

Abstract
The μ-opioid receptor (MOR, encoded by Oprm1) agonists are the mainstay analgesics for treating moderate to severe pain. Nerve injury causes down-regulation of MORs in the dorsal root ganglion (DRG) and diminishes the opioid effect on neuropathic pain. However, the epigenetic mechanisms underlying the diminished MOR expression caused by nerve injury are not clear. G9a (encoded by Ehmt2), a histone 3 at lysine 9 methyltransferase, is a key chromatin regulator responsible for gene silencing. In this study, we determined the role of G9a in diminished MOR expression and opioid analgesic effects in animal models of neuropathic pain. We found that nerve injury in rats induced a long-lasting reduction in the expression level of MORs in the DRG but not in the spinal cord. Nerve injury consistently increased the enrichment of the G9a product histone 3 at lysine 9 dimethylation in the promoter of Oprm1 in the DRG. G9a inhibition or siRNA knockdown fully reversed MOR expression in the injured DRG and potentiated the morphine effect on pain hypersensitivity induced by nerve injury. In mice lacking Ehmt2 in DRG neurons, nerve injury failed to reduce the expression level of MORs and the morphine effect. In addition, G9a inhibition or Ehmt2 knockout in DRG neurons normalized nerve injury-induced reduction in the inhibitory effect of the opioid on synaptic glutamate release from primary afferent nerves. Our findings indicate that G9a contributes critically to transcriptional repression of MORs in primary sensory neurons in neuropathic pain. G9a inhibitors may be used to enhance the opioid analgesic effect in the treatment of chronic neuropathic pain.
Keywords: electrophysiology, epigenetics, neuron, neurophysiology, opiate opioid, pain
Introduction
Chronic neuropathic pain resulting from damage to the peripheral or central nervous system causes agonizing suffering and reduced quality of life. Neuropathic pain is often resistant to conventional analgesic treatments and remains a major therapeutic challenge. Opioid drugs such as morphine produce their therapeutic effects through binding to the μ-opioid receptors (MORs,3 encoded by Oprm1) (1, 2) and are widely used to treat moderate to severe pain. In patients with neuropathic pain, however, the analgesic potency of MOR agonists is reduced (3, 4). The opioid effects are also diminished in animal models of neuropathic pain (5–7). MORs expressed at primary sensory neurons and their central terminals in the spinal dorsal horn are essential for the analgesic effects of opioids (8–10). Peripheral nerve injury reduces the expression level of MORs in the dorsal root ganglion (DRG), contributing to the loss of opioid analgesic efficacy in neuropathic pain (6, 11, 12). However, the epigenetic mechanisms by which nerve injury leads to diminished MOR expression in the DRG remain unclear.
Transcriptional homeostasis is largely maintained by the dynamic balance between positive and negative regulation of gene transcription. Gene expression is critically controlled by chromatin structure and the modification status of histone tails (13–15). DNA methylation and histone modifications are two major components of the epigenetic mechanisms of gene expression. The euchromatin histone methyltransferases are a family of evolutionarily conserved proteins that write part of the epigenetic code through methylation of histone 3 at lysine 9 (H3K9). H3K9 dimethylation (H3K9me2) is commonly associated with gene silencing (16–18). H3K9me2 is catalyzed by an enzymatic complex comprised of the histone H3K9 dimethyltransferases G9a (encoded by Ehmt2) and G9a-like protein (GLP) (17–20). G9a and GLP form a heterodimeric complex. Genetic ablation of either of these proteins results in the loss of H3K9me2 and produces nearly identical phenotypes, such as early embryonic lethality and abnormal gene transcription (18, 20). G9a is present in the nucleus of DRG neurons, and nerve injury increases the protein level of G9a and H3K9me2 in the DRG tissue, accounting for the down-regulation of various potassium channels in neuropathic pain (21). However, it is not clear whether or to what extent G9a is involved in nerve injury-induced transcriptional repression of Oprm1 in the DRG.
In this study, we investigated the role of G9a in the epigenetic silencing of the Oprm1 gene in the DRG and the diminished opioid effects in rodent models of neuropathic pain. We found that nerve injury consistently increased the enrichment of H3K9me2 in the promoter region of Oprm1. Remarkably, G9a inhibition or selective deletion of Ehmt2 in DRG neurons completely restored nerve injury-induced reduction in MOR expression and in the opioid effects on nociception and synaptic transmission between primary afferent nerves and spinal dorsal horn neurons. Thus, our study reveals that G9a-mediated H3K9me2 has a critical function in the transcriptional silencing of Oprm1 in injured DRGs and in the diminished opioid analgesic effect on neuropathic pain.
Experimental Procedures
Rat Model of Neuropathic Pain and Drug Treatment
We used male Sprague-Dawley rats (9–10 weeks old; Harlan, Indianapolis, IN) in this study. All of the experimental protocols were approved by the Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center and conformed to the National Institutes of Health guidelines for the ethical use of animals. Spinal nerve ligation (SNL) was used as an experimental model of neuropathic pain, as described previously (22, 23). Briefly, we induced anesthesia with 2–3% isoflurane, isolated the left L5 and L6 spinal nerves under a surgical microscope, and ligated them with a 6–0 silk suture. Control rats underwent a sham surgical procedure without nerve ligation. In the SNL model, stable pain hypersensitivity is typically established 10–14 days after SNL and lasts for at least 8 weeks. Final morphine analgesic testing and electrophysiological recordings were performed 3–4 weeks after SNL.
Two weeks after surgery, intrathecal catheters were implanted in some SNL rats during isoflurane-induced anesthesia. Briefly, we made a small incision at the back of the neck of the animal. Next, we made a small opening in the atlanto-occipital membrane of the cisterna magna and inserted a PE-10 catheter (∼8.0 cm) so that the caudal tip reached the lumbar spinal cord (8, 24). Rats displaying motor or neurological dysfunction after catheter insertion were immediately euthanized.
The G9a/GLP inhibitor UNC0638 (Sigma-Aldrich, St. Louis, MO) or dimethyl sulfoxide vehicle was injected intrathecally at a volume of 10 μl, followed by a 5-μl saline flush. Drug treatment started 3 weeks after SNL, when chronic pain had become well established. Morphine (West-Ward Pharmaceuticals, Eatontown, NJ) was used for testing the opioid analgesic effect in vivo, and [d-Ala2,N-Me-Phe4,Gly-ol]-enkephalin (DAMGO, Sigma-Aldrich) was used for assessing the opioid effect on synaptic transmission in spinal cord slices. In some SNL rats, G9a-specific siRNA (4 μg) or the negative control siRNA was administered intrathecally. G9a-specific siRNA (AGUAACGGGCAUCAAUGC) or universal negative control siRNA (SIC001, Sigma-Aldrich) was mixed with i-Fect (Neuromics, Edina, MN) to a final concentration of 400 mg/liter for the intrathecal injections (21, 25).
G9a Conditional Knockout Mice
Wild-type mice (8 weeks of age, both males and females, sex- and age-matched) with a C57/BL6J genetic background were obtained from The Jackson Laboratory (Bar Harbor, ME). We selectively deleted the Ehmt2 gene, which codes for G9a, in the DRG neurons by crossing female mice with a loxP-flanked Ehmt2 gene (Ehmt2flox/flox mice) with male mice of the primary sensory neuron-specific Cre line AdvillinCre/+ (obtained from Dr. F. Wang at Duke University) (26). From the first cross, we obtained male AdvillinCre/+;Ehmt2flox/+ mice, which were crossed again to female Ehmt2flox/flox mice to generate AdvillinCre/+;Ehmt2flox/flox mice, referred to henceforth as G9a conditional (KO) mice.
Neuropathic pain was induced in the mice using the spared nerve injury (SNI) model, as described previously (27). The mice were anesthetized with 2% isoflurane, and a small incision was made on the left lateral thigh to expose the sciatic nerve. We ligated and sectioned the common peroneal and tibial nerves, leaving the sural nerve intact, under a surgical microscope. The sham procedure consisted of the same surgery without nerve ligation and sectioning. Final behavioral testing and spinal cord slice recordings were performed 2–3 weeks after surgery. The animals were killed with 5% isoflurane and exsanguination at the end of the experiments.
Behavioral Assessment of Tactile Allodynia and Hyperalgesia
Behavioral tests were conducted as described previously (24, 28). In brief, to detect tactile allodynia, we applied von Frey filaments to the left hind paw of the animals (ipsilateral to SNL or SNI). We then placed the rats or mice individually in suspended chambers on a mesh floor. After an acclimation period of 30 min, we applied a series of calibrated von Frey filaments (Stoelting, Wood Dale, IL) perpendicularly to the plantar surface of the hind paw with sufficient force to bend the filament for 6 s. Brisk withdrawal or paw flinching was considered to be a positive response. After a response, the filament of the next lower force was applied. If there was no response, the filament of the next greater force was applied. We calculated the tactile stimulus force that produced a 50% likelihood of a withdrawal response using the “up-down” method (22, 24).
To quantify the mechanical nociceptive threshold in both the rats and the mice, we performed the paw pressure test on the left hind paw using an analgesiometer (Ugo Basile, Varese, Italy). To activate the device, a foot pedal was pressed, triggering a motor that applied a constantly increasing force on a linear scale. When the animal displayed pain by either withdrawing its paw or vocalizing, the pedal was immediately released, and the nociceptive threshold of the animal was read on the scale (24, 28, 29). Each trial was repeated two or three times at ∼2-min intervals, and the mean value was used as the force to produce withdrawal responses.
Quantitative PCR
Total RNA was extracted from the DRG and spinal cord tissues at the L5 and L6 levels using TRIzol-chloroform and then treated with DNase I (Invitrogen). cDNA was prepared by using the Superscript III first-strand synthesis kit and then treated with RNase H (Invitrogen). Quantitative PCR was performed using the iQ5 real-time PCR detection system (Bio-Rad) and SYBR Green (Bioline, Taunton, MA). The thermal cycling conditions used were as follows: one cycle at 95 °C for 1 min, 40 cycles at 95 °C for 15 s, and one cycle at 60 °C for 15 s. The following primers were used: rat Oprm1 forward, CCTCTTCGGAAACTTCCTGG; rat Oprm1 reverse, GCCATGTTCCCATCAGGTAG; rat Gapdh forward, TGCCACTCAGAAGACTGTGG; rat Gapdh reverse, TTCAGCTCTGGGATGACCTT; mouse Oprm1 forward, TGAAGACTGCCACCAACATC; mouse Oprm1 reverse, CCACGTTCCCATCAGGTAGT; mouse Gapdh forward, GGGTGTGAACCACGAGAAAT; and mouse Gapdh reverse, CCTTCCACAATGCCAAAGTT. Relative mRNA levels were calculated using the 2−ΔΔCT method and normalized by Gapdh in the same sample.
Western Immunoblotting
The excised DRG and spinal cord tissues were homogenized with a sonicator (Qsonica, Newtown, CT) in ice-cold radioimmunoprecipitation assay buffer containing protease mixture II (Sigma-Aldrich). Proteins were separated by polyacrylamide gel electrophoresis. The following primary antibodies were used: MOR1 (50 kDa, 1:400 dilution, catalog no. sc-7488, Santa Cruz Biotechnology, Dallas, TX) (30) and GAPDH (37 kDa, 1:5000 dilution, catalog no. 5174, Cell Signaling Technology, Danvers, MA). ECL Plus Western blotting substrate (ThermoFisher, Rockford, IL) was used for detecting immunoreactive signals. The amount of proteins was normalized by GAPDH, which was used as a loading control on the same gel.
ChIP
The ChIP assay was carried out using a Magna ChIP G tissue kit (catalog no. 17-20000, Millipore, Billerica, MA) according to the instructions of the manufacturer. Briefly, fresh DRG tissues (DRGs from two rats were pooled for each sample) were stabilized for 3 min using stabilization buffer. The DRGs were then incubated in 2% formaldehyde for 20 min at 25 °C. After being washed three times with PBS, the DRGs were incubated in lysis buffer for 15 min on ice. Finally, the DRGs were sonicated in the dilution buffer using a sonicator with a microprobe (Qsonica) at 4 °C. Chromatin was pulled down using the following antibodies: IgG (catalog no. ab124055, Abcam, Cambridge, UK), H3 (catalog no. 2650s, Cell Signaling Technology), H3K4me3 (catalog no. 9751s, Cell Signaling Technology) (21), H3K9ac (catalog no. 39917, Active Motif, Carlsbad, CA) (31), H3K9me2 (catalog no. ab1220, Abcam) (21, 32), and H3K27me3 (catalog no. 9773s, Cell Signaling Technology) (33). Real-time PCR was performed on three separate promoter regions of Oprm1. The PCR thermal conditions were as follows: one cycle at 95 °C for 1 min, 40 cycles at 95 °C for 15 s, and once cycle at 60 °C for 15 s. The following primers were used: Oprm1 (−43/+63 bp, around the transcription start site (TSS)), CACTCCTCTCCTCCCTCCTC (forward) and CAGAGGCGCATCTCTAGCTT (reverse); Oprm1 (−411/−302 bp, upstream of the TSS), AACAGGTTTGTGGGGTTGAA (forward) and AACAGGTTTGTGGGGTTGAA (reverse); and Oprm1 (+376/+495 bp, downstream of the TSS), CAACTTGTCCCACGTTGATG (forward) and TAATGGCTGTGACCATGGAA (reverse).
Spinal Cord Slice Preparation and Electrophysiological Recordings
We removed the lumbar spinal cord from rats or mice via laminectomy during isoflurane-induced anesthesia. The spinal cords at the L5 and L6 levels were placed in ice-cold sucrose artificial cerebrospinal fluid containing 234.0 mm sucrose, 3.6 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 1.2 mm NaH2PO4, 12.0 mm glucose, and 25.0 mm NaHCO3 presaturated with 95% O2 and 5% CO2. The spinal cord tissue was then placed in a shallow groove formed in an agar block and glued on the stage of a vibratome. Transverse slices (400 μm thick) of the spinal cords were cut in ice-cold sucrose artificial cerebrospinal and preincubated in Krebs solution (117.0 mm NaCl, 3.6 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 1.2 mm NaH2PO4, 11.0 mm glucose, and 25.0 mm NaHCO3) oxygenated with 95% O2 and 5% CO2 at 34 °C for at least 1 h before being transferred to the recording chamber. The spinal cord slices were placed in a glass-bottomed chamber (Warner Instruments, Hamden, CT) and continuously perfused with Krebs solution at 3.0 ml/min at 34 °C, maintained by an inline solution heater and a temperature controller.
The spinal lamina II, a translucent region in the superficial dorsal horn, was identified on an upright fixed-stage microscope using differential interference contrast/infrared illumination. The lamina II outer neurons were visualized and selected for whole-cell patch clamp recordings because they predominantly receive nociceptive input (34–36). We filled a glass pipette (5–10 MΩ) with an internal solution containing 135.0 mm potassium gluconate, 5.0 mm tetraethylammonium chloride, 2.0 mm MgCl2, 0.5 mm CaCl2, 5.0 mm HEPES, 5.0 mm EGTA, 5.0 mm ATP-Mg, 0.5 mm Na-GTP, 1.0 mm guanosine 5′-O-(2-thiodiphosphate), and 10.0 mm lidocaine N-ethyl bromide (adjusted to pH 7.2–7.4 with 1 m KOH, 290–300 mosmol). Guanosine 5′-O-(2-thiodiphosphate) was included in the pipette recording solution to block the postsynaptic effect of the opioids (10, 28). The input resistance was monitored, and the recording was abandoned if it changed more than 15%. Signals were recorded using an amplifier (MultiClamp700B, Axon Instruments Inc., Union City, CA), filtered at 1–2 kHz, digitized at 10 kHz, and stored for offline analysis. We used electrical stimulation (0.2–0.4 ms, 0.6 mA, and 0.1 Hz) of the dorsal root to evoke excitatory postsynaptic currents (EPSCs). EPSCs were recorded at a holding potential of −60 mV, and monosynaptic EPSCs were identified on the basis of the constant latency and the absence of conduction failure of evoked EPSCs in response to a 20-Hz electrical stimulation, as we described previously (10, 37). In the electrophysiological experiments, at least three animals were used for each group.
Statistical Analysis
All data were expressed as the mean ± S.E. Analyses of the opioid effect on the amplitude of evoked EPSCs were performed using Clampfit (Axon Instruments). Statistical analysis was performed using Prism software (GraphPad Software Inc., La Jolla, CA). We used Student's t test to compare two groups and one-way or two-way analysis of variance followed by Dunnett's or Tukey's post hoc test to compare more than two groups. We used nonparametric analyses (i.e. Mann-Whitney or Kruskal-Wallis test) when data were not normally distributed. The level of significance was set at p < 0.05.
Results
Nerve Injury Diminishes MOR Expression in the DRG
SNL is a commonly used animal model of neuropathic pain (22, 24). Using quantitative PCR, we determined the mRNA level of Oprm1 in the L5 and L6 DRGs at 5, 10, and 21 days after SNL. SNL, but not the sham procedure, caused a large reduction in the mRNA level of Oprm1 in DRGs at all three time points after surgery (Fig. 1A). Western immunoblotting showed that SNL also profoundly reduced the MOR protein level in the DRGs 21 days after surgery (Fig. 1B). However, SNL did not significantly change the Oprm1 mRNA level or the MOR protein level in the dorsal spinal cords (Fig. 1, C and D).
FIGURE 1.

Nerve injury causes a sustained reduction in MOR expression in the DRG. A, the mRNA levels of Oprm1 in the ipsilateral L5 and L6 DRG at 5, 10, and 21 days after sham or SNL surgery (n = 8 rats/group). B, original gel images and mean data (n = 6 rats/group) show the MOR protein level (∼55 kDa) in the DRG 21 days after sham or SNL surgery (n = 6 rats/group). C, the mRNA level of Oprm1 in the dorsal spinal cord at L5 and L6 levels 21 days after sham (n = 7 rats) or SNL surgery (n = 8 rats). D, original gel images and mean data show the MOR protein level in the dorsal spinal cord 21 days after sham or SNL surgery (n = 6 rats/group). The Oprm1 mRNA level was normalized to GAPDH. For Western immunoblotting, GAPDH (37 kDa) was used as a loading control. The mean value of MOR levels in sham control rats was considered to be 1. Data are shown as means ± S.E. **, p < 0.01 compared with the sham group. Two-way ANOVA (A) or Student's t test (B) was used.
Nerve Injury Consistently Increases H3K9me2 Levels in the Oprm1 Promoter in the DRG
Histone modifications associated with transcriptionally inactive chromatin include H3K9ac, H3K9me2, and H3K27me3, which are catalyzed by histone deacetylases, G9a and GLP, and enhancer of zeste homolog 2 (EZH2), respectively (13, 14, 38). To determine which histone modification is involved in the silencing of Oprm1 expression after nerve injury, we performed ChIP followed by quantitative PCR (ChIP-PCR) to determine how SNL alters different histone marks at the promoter regions of Oprm1 in the DRG. We selected two activating histone marks, H3K4me3 and H3K9ac (39–41), and two repressive histone marks, H3K9me2 and H3K27me3 (42, 43), because these four marks are predictive of gene expression in the DRG (21, 25). SNL caused a large increase in the occupancy of H3K9me2 in all three promoter regions of Oprm1 in the DRG (Fig. 2). In contrast, SNL significantly altered the enrichment of H3K9ac and H3K27me2 around the TSS, but not in the two other promoter regions, of Oprm1 in the DRG (Fig. 2). Also, SNL did not consistently affect the occupancy of H3K4me3 in the three promoter regions of Oprm1 in the DRG (Fig. 2). These data suggest that nerve injury-induced Oprm1 silencing in the DRG is predominantly associated with increased enrichment of H3K9me2.
FIGURE 2.

Nerve injury consistently increases H3K9me2 levels at the Oprm1 promoter in the DRG. A–C, ChIP-PCR quantification data shows the enrichment of histones H3K4me3, H3K9ac, H3K9me2, and H3K27me3 at the three different promoter regions of Oprm1: −43/+63 bp (around the TSS, A), −411/−302 bp (upstream of the TSS, B), and +376/+495 bp (downstream of the TSS, C). The ipsilateral L5 and L6 DRGs were obtained from sham and SNL rats 21 days after surgery (n = 7 independent experiments with 14 rats/group). Data were normalized to the total H3 values and presented as means ± S.E. *, p < 0.05; **, p < 0.01 compared with the sham group (Mann-Whitney test).
Inhibition of G9a Restores MOR Expression and Rescues the Morphine Analgesic Effect in Neuropathic Pain
We next examined whether inhibition of G9a activity could reverse diminished MOR expression in the injured DRG. Because intrathecal injections bring agents in direct contact with DRG neurons (21, 25), we treated the animals with daily intrathecal injections of 10 μg of UNC0638, a highly specific G9a/GLP inhibitor (44), or vehicle for 7 days. The efficacy and specificity of intrathecal UNC0638 had been confirmed by quantifying H3K9me2 and other histone marks in our recent in vivo study (21). As expected, treatment with UNC0638 normalized the mRNA and MOR protein levels in the injured DRGs (Fig. 3, A and B) and significantly attenuated pain hypersensitivity in SNL rats before morphine treatment (Fig. 3, C and D).
FIGURE 3.

G9a inhibition rescues MOR expression in the DRG and the morphine analgesic effect diminished by nerve injury. A and B, the mRNA and protein levels of MORs in the DRG from sham or SNL rats treated with UNC0638 (UNC) or vehicle (Veh) (n = 7 rats/group). UNC0638 treatment began 21 days after nerve ligation. The ipsilateral L5 and L6 DRG tissues were obtained 24 h after the last UNC0638 injection. The amount of MOR mRNA and protein was normalized to GAPDH in the same samples, and the mean value of MOR levels in sham control rats was considered to be 1. C, time course of the effect of intrathecal morphine on the tactile and pressure withdrawal thresholds in SNL rats treated with UNC0638 or vehicle (n = 10 rats/group). D, time course of the effects of intraperitoneal morphine on the tactile and pressure withdrawal thresholds in SNL rats treated with UNC0638 or vehicle (n = 9 rats/group). The withdrawal thresholds 21 days after SNL were plotted as the baseline control (BL). Data are shown as means ± S.E. *, p < 0.05; **, p < 0.01 compared with the sham group or respective baseline controls (time 0). #, p < 0.01 compared with the respective baseline. Student's t test (A and B) or one-way ANOVA (C and D) was used.
To directly determine whether increased G9a activity is involved in the diminished morphine analgesic effect in neuropathic pain, we measured the effect of two different doses of morphine administered intrathecally or intraperitoneally on nociception in SNL rats 7 days after treatment with UNC0638 or vehicle. In vehicle-treated SNL rats, neither dose of intrathecal morphine (0.3 and 3.0 μg) had a substantial effect on tactile allodynia, measured with von Frey filaments, or mechanical hyperalgesia, tested with a noxious pressure stimulus. In contrast, the analgesic effects of intrathecal morphine on allodynia and hyperalgesia were significantly potentiated in UNC0638-treated SNL rats (Fig. 3C). Similarly, intraperitoneal injection of morphine at both 2.5 and 5.0 mg/kg produced only small effects on allodynia and hyperalgesia in vehicle-treated SNL rats. By comparison, treatment with UNC0638 largely enhanced the analgesic effect of systemic morphine in SNL rats (Fig. 3D). These results suggest that increased G9a activity has an important function in Oprm1 silencing in injured DRGs and the diminished opioid analgesic effect in neuropathic pain.
siRNA Knockdown of G9a Rescues the MOR Expression Levels and the Morphine Analgesic Effect in Neuropathic Pain
Because our ChIP-PCR data suggested a dominant role for G9a-mediated H3K9me2 in nerve injury-induced Oprm1 silencing, we used an siRNA targeting the G9a-encoding gene Ehmt2 to validate the results obtained with UNC0638. The efficacy and specificity of the G9a siRNA in the rat DRG had been validated in our previous study (21). Intrathecal injection of G9a-specific siRNA, but not control siRNA, for 4 days significantly increased the Oprm1 mRNA and MOR protein levels in the injured DRGs (Fig. 4, A and B) and significantly reduced tactile allodynia and mechanical hyperalgesia before morphine treatment (Fig. 4C) (21). Furthermore, behavioral tests showed that the effects of morphine on tactile allodynia and mechanical hyperalgesia were much greater in G9a-specific siRNA-treated SNL rats than in control siRNA-treated SNL rats (Fig. 4C).
FIGURE 4.

G9a knockdown with siRNA reverses the MOR expression in the DRG and the morphine analgesic effect diminished by nerve injury. A and B, quantitative PCR (A) and Western blotting (B) analyses show the mRNA and protein levels of MORs in the DRGs of sham and SNL rats treated with control (Ctl) or G9a-specific siRNA (n = 10 rats/group). The ipsilateral L5 and L6 DRG tissues were removed 24 h after the last siRNA injection. The amount of MOR mRNA and protein was normalized to GAPDH in the same samples, and the mean value of MOR levels in sham control rats was considered to be 1. C, time course of the intrathecal morphine effects on the tactile and pressure withdrawal thresholds in sham and SNL rats treated with G9a-specific siRNA or negative control siRNA (n = 9 rats/group). The withdrawal thresholds after the last siRNA injection were plotted as the baseline control (BL). Data are shown as means ± S.E. *, p < 0.05; **, p < 0.01 compared with the sham group or respective baseline control (time 0). #, p < 0.01 compared with the respective baseline (one-way ANOVA).
Nerve Injury Has No Effect on MOR Expression and Opioid Analgesia in G9a Conditional KO Mice
To directly demonstrate the role of G9a in DRG neurons in the diminished MOR expression and opioid analgesia caused by nerve injury, we selectively deleted Ehmt2 in DRG neurons by crossing Ehmt2flox/flox mice with the primary sensory neuron-specific Cre mouse line AdvillinCre/+ (21, 26), producing G9a conditional KO mice. In wild-type control mice, SNI significantly decreased the tactile and pressure withdrawal thresholds and reduced the mRNA and protein levels of MORs in the DRG (Fig. 5). In contrast, in the G9a conditional KO mice, SNI did not produce chronic pain hypersensitivity (21) and failed to reduce the mRNA and protein levels of MORs in the DRG (Fig. 5). Also, the analgesic effect of systemic morphine at both 2.5 and 5.0 mg/kg was markedly greater in G9a conditional KO mice than in littermate control mice 2 weeks after SNI surgery (Fig. 5). These results clearly indicate that G9a in DRG neurons is essential for the nerve injury-induced reduction in MOR expression and the opioid analgesic effect.
FIGURE 5.

Selective deletion of Ehmt2 in DRG neurons prevents nerve injury-induced MOR down-regulation and the diminished morphine analgesic effect. A and B, quantitative PCR and Western blotting analyses show the effect of nerve injury on the mRNA and protein levels of MORs in the DRG of G9a conditional KO and WT control mice (n = 8 mice/group). The amount of MOR mRNA and protein was normalized to GAPDH in the same samples, and the mean value of MOR levels in sham control rats was considered to be 1. The ipsilateral L5 and L6 DRG tissues were removed 2 weeks after surgery. C and D, time course of the effects of intraperitoneal morphine (2.5 and 5 mg/kg) on the tactile and pressure withdrawal threshold of SNI mice (n = 9 mice/group). Data are shown as means ± S.E. *, p < 0.05; **, p < 0.01 compared with the sham group or respective baseline control at 0 min. #, p < 0.01 compared with the wild-type sham group at baseline (time 0). Student's t test (A and B) or one-way ANOVA (C and D) was used.
Inhibition of G9a Restores the Inhibitory Effect of DAMGO on Glutamate Release from Primary Afferent Nerves
Activation of MORs has a profound inhibitory effect on glutamatergic synaptic transmission between primary afferent nerves and spinal dorsal horn neurons (9, 10). Thus, we conducted electrophysiological experiments to determine whether G9a inhibition could restore the effect of the MOR agonist on synaptic glutamate release from primary afferent nerves in SNL rats. DAMGO, a highly specific MOR agonist (10, 45), was used for the spinal cord slice recordings. To measure glutamate release from presynaptic terminals of primary afferent nerves, we recorded EPSCs of spinal dorsal horn neurons evoked monosynaptically by electrical stimulation of the attached dorsal root (10, 37). Bath application of DAMGO at 0.2–2 μm reduced the amplitude of evoked EPSCs of dorsal horn neurons from sham-operated rats in a concentration-dependent manner (Fig. 6). The inhibitory effect of DAMGO on evoked EPSCs was profoundly reduced in SNL rats compared with sham control rats. Treatment with the G9a/GLP inhibitor UNC0638 fully restored the inhibitory effect of DAMGO on EPSCs in SNL rats to the level observed in the sham control group (Fig. 6). These data further validate the finding that increased G9a activity is significant in the diminished MOR expression and function in primary sensory neurons in neuropathic pain.
FIGURE 6.
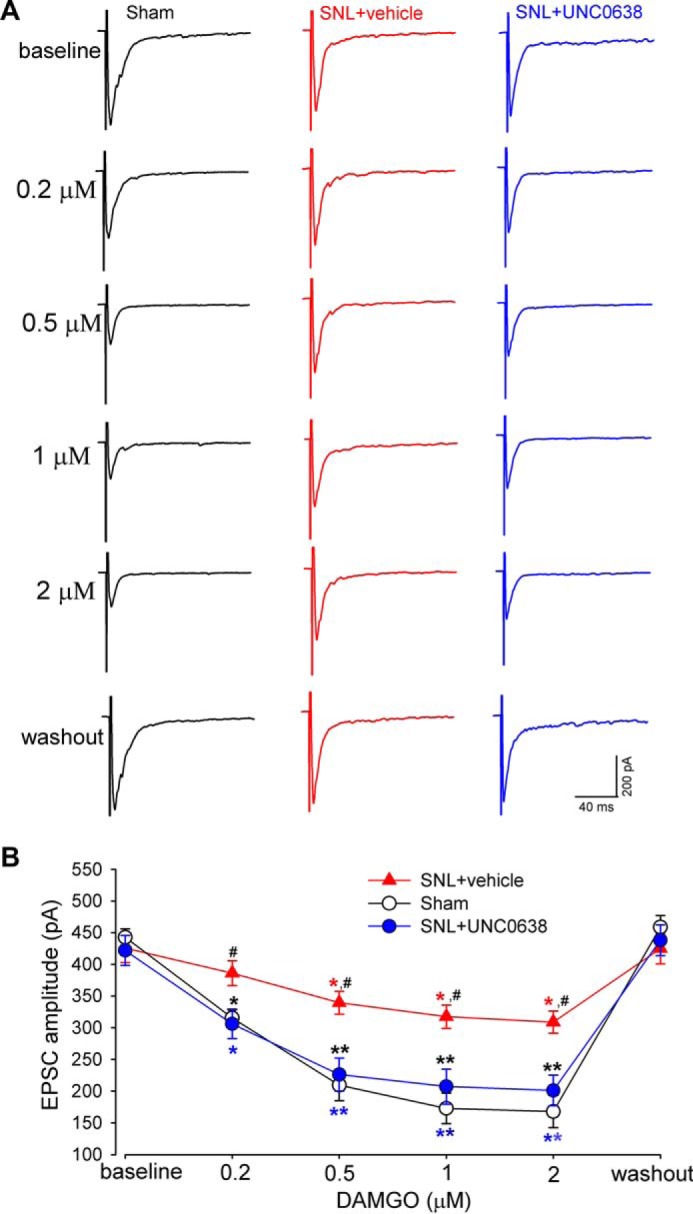
G9a inhibition normalizes the inhibitory effect of DAMGO on glutamate release from presynaptic terminals of primary afferent nerves in SNL rats. A and B, original recording traces (A) and mean data (B) show the effect of 0.2–2 μm DAMGO on the amplitude of monosynaptic EPSCs of dorsal horn neurons evoked from stimulation of the dorsal root in sham control rats (n = 14 neurons) and SNL rats treated with UNC0638 (n = 14 neurons) or vehicle (n = 13 neurons). Data are shown as means ± S.E. *, p < 0.05; **, p < 0.01 compared with respective baseline values. #, p < 0.05 compared with the corresponding value in sham control rats (two-way ANOVA).
Ablation of G9a in DRG Neurons Preserves the Opioid Effect on Synaptic Glutamate Release from Primary Afferents after Nerve Injury
We next used G9a conditional KO mice to determine the role of G9a in DRG neurons in nerve injury-induced reduction in the opioid effect on glutamate release from primary afferent nerves. In wild-type control mice, nerve injury caused a large reduction in the inhibitory effect of 0.2–2 μm DAMGO on the amplitude of monosynaptically evoked EPSCs (Fig. 7). However, in G9a conditional KO mice, SNI failed to reduce the inhibitory effect of DAMGO on the amplitude of monosynaptic EPSCs of spinal dorsal horn neurons (Fig. 7). These results indicate that G9a in DRG neurons plays a key role in nerve injury-induced reduction in the opioid inhibition of primary afferent input to spinal dorsal horn neurons.
FIGURE 7.

Ablation of G9a in DRG neurons reverses the nerve injury-induced reduction in the inhibitory effect of DAMGO on glutamate release from primary afferent terminals. A and B, representative recording traces (A) and mean data (B) show the inhibitory effect of 0.2–2 μm DAMGO on the amplitude of EPSCs of dorsal horn neurons evoked monosynaptically from dorsal root stimulation in WT mice subjected to SNI (n = 15 neurons) or sham procedure (n = 15 neurons) and G9a conditional KO mice subjected to SNI (n = 14 neurons) or sham procedure (n = 12 neurons) 2 weeks after surgery. Data are shown as means ± S.E. *, p < 0.05; **, p < 0.01 compared with the respective baseline values. #, p < 0.05 compared with the corresponding value in WT sham mice (two-way ANOVA).
Discussion
A major finding of our study is that Oprm1 expression diminished by nerve injury was associated with increased levels of H3K9me2 in the Oprm1 promoter region in the DRG. We found that nerve injury caused a large and sustained reduction in MOR expression levels in the DRG. The opioid effect on synaptic glutamate release from primary afferent terminals was also significantly attenuated by nerve injury. Thus, when the level of MORs in DRG neurons and their central termini is reduced by nerve injury, activation of MORs cannot adequately reduce the excitatory input from nociceptors, thereby leading to the diminished opioid analgesic effect on neuropathic pain. G9a-mediated H3K9me2 is generally associated with transcriptional repression and gene silencing (16). We found that the enrichment of H3K9me2 occurred not only around the TSS but also at ∼500 bp upstream and ∼500 bp downstream of the TSS. This finding suggests that increased histone methylation by G9a is widely distributed in the promoter region of Oprm1 in the injured DRG. In contrast, nerve injury-induced changes of other histone marks (e.g. H3K4me3, H3K9ac, and H3K27me3) occurred predominantly at the Oprm1 TSS in the DRG. Thus, we conclude that G9a-controlled H3K9me2 has a dominant function in the silencing of Oprm1 by nerve injury in the DRG. These data support the general notion that a closed chromatin structure mediated by G9a contributes to nerve injury-induced transcriptional repression of Oprm1 in the DRG.
By using complementary approaches, our study provides unequivocal evidence linking G9a-mediated Oprm1 gene silencing in the DRG to the diminished MOR expression and reduced opioid analgesic effect found in neuropathic pain. We found that treatment with either a G9a/GLP inhibitor or a G9a-specific siRNA fully reversed the decrease in MOR expression level in the injured DRG and in the opioid analgesic effect diminished by nerve injury. Also, nerve injury failed to reduce MOR expression and the opioid analgesic effect in G9a conditional KO mice in which Ehmt2 had been specifically deleted in DRG neurons (21). Furthermore, the inhibitory effect of DAMGO on glutamate release from presynaptic terminals of primary afferent nerves in the spinal dorsal horn was normalized both by treatment with a G9a/GLP inhibitor and in G9a conditional KO mice. Therefore, our study identified an essential role for G9a in nerve injury-induced silencing of MOR expression in DRG neurons and in the diminished opioid analgesic effect in neuropathic pain.
G9a is a crucial chromatin regulator that could link histone modifications, DNA methylation, and transcriptional repressors in the epigenetic silencing of MORs in the DRG after nerve injury. G9a may interact with DNA methyltransferases to coordinate DNA and histone methylation (46). DNA methylation may also contribute to the diminished MOR expression caused by nerve injury in the mouse DRG (47). Transcription factors, such as repressor element 1-silencing transcription factor may bind to the Oprm1 promoter to inhibit Oprm1 expression through histone deacetylases. Both histone deacetylase inhibitors and repressor element 1-silencing transcription factor knockdown can increase MOR expression levels and the morphine analgesic effect reduced by nerve injury (12, 48), supporting this possibility. Because G9a is essential for silencing of repressor element 1-silencing transcription factor-regulated genes (49) and can regulate polycomb repressive complex 2 on histone H3K9 to mediate gene silencing (38), it is possible that G9a forms a repressive complex with repressor element 1-silencing transcription factor, histone deacetylases, DNA methyltransferases, and EZH2 at the Oprm1 promoter in the injured DRG. Although our study indicates that G9a has an essential function in nerve injury-induced Oprm1 silencing in the DRG, further studies are warranted to define its complex interactions with other epigenetic components in coordinating gene expression in neuropathic pain.
In summary, our findings provide new insights into the epigenetic mechanism regulating MOR expression in primary sensory neurons in neuropathic pain. Our multidisciplinary approach provides conclusive evidence for G9a as a key chromatin regulator responsible for MOR down-regulation in the DRG and the analgesic efficacy of opioids reduced by nerve injury. A better understanding of the epigenetic mechanisms underlying nerve injury-induced down-regulation of MORs in primary sensory neurons could help improve the analgesic efficacy of opioids for treating chronic neuropathic pain. G9a inhibitors could be used to enhance the opioid analgesic effect and reduce opioid consumption in patients with chronic neuropathic pain.
Author Contributions
Y. Z., S. R. C., and H. L. P. participated in the study design. Y. Z., S. R. C., G. L., and H. C. conducted the experiments. Y. Z., S. R. C., G. L., and H. L. P. performed the data analysis. H. L. P. wrote the manuscript with input from the other authors.
Acknowledgments
We thank Dr. Z. Pan (MD Anderson Cancer Center) and Dr. F. Wang (Duke University) for the Ehmt2flox/flox and AdvillinCre mice, respectively.
This work was supported by grants from the National Institutes of Health (DE022015 and NS073935) and by the N. G. and Helen T. Hawkins Endowment (to H. L. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MOR
- μ-opioid receptor
- DRG
- dorsal root ganglion
- H3K9
- histone 3 at lysine 9
- H3K9me2
- histone 3 at lysine 9 dimethylation
- H3K4me3
- histone 3 at lysine 4 trimethylation
- H3K9ac
- histone 3 at lysine 9 acetylation
- H3K27me3
- histone 3 at lysine 27 trimethylation
- GLP
- G9a-like protein
- SNL
- spinal nerve ligation
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly-ol]-enkephalin
- SNI
- spared nerve injury
- TSS
- transcription start site
- EPSC
- excitatory postsynaptic current
- ANOVA
- analysis of variance.
References
- 1. Matthes H. W., Maldonado R., Simonin F., Valverde O., Slowe S., Kitchen I., Befort K., Dierich A., Le Meur M., Dollé P., Tzavara E., Hanoune J., Roques B. P., and Kieffer B. L. (1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature 383, 819–823 [DOI] [PubMed] [Google Scholar]
- 2. Sora I., Takahashi N., Funada M., Ujike H., Revay R. S., Donovan D. M., Miner L. L., and Uhl G. R. (1997) Opiate receptor knockout mice define μ receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. U.S.A. 94, 1544–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arnér S., and Meyerson B. A. (1988) Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain 33, 11–23 [DOI] [PubMed] [Google Scholar]
- 4. Kupers R. C., Konings H., Adriaensen H., and Gybels J. M. (1991) Morphine differentially affects the sensory and affective pain ratings in neurogenic and idiopathic forms of pain. Pain 47, 5–12 [DOI] [PubMed] [Google Scholar]
- 5. Ossipov M. H., Lopez Y., Nichols M. L., Bian D., and Porreca F. (1995) The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci. Lett. 199, 87–90 [DOI] [PubMed] [Google Scholar]
- 6. Rashid M. H., Inoue M., Toda K., and Ueda H. (2004) Loss of peripheral morphine analgesia contributes to the reduced effectiveness of systemic morphine in neuropathic pain. J. Pharmacol. Exp. Ther. 309, 380–387 [DOI] [PubMed] [Google Scholar]
- 7. Chen S. R., and Pan H. L. (2002) Hypersensitivity of spinothalamic tract neurons associated with diabetic neuropathic pain in rats. J. Neurophysiol. 87, 2726–2733 [DOI] [PubMed] [Google Scholar]
- 8. Chen S. R., and Pan H. L. (2006) Blocking μ opioid receptors in the spinal cord prevents the analgesic action by subsequent systemic opioids. Brain Res. 1081, 119–125 [DOI] [PubMed] [Google Scholar]
- 9. Kohno T., Kumamoto E., Higashi H., Shimoji K., and Yoshimura M. (1999) Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J. Physiol. 518, 803–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou H. Y., Chen S. R., Chen H., and Pan H. L. (2010) Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J. Neurosci. 30, 4460–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang X., Bao L., Shi T. J., Ju G., Elde R., and Hokfelt T. (1998) Down-regulation of μ-opioid receptors in rat and monkey dorsal root ganglion neurons and spinal cord after peripheral axotomy. Neuroscience 82, 223–240 [DOI] [PubMed] [Google Scholar]
- 12. Uchida H., Ma L., and Ueda H. (2010) Epigenetic gene silencing underlies C-fiber dysfunctions in neuropathic pain. J. Neurosci. 30, 4806–4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 14. Riccio A. (2010) Dynamic epigenetic regulation in neurons: enzymes, stimuli and signaling pathways. Nat. Neurosci. 13, 1330–1337 [DOI] [PubMed] [Google Scholar]
- 15. Ronan J. L., Wu W., and Crabtree G. R. (2013) From neural development to cognition: unexpected roles for chromatin. Nat. Rev. Genet. 14, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., and Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 17. Peters A. H., Kubicek S., Mechtler K., O'Sullivan R. J., Derijck A. A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., Martens J. H., and Jenuwein T. (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12, 1577–1589 [DOI] [PubMed] [Google Scholar]
- 18. Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H., and Shinkai Y. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tachibana M., Sugimoto K., Fukushima T., and Shinkai Y. (2001) Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 276, 25309–25317 [DOI] [PubMed] [Google Scholar]
- 20. Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., and Shinkai Y. (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laumet G., Garriga J., Chen S. R., Zhang Y., Li D. P., Smith T. M., Dong Y., Jelinek J., Cesaroni M., Issa J. P., and Pan H. L. (2015) G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nat. Neurosci. 18, 1746–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., and Yaksh T. L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 [DOI] [PubMed] [Google Scholar]
- 23. Chen S. R., Cai Y. Q., and Pan H. L. (2009) Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J. Neurochem. 110, 352–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen S. R., Zhou H. Y., Byun H. S., Chen H., and Pan H. L. (2014) Casein kinase II regulates N-methyl-d-aspartate receptor activity in spinal cords and pain hypersensitivity induced by nerve injury. J. Pharmacol. Exp. Ther. 350, 301–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y., Laumet G., Chen S. R., Hittelman W. N., and Pan H. L. (2015) Pannexin-1 up-regulation in the dorsal root ganglion contributes to neuropathic pain development. J. Biol. Chem. 290, 14647–14655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasegawa H., Abbott S., Han B. X., Qi Y., and Wang F. (2007) Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. J. Neurosci. 27, 14404–14414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Laedermann C. J., Pertin M., Suter M. R., and Decosterd I. (2014) Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y. L., Chen S. R., Chen H., and Pan H. L. (2012) Chronic opioid potentiates presynaptic but impairs postsynaptic N-methyl-d-aspartic acid receptor activity in spinal cords: implications for opioid hyperalgesia and tolerance. J. Biol. Chem. 287, 25073–25085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen S. R., and Pan H. L. (2006) Loss of TRPV1-expressing sensory neurons reduces spinal μ opioid receptors but paradoxically potentiates opioid analgesia. J. Neurophysiol. 95, 3086–3096 [DOI] [PubMed] [Google Scholar]
- 30. Sanna M. D., Ghelardini C., and Galeotti N. (2015) Activation of JNK pathway in spinal astrocytes contributes to acute ultra-low-dose morphine thermal hyperalgesia. Pain 156, 1265–1275 [DOI] [PubMed] [Google Scholar]
- 31. Ringel A. E., Cieniewicz A. M., Taverna S. D., and Wolberger C. (2015) Nucleosome competition reveals processive acetylation by the SAGA HAT module. Proc. Natl. Acad. Sci. U.S.A. 112, E5461–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jose C. C., Xu B., Jagannathan L., Trac C., Mallela R. K., Hattori T., Lai D., Koide S., Schones D. E., and Cuddapah S. (2014) Epigenetic dysregulation by nickel through repressive chromatin domain disruption. Proc. Natl. Acad. Sci. U.S.A. 111, 14631–14636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chan K. M., Fang D., Gan H., Hashizume R., Yu C., Schroeder M., Gupta N., Mueller S., James C. D., Jenkins R., Sarkaria J., and Zhang Z. (2013) The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27, 985–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pan H. L., Khan G. M., Alloway K. D., and Chen S. R. (2003) Resiniferatoxin induces paradoxical changes in thermal and mechanical sensitivities in rats: mechanism of action. J. Neurosci. 23, 2911–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pan Y. Z., and Pan H. L. (2004) Primary afferent stimulation differentially potentiates excitatory and inhibitory inputs to spinal lamina II outer and inner neurons. J. Neurophysiol. 91, 2413–2421 [DOI] [PubMed] [Google Scholar]
- 36. Santos S. F., Rebelo S., Derkach V. A., and Safronov B. V. (2007) Excitatory interneurons dominate sensory processing in the spinal substantia gelatinosa of rat. J. Physiol. 581, 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li D. P., Chen S. R., Pan Y. Z., Levey A. I., and Pan H. L. (2002) Role of presynaptic muscarinic and GABA(B) receptors in spinal glutamate release and cholinergic analgesia in rats. J. Physiol. 543, 807–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mozzetta C., Pontis J., Fritsch L., Robin P., Portoso M., Proux C., Margueron R., and Ait-Si-Ali S. (2014) The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 53, 277–289 [DOI] [PubMed] [Google Scholar]
- 39. Jenuwein T., and Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 40. Ng H. H., Robert F., Young R. A., and Struhl K. (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11, 709–719 [DOI] [PubMed] [Google Scholar]
- 41. Santos-Rosa H., Schneider R., Bannister A. J., Sherriff J., Bernstein B. E., Emre N. C., Schreiber S. L., Mellor J., and Kouzarides T. (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 [DOI] [PubMed] [Google Scholar]
- 42. Nakayama J., Rice J. C., Strahl B. D., Allis C. D., and Grewal S. I. (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113 [DOI] [PubMed] [Google Scholar]
- 43. Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., and Cavalli G. (2007) Genome regulation by polycomb and trithorax proteins. Cell 128, 735–745 [DOI] [PubMed] [Google Scholar]
- 44. Vedadi M., Barsyte-Lovejoy D., Liu F., Rival-Gervier S., Allali-Hassani A., Labrie V., Wigle T. J., Dimaggio P. A., Wasney G. A., Siarheyeva A., Dong A., Tempel W., Wang S. C., Chen X., Chau I., Mangano T. J., Huang X. P., Simpson C. D., Pattenden S. G., Norris J. L., Kireev D. B., Tripathy A., Edwards A., Roth B. L., Janzen W. P., Garcia B. A., Petronis A., Ellis J., Brown P. J., Frye S. V., Arrowsmith C. H., and Jin J. (2011) A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu Z. Z., Chen S. R., and Pan H. L. (2004) Differential sensitivity of N- and P/Q-type Ca2+ channel currents to a mu opioid in isolectin B4-positive and -negative dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 311, 939–947 [DOI] [PubMed] [Google Scholar]
- 46. Estève P. O., Chin H. G., Smallwood A., Feehery G. R., Gangisetty O., Karpf A. R., Carey M. F., and Pradhan S. (2006) Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 20, 3089–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou X. L., Yu L. N., Wang Y., Tang L. H., Peng Y. N., Cao J. L., and Yan M. (2014) Increased methylation of the MOR gene proximal promoter in primary sensory neurons plays a crucial role in the decreased analgesic effect of opioids in neuropathic pain. Mol. Pain 10, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uchida H., Matsushita Y., Araki K., Mukae T., and Ueda H. (2015) Histone deacetylase inhibitors relieve morphine resistance in neuropathic pain after peripheral nerve injury. J. Pharmacol. Sci. 128, 208–211 [DOI] [PubMed] [Google Scholar]
- 49. Roopra A., Qazi R., Schoenike B., Daley T. J., and Morrison J. F. (2004) Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell 14, 727–738 [DOI] [PubMed] [Google Scholar]