2/SCARB2 with LAMP1 at the limiting membrane of lamellar bodies (Fig. 4C). This supports the prediction that AP-3 is required for the delivery of LIMP-2/SCARB2 to lamellar bodies.
FIGURE 4.

Reduced targeting of LIMP-2/SCARB2 to lamellar bodies in pearl mice. Representative immunoblots (A) with densitometry (B) for LIMP-2/SCARB2, PRDX6, SP-A, and SP-B using lamellar body fractions (LB; 5 μg of protein) from wild type (WT) and pearl (pe) mice (n = 4 samples prepared from 2 to 4 mice each; mean ± S.D.). C, immunolocalization of LIMP-2/SCARB2 with LAMP1 identifying lamellar bodies in isolated alveolar type 2 cells from WT and pearl mice (representative of two experiments; bars represent 5 μm).
To determine whether LIMP-2/SCARB2 might be responsible for ferrying PRDX6 to lamellar bodies, we next examined the lung phenotype of Limp-2−/− mice. Consistent with our model, PRDX6 levels were reduced in lamellar body fractions of Limp-2−/− compared with WT controls, despite similar levels of SP-B (Fig. 5A). Moreover, as observed in both Prdx6−/− and pearl mice, total phospholipid, PC, and DSPC were increased in total lung homogenates from Limp-2−/− mice relative to WT (Fig. 5B), and levels of phospholipid, PC, and DSPC were also increased in the bronchoalveolar lavage of Limp-2−/− mice (Fig. 5C). Finally, immunofluorescence microscopy of Limp2−/− AT2 showed reduced intraluminal PRDX6 compared with WT littermate controls (Fig. 5D). These data support the notion that LIMP-2/SCARB2 is required for the AP-3-dependent delivery of PRDX6 to lamellar bodies.
FIGURE 5.

Limp-2−/− mice exhibit reduced lamellar body PRDX6 and increased surfactant phospholipid pool sizes. A, representative immunoblots of LIMP2/SCARB2, PRDX6, SP-B, and GAPDH in total lung (LH), alveolar type 2 (AT2) cells, and lamellar body fractions (LB) from wild type (WT) and Limp-2−/− mice (10 μg of total protein per lane). A light exposure of the SP-B immunoblot is presented as a loading control for lamellar body lanes; darker exposures demonstrate SP-B in lung homogenate and AT2 cell samples. B and C, total phospholipid, total phosphatidylcholine, and total DSPC were measured in total lung tissue (B) and bronchoalveolar lavage fluid from WT and Limp-2−/− mice (C) (n = 4 mice of each genotype, mean ± S.E.). D, immunolocalization of PRDX6 in isolated alveolar type 2 cells (mAT2) from Limp-2−/− mice and wild type littermates (WT) after depletion of cytosolic constituents, using LAMP1 immunostaining to identify lamellar bodies (representative of two experiments; bars represent 5 μm).
LIMP-2/SCARB2 and PRDX6 Proteins Directly Interact in Vitro and in Vivo
Our data are consistent with a model in which PRDX6 is trafficked to lamellar bodies by associating with LIMP2/SCARB2, which is targeted by virtue of its known interaction with AP-3. Like prior studies of LIMP2/SCARB2 interactions with β-glucocerebrosidase (41), we were unable to detect association between endogenous PRDX6 and LIMP2/SCARB2 in cell lysates, possibly due to either transient or pH-dependent interactions. To test for direct protein/protein interactions between PRDX6 and LIMP2/SCARB2, we performed in vitro co-immunoprecipitation of recombinant proteins (Fig. 6A). Recombinant protein A-His6-LIMP2 was immobilized to IgG-Sepharose beads, and recombinant PRDX6-His6 was added in buffers of pH 5 or pH 7. Bound material was analyzed by immunoblotting for the His6 tags. Although protein A-His6-LIMP2 was efficiently recovered under all conditions, PRDX6-His6 was detected only when incubations were performed at pH 5 but not at pH 7. This suggests that PRDX6 binds to LIMP2 only at low pH. To determine whether the in vitro interaction between LIMP2 and PRDX6 is relevant for LIMP2/SCARB2- and AP-3-dependent trafficking of PRDX6 to lamellar bodies in vivo, we performed a proximity ligation assay using antibodies to LIMP2/SCARB2 and PRDX6 in isolated AT2 from WT, pearl, and Limp-2−/− mice (Fig. 6B). By this method, proteins separated by <40 nm are detected through incorporation of fluorescent label into a growing PCR product that is dependent upon annealing of two oligonucleotides forming a continuous loop only upon the binding of antibody complexes to each target protein. Low numbers of PCR products were detected when LIMP2 antibody was omitted (Fig. 6B, upper panels) or in Limp-2−/− AT2 cells (data not shown). By contrast, numerous PCR products were detected when both antibodies were used in either WT or pearl AT2 (Fig. 6B, lower panels). Interestingly, although PCR products were detected in proximity to LB in both WT and pearl AT2, increased numbers of PCR products were detected in the cell periphery of pearl AT2, away from clusters of LB near the cell nuclei. Although this distribution is reminiscent of the endosomal network, we were unable to determine the precise subcellular localization of these complexes using immunofluorescence microscopy. Together, these data provide strong evidence for a specific protein/protein interaction between LIMP2/SCARB2 and PRDX6 in LBs and suggest that this interaction is prolonged in other subcellular structures in pearl AT2 cells.
FIGURE 6.
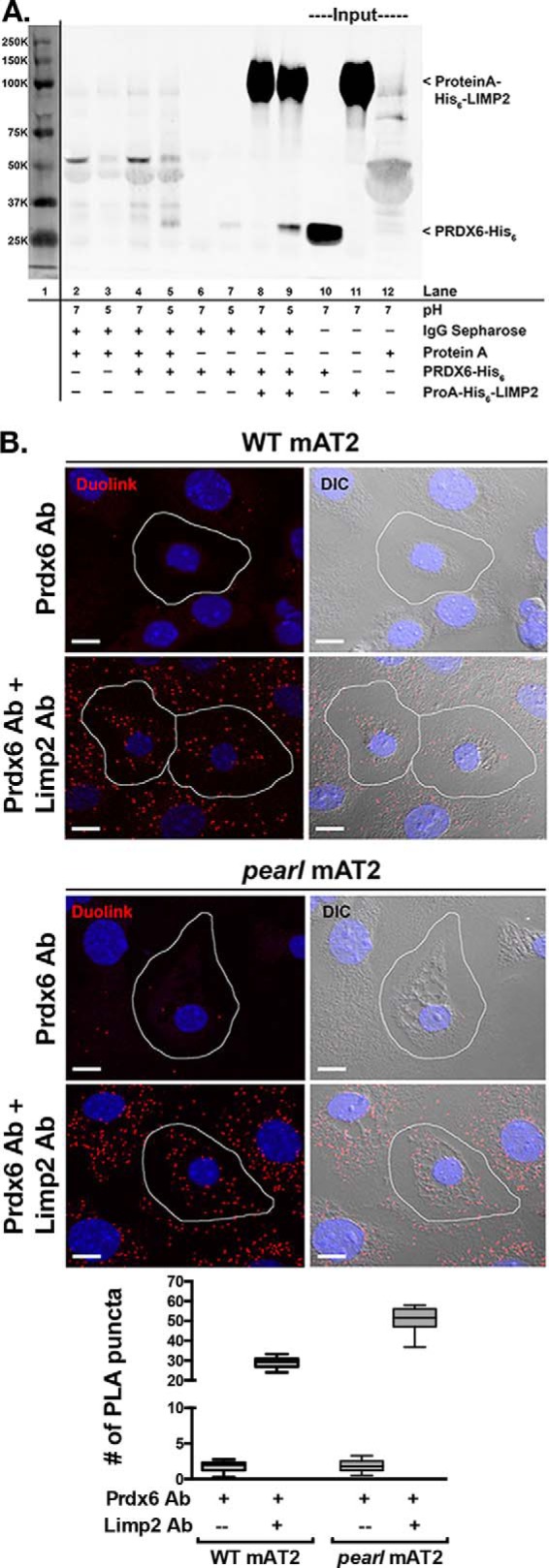
Protein/protein interaction between LIMP2/SCARB2 and PRDX6 in vitro and in vivo. A, co-immunoprecipitation of recombinant PRDX6-His6 by recombinant protein A-His6-LIMP2 is favored in buffer of pH 5 compared with buffer of pH 7 (lanes 8 and 9). Representative immunoblot from two separate experiments. Lane 1 demonstrates molecular weight markers visualized by scanning at 700 nm. Lanes 2–7 illustrate nonspecific interactions between IgG-Sepharose beads with protein A alone (lanes 2 and 3), protein A and PRDX6-His6 (lanes 4 and 5), or PRDX6-His6 alone (lanes 6 and 7), at pH 7 and 5. Lanes 10–12 contain input protein for PRDX6-His6 and protein A-His6-LIMP2. B, representative confocal images of the Duolink PLA interaction between mouse anti-PRDX6 and rabbit anti-LIMP2 (red puncta). For negative controls, mAT2 cells were incubated with mouse anti-PRDX6 alone. The nuclei are labeled with DAPI and differential interference contrast (DIC) images were captured to distinguish the lamellar bodies within mAT2 cells. The red puncta represent positive Duolink signals showing the interaction between PRDX6 and LIMP2; scale bars, 10 μm. Summary data are shown for average number of PLA products per AT2 cell and were derived from two separate experiments, each using lung epithelial cells isolated from four mice of each genotype with 20–25 single AT2 cell images analyzed per genotype per experiment for each of the conditions shown. The box and whiskers plot shows the median (center line), 75th and 25th percentiles (upper and lower limits of the box), and range of raw data (maximum to minimum as hinges). The data were analyzed by unpaired t test, which showed p < 0.0001 WT versus pearl using both antisera in the proximity ligation assay.
Discussion
Three subtypes of HPS, HPS1, -2, and -4, are associated with lethal pulmonary fibrosis and hyperplasia of AT2 cells, with enlarged lamellar bodies containing increased surfactant phospholipid (6, 17, 18, 20). Although it is generally thought that the changes in lamellar body size and phospholipid content ultimately lead to AT2 cell injury, downstream inflammation, and ultimately fibrosis, it has not been clear how specific protein trafficking defects, like those observed in HPS model melanocytes during melanosome biogenesis, might cause these morphological and content changes in lamellar bodies. Here, we show that HPS2 resulting from a defect in AP-3 is associated with increased lamellar body phospholipid due to impaired trafficking of the luminal enzyme PRDX6. This is the first demonstration linking trafficking of a lipid-modifying enzyme to a genetic disorder of lamellar body biogenesis and function. Moreover, our data make HPS one of a growing category of genetic disorders of lung function that reflects altered surfactant phospholipid metabolism.
Phospholipase A2 activity is an important component of the remodeling pathway for phospholipid synthesis in AT2 cells, and it has long been considered a mechanism for enriching DSPC within lamellar body surfactant. PRDX6 is the primary PLA2 in the lumen of lamellar bodies, placing this enzyme in the right place to modify phospholipid content (8). It has been underappreciated that lamellar bodies regulate phospholipid content, not just composition, through PRDX6 enzymatic activity. The modification of surfactant homeostasis in lungs from AP-3-deficient pearl mice is nearly identical to that of WT mouse lungs treated with the PLA2 inhibitor MJ33 and of lungs from Prdx6−/− mice (11, 27). Specifically, all three mouse models present with impaired degradation of endocytosed DPPC and impaired synthesis of DPPC via palmitate incorporation using the remodeling pathway. Although PRDX6-dependent lamellar body degradation may play a minor role in the daily management of phospholipid pools in the lung (42), our data support earlier observations that disruption of this mechanism over time can have a measurable pathological effect on lamellar body phospholipid content. In both pearl and Prdx6−/− mice, phospholipid accumulates in lung progressively as the animals age (11, 24). In the pearl mouse, phospholipid accumulation is associated with enlargement of lamellar bodies (25), and both are corrected when AP-3 function is restored only in AT2 cells (Fig. 3). The lack of lamellar body enlargement in Prdx6−/− mice is reflected by a more modest increase in phospholipid pools relative to WT mice (12), and this suggests that depletion of additional cargoes besides PRDX6 likely contribute to increased phospholipid accumulation in pearl mice. Our prior studies (24, 33) showed that depletion of AP-3 does not appear to impact the lamellar body localization of ABCA3, an ATP-binding cassette protein that is required to enrich lamellar body phospholipid content (43–45), but we predict that other transmembrane lipid transporters might require AP-3 for delivery to lamellar bodies. Delivery of PRDX6 and other phospholipid-metabolizing enzymes likely explain the more marked lamellar body defects in HPS type 2 patients and the pearl mouse model relative to Prdx6−/− mice.
Endocytosis is important for recycling of secreted surfactant phospholipids back to lamellar bodies via a clathrin-dependent pathway that relies on association with surfactant protein A and its receptor, CKAP4/p63 (46). Therefore, changes in endocytosis could impact the phospholipid content of lamellar bodies. Although AP-3 deficiency increases the surface expression of presumed transmembrane cargo proteins of lysosomes (26, 47, 48) and melanosomes (49), it does not appear to directly impact the rate of cargo endocytosis (49–51). Therefore, it is not surprising that [3H]DPPC uptake from the alveolar space was unaltered in pearl mice. Impaired secretion could similarly modify the phospholipid content of lamellar bodies. We previously reported a secretion defect in a mouse model of HPS that was developed from breeding the pearl and pale ear mouse models of HPS2 and HPS1, respectively, to homozygosity (22). Offspring developed massive enlargement of lamellar bodies that were associated with a 50% reduction in surfactant secretion (33). We now show that the secretion defect is not a primary manifestation of the pearl phenotype, and our prior results were likely either secondary to the massive enlargement of the lamellar body in this complex homozygous phenotype or a consequence of the pale ear phenotype.
Our data indicating that PRDX6 was targeted to lamellar bodies in an AP-3-dependent manner presented a conceptual challenge. Although AP-3 is well known to target specific cargo proteins such as LAMP1, TYR, OCA2, or PI4KIIa from early endosomes to lysosomes, LROs, or neurosecretory granules (50, 52–56), it does so by directly recognizing sorting signals on the cytosolic face of transmembrane proteins and sorting them into transport vesicles or tubules. As a cytosolic adaptor protein complex, AP-3 would thus be unable to interact directly with luminal PRDX6. Thus, AP-3-dependent targeting of PRDX6 would require an intermediary transmembrane protein that could interact in the lumen with PRDX6 and in the cytosol with AP-3. LIMP-2/SCARB2 was a strong candidate because (a) it harbors a well described cytoplasmic di-leucine-based sorting motif that directly binds the AP-3 core (39, 40), and (b) it is known to foster the movement of β-glucocerebrosidase to lysosomes via direct interactions with a luminal helical bundle of LIMP-2 (41, 57). Our data not only validate earlier reports suggesting that LIMP-2/SCARB2 is a lamellar body integral membrane protein (2) but also demonstrate that accumulation of LIMP2/SCARB2 at the limiting membrane of lamellar bodies is diminished in pearl mice, and efficient delivery to lamellar bodies requires AP-3. To our knowledge, this is the first demonstration that LIMP-2/SCARB2 trafficking to an LRO requires interaction with AP-3.
Importantly, our data demonstrate that LIMP2 is necessary for PRDX6 trafficking to lamellar bodies. PRDX6 content was reduced in lamellar body fractions from Limp-2−/− mice, and total phospholipid, PC, and DSPC were reduced in whole lung and bronchoalveolar lavage fractions, similarly to the pearl mouse model. Immunolabeling demonstrated a depletion of LIMP2 and PRDX6 from lamellar bodies of pearl AT2, and similarly, PRDX6 was depleted from lamellar bodies in Limp-2−/− AT2. Moreover, our data demonstrate direct interactions between LIMP2 and PRDX6 both in vitro and in vivo. Based on prior studies of β-glucocerebrosidase interactions, we expected that a LIMP2/PRDX6 interaction would be favored at neutral pH and not at an acidic pH (41). To our surprise, the LIMP2/PRDX6 interaction, although indeed pH-dependent, favored binding at low pH. The pH of early sorting endosomes is generally thought to be ∼6.2 and that of late endosomes ∼5.5, although in selected cells like melanocytes early endosomes are more acidic (58). Based on compartment pH alone, it is possible that loading of PRDX6 onto LIMP2 might occur in the endosomal network, which is supported by the distribution of PRDX6 and LIMP2 by immunostaining and by proximity ligation assay in pearl AT2. Early studies indicated an internal pH of 5.6 in lamellar bodies (59), which would predict that PRDX6 would remain bound to LIMP2 within the lamellar body unless displaced by some other mechanism. This might facilitate the interaction of PRDX6 with phospholipid, for which it has substantial affinity (60), perhaps via phospholipid transport through a hydrophobic tunnel in LIMP2/SCARB2 that is homologous to a cholesterol-binding domain in other CD36 family members (57). Results from proximity ligation assays support a close proximity of PRDX6 with LIMP2/SCARB2 in AT2 cells, even in the absence of AP-3. The high frequency of proximity PCR products in AT2 regions lacking lamellar bodies supports impaired lamellar body targeting of the LIMP2/SCARB2-PRDX6 complex, and it suggests that the complex may form in endosomes. Together, our data clearly implicate LIMP-2/SCARB2 and AP-3 in a pathway for targeting PRDX6 to lamellar bodies. A similar interaction with LIMP2/SCARB2 or another AP-3-dependent receptor might also explain the failure to deliver other luminal cargoes to LROs, such as elastase to the primary granules of neutrophils (61).
Our data indicate that the AP-3-dependent pathway is necessary for efficient targeting of LIMP2/SCARB2 and thus PRDX6 to lamellar bodies. However, we demonstrate substantial reductions in these two proteins in lamellar bodies but not a complete absence, and immunolabeling of lamellar bodies also shows variability of PRDX6 content. This suggests that the AP-3-dependent pathway for lamellar body targeting might be partially redundant with other pathways. For example, in melanocytes two distinct pathways mediate cargo sorting to melanosomes, and for some cargoes (such as tyrosinase) loss of AP-3 in one pathway can be partially compensated by the second AP-1- and BLOC-1-dependent pathway (62, 63). Mutations of Rab38 in chocolate mice, Long-Evans Cinnamon rats and Fawn-Hooded hypertension rats lead to enlarged LBs with increased phospholipid content (64–66) like AP-3-deficient pearl mice, and RAB38 has been suggested to interact with AP-3 (67). However, defects in LB-associated PRDX6 in these models have not been tested. Thus, it is possible that other lysosome-related organelle or membrane trafficking pathways might work in coordination with AP-3 to promote trafficking of PRDX6 and possibly other lamellar body cargo to lamellar bodies.
In summary, this is the first description of a membrane transport mechanism by which surfactant-related cargo is targeted to the lamellar body. That this mechanism is AP-3-dependent adds to a growing body of evidence that AP-3 is a component of a common targeting mechanism for selected LRO cargoes. Our data also provide the first evidence that LIMP-2/SCARB2, which is known to harbor a cytoplasmic AP-3 binding signal (39) and was identified in the lamellar body proteome (2), requires AP-3 for targeting to lamellar bodies. Its novel role in ferrying PRDX6 to lamellar bodies expands the repertoire of LIMP2/SCARB2 as a mediator of trafficking luminal proteins through the secretory pathway. Importantly, AP-3-mediated targeting of a phospholipid-modifying enzyme to lamellar bodies is significant for lung disease. Loss of targeting of PRDX6 in pearl mice correlates with phospholipid accumulation and lamellar body enlargement that are common features of human HPS2, and it is reminiscent of the failure to deliver cargoes like TYR and OCA2 to melanosomes (resulting in reduced pigmentation) or CD63 to Weibel-Palade bodies (impairing the capacity of endothelial cells to recruit leukocytes in the absence of AP-3). The greater phospholipid accumulation and lamellar body enlargement in pearl mice relative to Prdx6−/− mice further suggest that there are additional AP-3-dependent cargoes that have a role in surfactant phospholipid homeostasis and that use AP-3 for targeting to lamellar bodies. Moreover, the additive effects on phospholipid accumulation in the pale ear/pearl mouse model (24, 33) suggest that other HPS-associated complexes are also involved in targeting lipid-modifying cargoes to lamellar bodies. Thus, altered lamellar body cargo delivery may be an important driver of the AT2 cell injury in HPS that in turn mediates long term lung damage.
Author Contributions
S. K. (Figs. 1–6), P. W. (Figs. 1–6), and L. G. (Figs. 1–4) performed and analyzed experiments. L. Y. provided mouse models that she designed and constructed for experiments represented in Fig. 3, assisted in the interpretation of data from these experiments, and provided critical revision of the manuscript. M. S. and P. S. provided the Limp-2−/− mice and LIMP-2 reagents that they designed and constructed and provided critical revision of the manuscript. D. N. conceived the experiments, interpreted data, and provided methods for experiments in Fig. 6. X. W. and Y. M. developed and executed these experiments. M. S. M. provided significant input into the interpretation of data in Figs. 3–5, assisted in writing portions of the manuscript, and provided critical revision of the manuscript. M. F. B. assisted in the interpretation of experiments in Figs. 1–3, assisted in writing portions of the manuscript, and in critical revisions of the paper. S. G. conceived and coordinated the studies, acquired the funding for the studies, interpreted data from the experiments, prepared figures, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful for the generous experimental guidance and technical assistance of Aron Fisher, Chandra Dodia, and Elena Sorokina in selected experiments in Figs. 1–5. We also thank Dawn C. Harper, Yaniv Tomer, and Peggy Zhang for their assistance with animal husbandry.
This work was supported by American Heart Association Grants 12GRNT 12050265 (to S. G.) and 13GRNT17070104 (to M. F. B.), National Institutes of Health Grants HL059959 (to S. G.), HL119503 (to L. Y.), HL119436 (to M. F. B.), and GM108807 (to S. G. and M.M.), Department of Veterans Affairs Merit Award BX001176 (to M. F. B.), the Hermansky-Pudlak Syndrome Network, and Deutsche Forschungsgemeinschaft Grant GRK1459. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DSPC
- disaturated phosphatidylcholine
- LRO
- lysosome-related organelle
- LB
- lamellar body
- HPS
- Hermansky Pudlak syndrome
- DPPC
- dipalmitoyl phosphatidylcholine
- PLA2
- phospholipase A2
- PC
- phosphatidylcholine
- PRDX6
- peroxiredoxin 6
- lyso-PC
- lysophosphatidylcholine
- TG
- transgene
- PLA
- proximity ligation assay.
References
- 1. Whitsett J. A., Wert S. E., and Weaver T. E. (2015) Diseases of pulmonary surfactant homeostasis. Annu. Rev. Pathol. 10, 371–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ridsdale R., Na C.-L., Xu Y., Greis K. D., and Weaver T. (2011) Comparative proteomic analysis of lung lamellar bodies and lysosome-related organelles. PLoS ONE 6, e16482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wei M. L. (2006) Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 19, 19–42 [DOI] [PubMed] [Google Scholar]
- 4. Raposo G., and Marks M. S. (2007) Melanosomes–dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 8, 786–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marks M. S., Heijnen H. F., and Raposo G. (2013) Lysosome-related organelles: unusual compartments become mainstream. Curr. Opin. Cell Biol. 25, 495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei A.-H., and Li W. (2013) Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res. 26, 176–192 [DOI] [PubMed] [Google Scholar]
- 7. Sitaram A., and Marks M. S. (2012) Mechanisms of protein delivery to melanosomes in pigment cells. Physiology 27, 85–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fisher A. B. (2011) Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid. Redox Signal. 15, 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manevich Y., Shuvaeva T., Dodia C., Kazi A., Feinstein S. I., and Fisher A. B. (2009) Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A(2) activities. Arch. Biochem. Biophys. 485, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rooney S. A., Young S. L., and Mendelson C. R. (1994) Molecular and cellular processing of lung surfactant. FASEB J. 8, 957–967 [DOI] [PubMed] [Google Scholar]
- 11. Fisher A. B., Dodia C., Feinstein S. I., and Ho Y.-S. (2005) Altered lung phospholipid metabolism in mice with targeted deletion of lysosomal-type phospholipase A2. J. Lipid Res. 46, 1248–1256 [DOI] [PubMed] [Google Scholar]
- 12. Fisher A. B., Dodia C., Yu K., Manevich Y., and Feinstein S. I. (2006) Lung phospholipid metabolism in transgenic mice overexpressing peroxiredoxin 6. Biochim. Biophys. Acta 1761, 785–792 [DOI] [PubMed] [Google Scholar]
- 13. Sorokina E. M., Feinstein S. I., Milovanova T. N., and Fisher A. B. (2009) Identification of the amino acid sequence that targets peroxiredoxin 6 to lysosome-like structures of lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L871–L80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sorokina E. M., Feinstein S. I., Zhou S., and Fisher A. B. (2011) Intracellular targeting of peroxiredoxin 6 to lysosomal organelles requires MAPK activity and binding to 14-3-3ϵ. Am. J. Physiol. Cell Physiol. 300, C1430–C1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okado-Matsumoto A., Matsumoto A., Fujii J., and Taniguchi N. (2000) Peroxiredoxin IV is a secretable protein with heparin-binding properties under reduced conditions. J. Biochem. 127, 493–501 [DOI] [PubMed] [Google Scholar]
- 16. Kakihana T., Araki K., Vavassori S., Iemura S., Cortini M., Fagioli C., Natsume T., Sitia R., and Nagata K. (2013) Dynamic regulation of Ero1α and peroxiredoxin 4 localization in the secretory pathway. J. Biol. Chem. 288, 29586–29594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakatani Y., Nakamura N., Sano J., Inayama Y., Kawano N., Yamanaka S., Miyagi Y., Nagashima Y., Ohbayashi C., Mizushima M., Manabe T., Kuroda M., Yokoi T., and Matsubara O. (2000) Interstitial pneumonia in Hermansky-Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchows Arch. 437, 304–313 [DOI] [PubMed] [Google Scholar]
- 18. Bachli E. B., Brack T., Eppler E., Stallmach T., Tr Eb R. M., Huizing M., and Gahl W. A. (2004) Hermansky-Pudlak syndrome type 4 in a patient from Sri Lanka with pulmonary fibrosis. Am. J. Med. Genet. 127A, 201–207 [DOI] [PubMed] [Google Scholar]
- 19. Tang X., Yamanaka S., Miyagi Y., Nagashima Y., and Nakatani Y. (2005) Lung pathology of pale ear mouse (model of Hermansky-Pudlak syndrome 1) and beige mouse (model of Chediak-Higashi syndrome): severity of giant lamellar body degeneration of type II pneumocytes correlates with interstitial inflammation. Pathol. Int. 55, 137–143 [DOI] [PubMed] [Google Scholar]
- 20. Gochuico B. R., Huizing M., Golas G. A., Scher C. D., Tsokos M., Denver S. D., Frei-Jones M. J., and Gahl W. A. (2012) Interstitial lung disease and pulmonary fibrosis in Hermansky-Pudlak syndrome type 2, an adaptor protein-3 complex disease. Mol. Med. 18, 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGarry M. P., Reddington M., Novak E. K., and Swank R. T. (1999) Survival and lung pathology of mouse models of Hermansky-Pudlak syndrome and Chediak-Higashi syndrome. Proc. Soc. Exp. Biol. Med. 220, 162–168 [DOI] [PubMed] [Google Scholar]
- 22. Lyerla T. A., Rusiniak M. E., Borchers M., Jahreis G., Tan J., Ohtake P., Novak E. K., and Swank R. T. (2003) Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 285, L643–L653 [DOI] [PubMed] [Google Scholar]
- 23. Gautam R., Novak E. K., Tan J., Wakamatsu K., Ito S., and Swank R. T. (2006) Interaction of Hermansky-Pudlak Syndrome genes in the regulation of lysosome-related organelles. Traffic 7, 779–792 [DOI] [PubMed] [Google Scholar]
- 24. Atochina-Vasserman E. N., Bates S. R., Zhang P., Abramova H., Zhang Z., Gonzales L., Tao J.-Q., Gochuico B. R., Gahl W., Guo C.-J., Gow A. J., Beers M. F., and Guttentag S. (2011) Early alveolar epithelial dysfunction promotes lung inflammation in a mouse model of Hermansky Pudlak syndrome. Am. J. Respir. Crit. Care Med. 184, 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young L. R., Gulleman P. M., Bridges J. P., Weaver T. E., Deutsch G. H., Blackwell T. S., and McCormack F. X. (2012) The alveolar epithelium determines susceptibility to lung fibrosis in Hermansky-Pudlak syndrome. Am. J. Respir. Crit. Care Med. 186, 1014–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dell'Angelica E. C., Shotelersuk V., Aguilar R. C., Gahl W. A., and Bonifacino J. S. (1999) Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol. Cell 3, 11–21 [DOI] [PubMed] [Google Scholar]
- 27. Fisher A. B., and Dodia C. (2001) Lysosomal-type PLA2 and turnover of alveolar DPPC. Am. J. Physiol. Lung Cell Mol. Physiol. 280, L748–L754 [DOI] [PubMed] [Google Scholar]
- 28. Marinetti G. V. (1962) Hydrolysis of lecithin with sodium methoxide. Biochemistry 1, 350–353 [DOI] [PubMed] [Google Scholar]
- 29. Fisher A. B., Dodia C., and Chander A. (1987) Degradation and reutilization of alveolar phosphatidylcholine by rat lungs. J. Appl. Physiol. 62, 2295–2299 [DOI] [PubMed] [Google Scholar]
- 30. Kim T. S., Dodia C., Chen X., Hennigan B. B., Jain M., Feinstein S. I., and Fisher A. B. (1998) Cloning and expression of rat lung acidic Ca2+-independent PLA2 and its organ distribution. Am. J. Physiol. 274, L750–L761 [DOI] [PubMed] [Google Scholar]
- 31. Manevich Y., Sweitzer T., Pak J. H., Feinstein S. I., Muzykantov V., and Fisher A. B. (2002) 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc. Natl. Acad. Sci. U.S.A. 99, 11599–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fisher A. B., Dodia C., Chander A., and Jain M. (1992) A competitive inhibitor of phospholipase A2 decreases surfactant phosphatidylcholine degradation by the rat lung. Biochem. J. 288, 407–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guttentag S. H., Akhtar A., Tao J.-Q., Atochina E., Rusiniak M. E., Swank R. T., and Bates S. R. (2005) Defective surfactant secretion in a mouse model of Hermansky-Pudlak syndrome. Am. J. Respir. Cell Mol. Biol. 33, 14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Z., Michael I. P., Zhou D., Nagy A., and Rini J. M. (2013) Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc. Natl. Acad. Sci. U.S.A. 110, 5004–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feng L., Seymour A. B., Jiang S., To A., Peden A. A., Novak E. K., Zhen L., Rusiniak M. E., Eicher E. M., Robinson M. S., Gorin M. B., and Swank R. T. (1999) The β3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum. Mol. Genet. 8, 323–330 [DOI] [PubMed] [Google Scholar]
- 36. Weaver T. E., Na C.-L., and Stahlman M. (2002) Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin. Cell Dev. Biol. 13, 263–270 [DOI] [PubMed] [Google Scholar]
- 37. Manevich Y., and Fisher A. B. (2005) Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic. Biol. Med. 38, 1422–1432 [DOI] [PubMed] [Google Scholar]
- 38. Dell'Angelica E. C. (2009) AP-3-dependent trafficking and disease: the first decade. Curr. Opin. Cell Biol. 21, 552–559 [DOI] [PubMed] [Google Scholar]
- 39. Höning S., Sandoval I. V., and von Figura K. (1998) A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J. 17, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Janvier K., Kato Y., Boehm M., Rose J. R., Martina J. A., Kim B.-Y., Venkatesan S., and Bonifacino J. S. (2003) Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 γ-σ1 and AP-3 δ-σ3 hemicomplexes. J. Cell Biol. 163, 1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reczek D., Schwake M., Schröder J., Hughes H., Blanz J., Jin X., Brondyk W., Van Patten S., Edmunds T., and Saftig P. (2007) LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131, 770–783 [DOI] [PubMed] [Google Scholar]
- 42. Postle A. D., Henderson N. G., Koster G., Clark H. W., and Hunt A. N. (2011) Analysis of lung surfactant phosphatidylcholine metabolism in transgenic mice using stable isotopes. Chem. Phys. Lipids 164, 549–555 [DOI] [PubMed] [Google Scholar]
- 43. Cheong N., Zhang H., Madesh M., Zhao M., Yu K., Dodia C., Fisher A. B., Savani R. C., and Shuman H. (2007) ABCA3 is critical for lamellar body biogenesis in vivo. J. Biol. Chem. 282, 23811–23817 [DOI] [PubMed] [Google Scholar]
- 44. Fitzgerald M. L., Xavier R., Haley K. J., Welti R., Goss J. L., Brown C. E., Zhuang D. Z., Bell S. A., Lu N., McKee M., Seed B., and Freeman M. W. (2007) ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J. Lipid Res. 48, 621–632 [DOI] [PubMed] [Google Scholar]
- 45. Ban N., Matsumura Y., Sakai H., Takanezawa Y., Sasaki M., Arai H., and Inagaki N. (2007) ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J. Biol. Chem. 282, 9628–9634 [DOI] [PubMed] [Google Scholar]
- 46. Bates S. R., Kazi A. S., Tao J.-Q., Yu K. J., Gonder D. S., Feinstein S. I., and Fisher A. B. (2008) Role of P63 (CKAP4) in binding of surfactant protein-A to type II pneumocytes. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L658–L669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Le Borgne R., Alconada A., Bauer U., and Hoflack B. (1998) The mammalian AP-3 adaptor-like complex mediates the intracellular transport of lysosomal membrane glycoproteins. J. Biol. Chem. 273, 29451–29461 [DOI] [PubMed] [Google Scholar]
- 48. Yang W., Li C., Ward D. M., Kaplan J., and Mansour S. L. (2000) Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J. Cell Sci. 113, 4077–4086 [DOI] [PubMed] [Google Scholar]
- 49. Setty S. R., Tenza D., Truschel S. T., Chou E., Sviderskaya E. V., Theos A. C., Lamoreux M. L., Di Pietro S. M., Starcevic M., Bennett D. C., Dell'Angelica E. C., Raposo G., and Marks M. S. (2007) BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol. Biol. Cell 18, 768–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peden A. A., Oorschot V., Hesser B. A., Austin C. D., Scheller R. H., and Klumperman J. (2004) Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 164, 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Janvier K., and Bonifacino J. S. (2005) Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16, 4231–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Theos A. C., Berson J. F., Theos S. C., Herman K. E., Harper D. C., Tenza D., Sviderskaya E. V., Lamoreux M. L., Bennett D. C., Raposo G., and Marks M. S. (2006) Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell 17, 3598–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chapuy B., Tikkanen R., Mühlhausen C., Wenzel D., von Figura K., and Höning S. (2008) AP-1 and AP-3 mediate sorting of melanosomal and lysosomal membrane proteins into distinct post-Golgi trafficking pathways. Traffic 9, 1157–1172 [DOI] [PubMed] [Google Scholar]
- 54. Sitaram A., Dennis M. K., Chaudhuri R., De Jesus-Rojas W., Tenza D., Setty S. R., Wood C. S., Sviderskaya E. V., Bennett D. C., Raposo G., Bonifacino J. S., and Marks M. S. (2012) Differential recognition of a dileucine-based sorting signal by AP-1 and AP-3 reveals a requirement for both BLOC-1 and AP-3 in delivery of OCA2 to melanosomes. Mol. Biol. Cell 23, 3178–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Faúndez V., Horng J. T., and Kelly R. B. (1998) A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell 93, 423–432 [DOI] [PubMed] [Google Scholar]
- 56. Salazar G., Love R., Styers M. L., Werner E., Peden A., Rodriguez S., Gearing M., Wainer B. H., and Faundez V. (2004) AP-3-dependent mechanisms control the targeting of a chloride channel (ClC-3) in neuronal and non-neuronal cells. J. Biol. Chem. 279, 25430–25439 [DOI] [PubMed] [Google Scholar]
- 57. Neculai D., Schwake M., Ravichandran M., Zunke F., Collins R. F., Peters J., Neculai M., Plumb J., Loppnau P., Pizarro J. C., Seitova A., Trimble W. S., Saftig P., Grinstein S., and Dhe-Paganon S. (2013) Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 504, 172–176 [DOI] [PubMed] [Google Scholar]
- 58. Raposo G., Tenza D., Murphy D. M., Berson J. F., and Marks M. S. (2001) Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 152, 809–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chander A., Johnson R. G., Reicherter J., and Fisher A. B. (1986) Lung lamellar bodies maintain an acidic internal pH. J. Biol. Chem. 261, 6126–6131 [PubMed] [Google Scholar]
- 60. Manevich Y., Reddy K. S., Shuvaeva T., Feinstein S. I., and Fisher A. B. (2007) Structure and phospholipase function of peroxiredoxin 6: identification of the catalytic triad and its role in phospholipid substrate binding. J. Lipid Res. 48, 2306–2318 [DOI] [PubMed] [Google Scholar]
- 61. Meng R., Bridgman R., Toivio-Kinnucan M., Niemeyer G. P., Vernau W., Hock T., and Lothrop C. D. (2010) Neutrophil elastase-processing defect in cyclic hematopoietic dogs. Exp. Hematol. 38, 104–115 [DOI] [PubMed] [Google Scholar]
- 62. Theos A. C., Tenza D., Martina J. A., Hurbain I., Peden A. A., Sviderskaya E. V., Stewart A., Robinson M. S., Bennett D. C., Cutler D. F., Bonifacino J. S., Marks M. S., and Raposo G. (2005) Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol. Biol. Cell 16, 5356–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Setty S. R., Tenza D., Sviderskaya E. V., Bennett D. C., Raposo G., and Marks M. S. (2008) Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature 454, 1142–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Osanai K., Oikawa R., Higuchi J., Kobayashi M., Tsuchihara K., Iguchi M., Jongsu H., Toga H., and Voelker D. R. (2008) A mutation in Rab38 small GTPase causes abnormal lung surfactant homeostasis and aberrant alveolar structure in mice. Am. J. Pathol. 173, 1265–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Osanai K., Higuchi J., Oikawa R., Kobayashi M., Tsuchihara K., Iguchi M., Huang J., Voelker D. R., and Toga H. (2010) Altered lung surfactant system in a Rab38-deficient rat model of Hermansky-Pudlak syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L243–L251 [DOI] [PubMed] [Google Scholar]
- 66. Zhang L., Yu K., Robert K. W., DeBolt K. M., Hong N., Tao J.-Q., Fukuda M., Fisher A. B., and Huang S. (2011) Rab38 targets to lamellar bodies and normalizes their sizes in lung alveolar type II epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 301, L461–L477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bultema J. J., Ambrosio A. L., Burek C. L., and Di Pietro S. M. (2012) BLOC-2, AP-3, and AP-1 proteins function in concert with Rab38 and Rab32 proteins to mediate protein trafficking to lysosome-related organelles. J. Biol. Chem. 287, 19550–19563 [DOI] [PMC free article] [PubMed] [Google Scholar]