

Abstract
Mutations in TSC1 or TSC2 cause tuberous sclerosis complex (TSC), an autosomal dominant disorder characterized by the occurrence of benign tumors in various vital organs and tissues. TSC1 and TSC2, the TSC1 and TSC2 gene products, form the TSC protein complex that senses specific cellular growth conditions to control mTORC1 signaling. TBC1D7 is the third subunit of the TSC complex, and helps to stabilize the TSC1-TSC2 complex through its direct interaction with TSC1. Homozygous inactivation of TBC1D7 causes intellectual disability and megaencephaly. Here we report the crystal structure of a TSC1-TBC1D7 complex and biochemical characterization of the TSC1-TBC1D7 interaction. TBC1D7 interacts with the C-terminal region of the predicted coiled-coil domain of TSC1. The TSC1-TBC1D7 interface is largely hydrophobic, involving the α4 helix of TBC1D7. Each TBC1D7 molecule interacts simultaneously with two parallel TSC1 helices from two TSC1 molecules, suggesting that TBC1D7 may stabilize the TSC complex by tethering the C-terminal ends of two TSC1 coiled-coils.
Keywords: crystal structure, isothermal titration calorimetry (ITC), mTOR complex (mTORC), protein complex, tuberous sclerosis complex (TSC), TBC1D7, TSC1
Introduction
Tuberous sclerosis complex (TSC)4 is a multisystem genetic disease that is characterized by the development of hamartomas or benign tumors in various organs, including skin, brain, and kidneys, and is frequently associated with epilepsy, intellectual disability, and autism (1, 2). TSC is caused by mutations to either of two genes, TSC1 and TSC2, which encode for proteins hamartin (TSC1) and tuberin (TSC2), respectively. TSC1 and TSC2 form a protein complex that acts as a GTPase-activating protein (GAP) for the small G-protein Rheb, which in turn serves as a crucial activator of the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1). The TSC complex is a tumor suppressor, inhibiting the Rheb-mTORC1-dependent stimulation of anabolic metabolism and cell growth (2–11). TSC2 contains a GAP domain and is the catalytic subunit of the complex. TSC1 is believed to enhance TSC2 function by activating TSC2 GAP activity, stabilizing TSC2, and/or maintaining the correct intracellular localization of the TSC complex (12–19). Recent work indicates that TSC1 might also have TSC2-independent activities in the cell (20–22).
Tre2-Bub2-Cdc16 domain family member 7 (TBC1D7) is a TSC1-binding protein (23–25), and has been recently identified as the third subunit of the TSC complex (25). TSC1, TSC2, and TBC1D7 form the core of the TSC protein complex in the cell, which migrates in size exclusion chromatography with a peak mass of ∼2 MDa (26). Consistent with its important role in the TSC protein complex, knockdown of TBC1D7 results in decreased association of TSC1 and TSC2, leading to decreased Rheb-GAP activity, increased mTORC1 signaling, delayed induction of autophagy, and enhanced cell growth under poor growth conditions (25). Intriguingly, overexpression of TBC1D7 was also reported to increase mTORC1 activity (23). TBC1D7 mutations have not been found in TSC patients, but homozygous loss of TBC1D7 causes intellectual disability and megalencephaly, a developmental disorder associated with mTORC1 activation (27). Genome-wide brain tissue expression analysis also identified TBC1D7 as a susceptibility locus for migraine (28). More recently, loss-of-function TBC1D7 mutations were associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease (29). Interestingly, elevated expression of TBC1D7 is associated with lung cancer. In this situation, TSC1 may stabilize TBC1D7 and thus promote tumorigenesis (24).
TBC1D7 is a 293-residue member of the TBC family of proteins. TBC domains usually function as GAPs for members of the Rab family of small G-proteins using a dual-finger mechanism (30, 31). Although the TBC1D7 TBC domain (residues 50–231) does not contain the two most critical catalytic (“finger”) residues (25, 31), TBC1D7 was shown to specifically inactivate Rab17 and to be involved in primary cilia formation (32). Thus it remains unclear as to whether TBC1D7 might serve as a GAP.
TSC1 contains a folded N-terminal domain (residues 1–265) that is important for TSC1 folding and stability (33, 34). Mutations affecting this domain account for the majority of pathogenic TSC1 missense mutations. In addition to the N-terminal domain, TSC1 contains a large coiled-coil domain (residues 725–988). The N-terminal region of this domain (residues 681–937), along with a discrete more N-terminal region (residues 302–430), were shown to be important for TSC2 binding (35). The TBC1D7-binding site of TSC1 was mapped to the C-terminal region of the coiled-coil domain (residue 939–977) (35). To understand the molecular mechanism of the TSC1-TBC1D7 interaction and TSC complex function, we have determined the crystal structure of a TSC1-TBC1D7 complex. Our work provides new insight into the formation and structure of the TSC complex and will facilitate future studies aimed at understanding the pathogenetic mechanisms underlying TSC1, TSC2, and TBC1D7 dysregulation in human disease.
Experimental Procedures
Expression, Purification, and Crystallization of the TSC1-TBC1D7 Complex
A bacterial expression plasmid of human TBC1D7 was constructed by cloning the full-length cDNA into a self-made vector named pCool. DNA sequences expressing residues 19–293 of TBC1D7 were inserted into a pGEX-4T-1 vector (Novagen). DNA sequences encoding residues 939–992 of human TSC1 were inserted into the pCool vector.
GST-tagged recombinant proteins were produced in the BL21(DE3) strain of Escherichia coli. Protein expression was induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside when the cell density reached A600 = 0.8, after which point the temperature was shifted to 18 °C for 20 h. GST-tagged TBC1D7 was first purified using glutathione-Sepharose beads (GE Healthcare), followed by anion exchange (MonoQ, GE Healthcare) and gel-filtration column chromatography (Superdex 200 10/300 GL, GE Healthcare). Selenomethionine-substituted TBC1D7(19–293) with a N-terminal GST tag was expressed using the autoinduction method. TBC1D7(19–293)-TSC1(939–992) complex crystals were grown by vapor diffusion in hanging drops at 25 °C in 200 mm NaCl, 20 mm CaCl2, and 15% PEG-3350. Single or multiple point mutations of TBC1D7 and TSC1 were generated by PCR and verified by DNA sequencing. Mutant proteins were expressed and purified following the same protocol as for the wild-type (wt) proteins.
Data Collection and Structure Determination
The 3.1-Å resolution native dataset and the 3.2-Å resolution single wavelength anomalous dispersion dataset were collected using the BL 8.2.1 and BL 8.2.2 beamlines of the Advanced Light Source in Berkeley. The datasets were processed with HKL2000 (36). The crystal belongs to the C2 space group and there are two TSC1-TBC1D7 complexes in one asymmetric unit. The selenium sites were searched by AutoSol in PHENIX (37), and all 16 potential selenium sites were found and used for phasing. Autobuilding was carried out with ARP/wARP in the CCP4 suite (38). The 1.9-Å resolution structure of human TBC1D7 (Protein Data Bank code 3QWL) was used to guide model building. The 3.1-Å resolution native dataset was used for refinement. Model building was performed in COOT (39) and iterative cycles of model building were fulfilled by Refmac5 in CCP4 (38) and PHENIX (37). NCS restraint and TLS were used throughout refinement. The electrostatic potential surfaces shown were generated by the APBS tool in PyMOL (40).
Isothermal Titration Calorimetry (ITC)
ITC was performed on a VP-ITC Microcal colorimeter (Microcal) at 25 °C. TBC1D7 and TSC1 fragments were buffer exchanged into 20 mm Tris HCl (pH 8.0), 150 mm NaCl, 1 mm DTT using size exclusion on a prepacked Superdex 200 column (GE Healthcare), eluting at a final concentration of ∼15–20 μm. Both ligand and protein were degassed before use. TBC1D7 concentrated to 200 μm was injected in 5-μl quantities every 5 min for a total of 25–40 injections into a 1.4218-ml protein chamber with TSC1, GST-TSC1, or GST separately. Data were analyzed using Origin 7.0.
In Vitro GST Pulldown
Purified TSC1(939–992) and TBC1D7(1–293) wt and mutant proteins were used in pulldown experiments. All binding and wash steps were carried out in a buffer containing 20 mm Tris-HCl (pH 8.0), 200 mm NaCl, 2 mm DTT and 0.1% Tween 20. All GST pulldowns were repeated three times and stained with Coomassie Blue. To semiquantify the GST pulldown results, we also performed technical triplicates for all TSC1 and TBC1D7 wt/mutant proteins and stained the SDS-PAGE gels using Oriole Fluorescent Gel Stain (Bio-Rad).
Analytical Ultracentrifugation Assay (AUC)
TSC1(939–992), TBC1D7(1–293), and different molar ratios of TBC1D7 and TSC1 were measured by analytical ultracentrifugation analysis performed on a XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA) with a four-cell An-60 Ti rotor. All proteins were in the same buffer, containing 20 mm Tris-HCl (pH 8.0), 200 mm NaCl, and 2 mm DTT. The absorbance data at 280 nm were collected at 20 °C. Rotor speed was set at 45,000 rpm for all samples. The analytical ultracentrifugation data were analyzed by the SEDFIT (version 14.81) software.
SEC-MALS
Size exclusion chromatography-coupled multi-angle light scattering (SEC-MALS) experiments were performed at room temperature by loading samples on a 24-ml size exclusion column with a TREOS MiniDAWN MALS detector (Wyatt Technology). TBC1D7 and TSC1 were purified on a Superdex 200 column, using a buffer containing 20 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 2 mm DTT. The light scattered by a protein was directly proportional to its weight-average molecular mass and concentration.
Co-immunoprecipitation
TBC1D7 and TSC1 variants were derived by site-directed mutagenesis and verified by sequencing of the complete open reading frame. Human embryonal kidney (HEK) 293T cells were transfected as described previously (35). The cells were lysed 24 h after transfection. Immunoprecipitation, immunoblotting, and signal quantification were performed as described previously (35).
Antibodies were purchased from Cell Signaling Technology (Danvers, MA) (9B11 anti-myc mouse monoclonal, number 2276; anti-HA tag rabbit polyclonal, number 3724), Covance (Princeton, NJ) (mouse anti-HA tag, MMS-101R), or Li-Cor Biosciences (Lincoln, NE) (goat anti-rabbit 680 nm, 926–68073; and goat anti-mouse 800 nm conjugates, 926–68072). Rabbit polyclonal antibodies raised against human TSC2 were described previously (4). Anti-myc (E6654) and anti-HA (E6779) affinity resins were purchased from Sigma.
Results
Biochemical Characterization of the TSC1-TBC1D7 Interaction
To map the TSC1-TBC1D7 interaction regions in each protein, we overexpressed and purified human TBC1D7 and various GST-tagged human TSC1 fragments. Pulldown experiments using the purified proteins showed that TSC1(939–992) was sufficient for interacting with TBC1D7 (Fig. 1a). TBC1D7 and TSC1(939–992) formed a stable complex and co-migrated in SEC. To minimize shape effects, we used SEC-MALS to measure the molecular weight of each protein and the respective complexes (Fig. 1c). Although the N-terminal half of TSC1(939–992) is part of the predicted TSC1 coiled-coil domain, TSC1(939–992) behaved as a monomer in solution. ITC analysis demonstrated that, when concentrated full-length TBC1D7 is titrated into TSC1(939–992), the apparent Kd for the TBC1D7-TSC1(939–992) interaction is 6.9 μm, with an N number ∼0.5, suggesting two TSC1(939–992) molecules interact with one TBC1D7 molecule (Fig. 1b). Because TSC1(939–992) is monomeric in solution, it is possible that the above ITC result is a mixed effect of TSC1(939–992) dimerization and TBC1D7 binding. Thus we performed ITC analysis using GST-TSC1(939–992), which forms a homodimer in the absence of TBC1D7 based on the SEC-MALS result (data not shown). Under similar ITC titration conditions, TBC1D7 has a Kd of 0.4 μm, which is ∼18-fold tighter than untagged TSC1(939–992) (Fig. 1b). These results indicate that preformed TSC1 dimers enhance TBC1D7 binding.
FIGURE 1.

Biochemical characterization of the TSC1-TBC1D7 interaction. a, mapping of the TBC1D7 binding site on TSC1 by GST pulldown assay. Human TSC1(939–992) is sufficient for TBC1D7 binding. The bands showing TBC1D7(19–293) pulled down are marked with red stars. The molecular mass markers above and below the GST-TSC1 and TBC1D7 bands are 45 and 31 kDa, respectively. b, ITC analysis. Homodimerization of TSC1(939–992) promoted by GST fusion enhances its interaction with TBC1D7. The flexible N-terminal tail of TBC1D7 is dispensable for the TSC1(939–992)-TBC1D7 interaction. c, SEC-MALS characterization of TSC1 fragments. The molar ratio of TSC1-TBC1D7 complexes appears to be 2:1. TSC1 fragments instead of TBC1D7 were added in excess for a better separation from the complex peak. All experiments in this figure were performed at least twice. All results were highly reproducible.
TBC1D7 residues 1–18 are not highly conserved and are predicted to be intrinsically disordered by the metaPrDOS server (41). ITC showed that full-length TBC1D7 and N-terminal truncated TBC1D7 (residues 19–293) interacted with TSC1(939–992) with comparable affinities: 0.4 and 0.2 μm, respectively (Fig. 1b). Thus we used TBC1D7(19–293) in our structural analysis. In contrast to monomeric TSC1(939–992), a longer TSC1 fragment, TSC1(871–992), existed predominantly as a homodimer (Fig. 1c). We examined the TSC1-TBC1D7 interaction in the SEC-MALS assay using three different TSC1 fragments: 837–992, 871–992, and 939–992. In each case the TSC1-TBC1D7 complex contained two TSC1 molecules and one TBC1D7 molecule (Fig. 1c).
Overall Structure of the TSC1-TBC1D7 Complex
To reveal how TBC1D7 interacts with TSC1, we determined the crystal structure of the TSC1 TBC1D7-binding domain (residues 939–992) in complex with near full-length TBC1D7(19–293), using the Se-Met single wavelength anomalous dispersion method (Table 1). In each crystallographic asymmetric unit, there were two TBC1D7 molecules and four TSC1(939–992) fragments. The first 39 residues of each TSC1 helix (residues 939–977) formed a single helix, whereas the last 15 residues were not visible in our electron density map. Two TSC1 helices, A and B, from two different TSC1 molecules, formed a parallel coiled-coil and interacted with one TBC1D7 molecule. Thus there were two 2:1 TSC1-TBC1D7 complexes in each asymmetric unit. Both complexes have almost identical structures, with a r.m.s. deviation value of 0.135 Å. From now on, we will focus on the analysis of the 2:1 TSC1(939–992)-TBC1D7 complex (Fig. 2).
TABLE 1.
Statistics of crystallographic analysis
Values in parentheses indicate the corresponding statistics in the highest-resolution shell.
| Dataset | SeMet | Native |
|---|---|---|
| Data collection | ||
| Space group | C2 | C2 |
| Cell dimensions | a = 180.96 Å, b = 66.04 Å, c = 98.67 Å, | a = 183.40 Å, b = 66.70 Å, c = 98.37 Å, |
| α = γ = 90.00°, β = 90.67° | α = γ = 90.00°, β = 91.91° | |
| Wavelength | 0.9792 Å | 1.0000 Å |
| Resolution | 50.00 Å - 3.20 Å (3.29Å – 3.20 Å) | 50.00 Å - 3.10Å (3.19 Å – 3.10 Å) |
| Unique reflections | 19,429 | 21,259 |
| Rsym | 9.1% (91.7%) | 10.8% (75.8%) |
| 〈I〉/〈SIGI〉 | 21.3 (1.40) | 11.7 (1.45) |
| Completeness | 99.7% (99.1%) | 99.7% (98.6%) |
| CC1/2a | (0.694) | (0.726) |
| Redundancy | 3.7 (3.6) | 3.5 (3.1) |
| Refinement | ||
| Rwork/Rfree | 21.8% / 23.8% | |
| Average B factor | 79.28Å2 | |
| R.m.s. deviation bond ideality | 0.0053Å | |
| R.m.s. deviation angle ideality | 0.9778° | |
| Ramachandran favored | 98.0% | |
| Ramachandran allowed | 2.0% | |
| Protein residues | 679 | |
| Water | 0 | |
a CC1/2 is the correlation coefficient between symmetry-related intensities taken from random halves of the data set.
FIGURE 2.
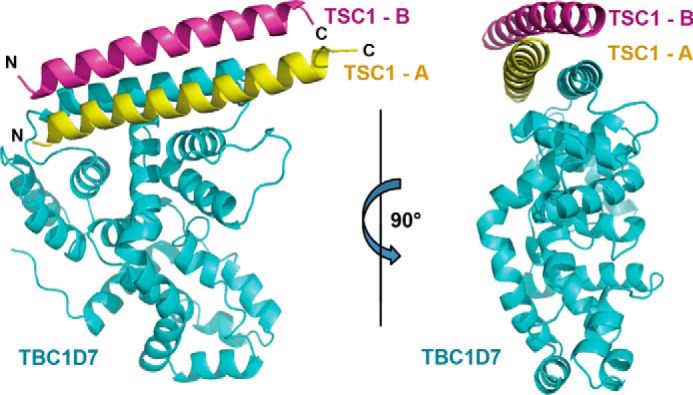
Overall structure of a TSC1-TBC1D7 complex. Two orthogonal schematic views of TSC1-TBC1D7 are shown. TBC1D7 and two TSC1 molecules are colored in cyan, pink, and yellow, respectively.
The TBC1D7 structure in our TSC1-TBC1D7 complex is almost identical to the previously solved unliganded TBC1D7 structure (PDB code 3QWL) (r.m.s. deviation = 0.497 Å). The two TSC1 helices predominantly dock onto the longest helix in the TBC1D7 structure (helix α4; residues 70–95; Fig. 2), which is located on a surface distinct from the Rab small G-protein binding sites on other TBC family members (see below).
The TSC1-TBC1D7 Interface
Both TSC1(939–992) A and B helices interact directly with TBC1D7. Helix A makes the majority of the interactions and lies roughly parallel to the long TBC1D7 α4 helix (residues 74–95). Additional interactions occur between the TSC1 A helix and three neighboring TBC1D7 helices and flanking loops. In contrast, the TSC1 B helix docks on top of the TSC1 A helix and the TBC1D7 α4 helix. The TSC1-TBC1D7 interaction covers a surface area of 2,711 Å2, with the TSC1 A helix accounting for >80% of this interface. Because both TSC1 helices interact differently with TBC1D7, for convenience, we add a prime to denote TSC1 B helix residues (e.g. Ile962′ is residue Ile962 from the B helix).
The two parallel TSC1 helices form a dimer that is largely symmetrical in terms of the main chain structure and the side chains involved in the dimerization. Other side chains are non-symmetrical due to the interaction with TBC1D7 on one side (see below). The interface between the two TSC1 helices is mainly formed by a number of hydrophobic interactions, involving Leu941, Phe958, Ile962, Leu965, and Leu969. Leu941 is located at the N-terminal side of the TSC1 helices, whereas Phe958, Ile962, Leu965, and Leu969 are located near the C-terminal end of the coiled-coil (Fig. 3, a and b). In addition, polar interactions stabilize the coiled-coil interaction. These include the hydrogen bonds between Gln951-Thr955′, Thr955-Gln951′, and Glu961-Tyr966′, and an Glu945-Arg947′ salt bridge.
FIGURE 3.

Details of the TSC1-TBC1D7 interface. a, electrostatic potential of the surface of TBC1D7. The core TSC1-binding surface of TBC1D7 is largely hydrophobic. b, hydrophobic interactions stabilizing the TSC1-TBC1D7 interaction. c, polar interactions on the “front” of the TSC1-TBC1D7 interface. D, polar interactions on the “back” of the TSC1-TBC1D7 interface. For clarity, the front interactions and hydrophobic interactions are not shown. Key interface residues (TSC1: Ile954 and Phe958 in the TSC1-A helix, Ile962′ and Tyr966′ in the TSC1-B helix; TBC1D7: Val94, Val95, Arg81, Gln84, and Arg96) are marked with red ovals.
The core of the TSC1-TBC1D7 interface is mediated by hydrophobic interactions and these are buttressed by surrounding hydrogen bonds and salt bridges (Fig. 3a). The hydrophobic interface is formed by TBC1D7 residues Ala91, Val94, Val95, Leu114, and Leu119; TSC1 A helix residues Ile954, Val957, and Phe958; and TSC1 B helix residues Ile962′ and Leu963′ (Fig. 3b). On one side of the TSC1-TBC1D7 complex, TBC1D7 Arg81 and Gln84 interact with the side chain and main chain carbonyl, respectively, of Ser946 of the TSC1 A helix. In addition, TBC1D7 Arg121 forms two salt bridges with Glu961 and Asp964 of the TSC1 A helix (Fig. 3c). On the opposite side of the TSC1-TBC1D7 complex, TBC1D7 residues Tyr80, Glu83, Asp87, His90, and Arg121 form multiple hydrogen bonds or salt bridges with both TSC1 helices (Fig. 3d). Sequence alignments indicate that the TSC1-TBC1D7 interface residues are conserved among mammals, zebrafish, and Drosophila, but not in nematodes (e.g. Brugia malayi and Loa loa; Fig. 4).
FIGURE 4.

Sequence alignment of TSC1 and TBC1D7 from different organisms. Conserved residues are boxed in red and similar residues are highlighted in red. Residues are framed in blue if more than 70% of its residues are similar according to physicochemical properties. For TBC1D7, helices are marked with squiggles, turns with TT letters, and interface residues with red stars. For the two TSC1 helices interacting with TBC1D7, red stars in the upper row represent TSC1-A residues involved in TBC1D7 interaction, red hashtags in the lower row indicate TSC1-B residues interacting with TBC1D7. For counting convenience, small black dots are added in every 10 residues on top of sequences.
Mutagenesis Analysis of the TSC1-TBC1D7 Interface: in Vitro Pulldown
To validate the crystal structure and characterize the TSC1-TBC1D7 interface, we purified 15 TSC1 mutants and 13 TBC1D7 mutants, and examined their binding using an in vitro GST pulldown assay. Overall, mutations of the hydrophobic interface residues had stronger effects than residues involved in salt bridge and hydrogen bond formation. For example, the TSC1 I954A, F958A, and I962A substitutions and TBC1D7 L114A and V94A/V95A substitutions abolished the TSC1-TBC1D7 interaction. Other mutants had no or moderate effect (Fig. 5a). Among the mutants involving hydrophilic side chains, TBC1D7 R96A had the strongest effect. The TBC1D7 Arg96 guanidinine group stacks onto the TSC1 Tyr966′ phenol ring (Figs. 3d and 5b).
FIGURE 5.

GST pulldown analysis of the TSC1-TBC1D7 interaction. a and b, in vitro GST pulldown using purified proteins. SDS-PAGE gels were stained with Coomassie Brilliant Blue. Three independent GST pulldown experiments were performed, and all results were highly reproducible. a, wild-type GST-TBC1D7(1–293) pulls down untagged wild-type TSC1(939–992) and TSC1(939–992) mutants. b, wild-type GST-TBC1D7(1–293) and GST-TBC1D7(1–293) mutants pull down untagged wild-type TSC1(939–992). c and d, semi-quantification of the in vitro GST pulldown analysis. Purified wt/mutant TSC1 and TBC1D7 proteins were used for the pulldown analysis and the SDS-PAGE gels were stained using Oriole Fluorescent Gel Stain (Bio-Rad). A plot of relative densitometry values of the TSC1 is shown. Error bars indicate the mean ± 1 S.D. from three separate experiments.
TSC1 residues Leu941, Leu965, and Leu969 are involved in TSC1 dimerization, but do not directly interact with TBC1D7. Interestingly, the TSC1 L941A/L965A/L969A triple mutation completely disrupted the TSC1-TBC1D7 interaction, suggesting that TSC1 dimerization is required for the TSC1-TBC1D7 interaction. This is consistent with our ITC result (Fig. 1b).
Co-immunoprecipitation (Co-IP) Analysis of the TSC1-TBC1D7 Interface Mutants
To validate the biological relevance of our crystal structure and evaluate the biological roles of the TSC1-TBC1D7 interface residues in the context of full-length proteins in human cells, we co-expressed TSC2, TSC1, and TBC1D7 variants in HEK 293T cells and isolated TSC complexes by co-IP. We quantified the TSC1, TSC2, and TBC1D7 signals and compared the signals of the co-immunoprecipitated proteins, relative to the wt control, after adjusting for the signal of the immunoprecipitated protein (TBC1D7 or TSC1) (Fig. 6). Overall, the results were consistent with our crystal structure and in vitro GST pulldown data (for a comparison see Table 2).
FIGURE 6.

IP analysis of the TSC1-TBC1D7 interaction. C-terminal myc-tagged wild-type TSC1, N-terminal HA-tagged wild-type TBC1D7, or the TSC1 and TBC1D7 variants were coexpressed with TSC2 in HEK 293T cells. TSC complexes were isolated by IP, either with anti-myc affinity beads (specific for exogenous TSC1) or anti-HA affinity beads (specific for exogenous TBC1D7). Four separate transfection experiments were performed. In 3 experiments, IPs were washed 4 times with 20 volumes of lysis buffer. All 3 experiments gave very similar results. In a subsequent experiment, IPs were washed 4 times with lysis buffer and twice with lysis buffer containing 1 m NaCl. a and b, immunoblots showing co-IP of TSC2 and the TBC1D7 variants with immunoprecipitated TSC1. Immunoprecipitates were either washed with lysis buffer only (a), or with high salt buffer (b). c and d, immunoblots showing co-IP of TSC2 and TSC1 with immunoprecipitated TBC1D7. Immunoprecipitates were washed with lysis buffer only (c) or high salt lysis buffer (d), as in A and B. e-h, quantification of the signals on the immunoblots from 3 separate co-IP experiments (no high salt washes). The relative signals of the co-immunoprecipitated proteins are shown relative to the wild-type control, after adjusting for the signal of the immunoprecipitated protein (either TBC1D7 or TSC1). Error bars correspond to the mean ± S.E.; values significantly reduced compared with the wild-type controls (p < 0.05 Student's paired t test) are indicated with an asterisk. e, co-IP of TBC1D7 with TSC1. f, co-IP of TSC2 with TSC1. g, co-IP of TSC1 with TBC1D7. H, co-IP of TSC2 with TBC1D7.
TABLE 2.
Comparison of TSC1-TBC1D7 binding (pulldown versus co-IP): TSC1 and TBC1D7 amino acid substitutions
| Effect on TSC1-TBC1D7 interaction |
|||
|---|---|---|---|
| Pulldown | TBC1D7 IP; TSC1 co-IP (Fig. 6g) | TSC1 IP; TBC1D7 co-IP (Fig. 6e) | |
| TSC1 substitution | |||
| I954A (helix A) | No interaction | Reduced interaction | Reduced interaction |
| F958A (helix A) | No interaction | Reduced interaction | Reduced interaction |
| I962A (helix B) | No interaction | Reduced interaction | Reduced interaction |
| L965A/L969A (helix A/B)a | Reduced interaction | Reduced interaction | No significant reduction |
| Y966A (helix B) | Reduced interaction | Reduced interaction | No significant reduction |
| TBC1D7 substitution | |||
| R81A/Q84A/R121A | No interaction | No interaction | Reduced interaction |
| H90A/R96A | No interaction | No significant reduction | Reduced interaction |
| V94A/V95A | No interaction | Reduced interaction | Reduced interaction |
| R96A | Reduced interaction | No significant reduction | No significant reduction |
| L114A | No interaction | No significant reduction | No significant reduction |
| R121A | No significant reduction | No significant reduction | No significant reduction |
a The two L965/L969 pairs from both TSC1 helices interact with each other and play a role in stabilizing TSC1 dimerization, but neither residue directly contacts TBC1D7.
The TBC1D7 R81A/Q84A/R121A triple substitution prevented the TSC1-TBC1D7 interaction; whereas the R81A/Q84A and V94A/V95A double substitutions clearly disrupted the interaction: co-IP of both TSC1 and TSC2 was reduced with these variants, and they were not effectively co-immunoprecipitated with TSC1. The TBC1D7 R96A, H90A/R96A, L114A, and R121A substitutions did not have a large effect. The L114A and H90A/R96A substitutions abolished TSC1-TBC1D7 binding in the pulldown assay but not in the co-IP assay (compare Figs. 5 and 6). Most likely, the interaction between the full-length proteins, as assessed by co-IP, is more resilient to the effects of amino acid substitutions than the interaction between the bacterially expressed protein fragments. Indeed, the addition of 1 m NaCl to the IP wash buffer did not greatly affect the TSC1-TBC1D7 interactions (compare Fig. 6, a and b, and c and d), suggesting that TBC1D7 and TSC1 are tightly bound in the cell, consistent with previous studies (25).
The TSC1 I954A, F958A, and I962A variants showed clearly reduced interactions with TBC1D7 but not with TSC2, whereas the L965A/L969A and Y966A substitutions had a more moderate effect (compare Fig. 6, e and g).
Discussion
The TSC1-TBC1D7 interaction is important for the stability and activity of the TSC complex, a crucial regulator of the mTORC1 pathway that controls cell growth (25). Here we reveal the structural basis of the TSC1-TBC1D7 interaction. Our results are consistent with previous biochemical analyses and provide a solid understanding of the TSC1-TBC1D7 interface. Sequence alignments suggest that this interface is most likely conserved from Drosophila to human, but not in nematodes (Fig. 3).
In our crystal structure, one TBC1D7 occupies one side of the largely symmetric TSC1 coiled-coil dimer. Under most of our experimental conditions, TSC1 fragments were added in excess prior to SEC, and we observed a predominantly 1:2 stoichiometry. However, AUC analysis of the TSC1(939–992)-TBC1D7 interaction with excess TBC1D7 demonstrated the presence of a 2:2 molar ratio complex (Fig. 7). We speculate that a second TBC1D7 molecule may interact with the opposite side of the TSC1(939–992) homodimer at high molar ratio/concentrations. In fact, in our SEC-MALS analysis, we also typically observed a single TSC1-TBC1D7 complex peak with a measured molecular weight between 1:2 and 2:2, depending on the molar ratio, protein concentration, and buffer conditions. It is likely that the binding of the second TBC1D7 molecule is quite dynamic, although binding of the first TBC1D7 molecule, as we observed in our crystal structure, is quite stable.
FIGURE 7.
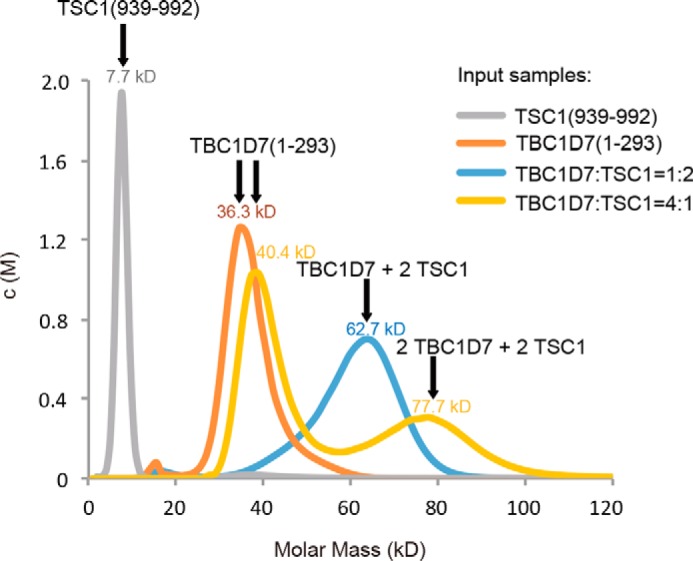
AUC analysis of the TSC1-TBC1D7 interaction. TSC1, TBC1D7, and mixtures of TBC1D7 and TSC1 with two different molar ratios were measured by analytical ultracentrifugation analysis using the sedimentation velocity method. TSC1 and TBC1D7 alone formed monomers. When TSC1 was in excess, the TSC1-TBC1D7 complex formed predominantly a 1:2 heterotrimer. In contrast, when TBC1D7 was in excess, 2:2 heterotetramers were also detected. All AUC measurements were performed at least twice, and all results were highly reproducible.
TBC1D7 belongs to the large TBC protein family that comprises over 30 members in humans. Many TBC family members have been shown to be GAPs for members of the Rab-GTPase family (31). TBC1D7 was shown to have specific GAP activity toward Rab17 (32), but it remains unclear whether TBC1D7 is a bona fide GAP, because it lacks residues that are essential for catalysis (Fig. 8). Superposition of our TSC1-TBC1D7 complex structure with a TBC/Rab-GTPase complex structure demonstrates that the TSC1 dimer binds to a surface distinct from the canonical Rab-GTPase binding site (Fig. 8a). In addition, superposition of our complex with the TBC1D7 apo structure indicates that TSC1 binding does not induce any significant conformational change in the canonical Rab binding site, suggesting that TSC1 binding would not induce or affect the GAP activity of TBC1D7. Comparison of the TBC1D7 structure with the TBC/Rab-GTPase complex clearly demonstrates not only that TBC1D7 lacks the two key catalytic residues, but also lacks the secondary structure components involved in Rab binding (Fig. 8b). Therefore, if TBC1D7 is a GAP, it most likely employs an unusual catalytic mechanism or requires additional factors (e.g. a cofactor and/or posttranslational modification) for activity.
FIGURE 8.

Structural comparison of the TSC1-TBC1D7 complex with a TBC-domain/Rab-GTPase complex. a, the TSC1-TBC1D7 complex structure superimposed with that of a Rab-TBC complex between Rab33 and the TBC domain of Gyp1p (PDB code 2G77). Colors of the individual molecules are indicated. b, a close-up view of the GAP active site. GDP-aluminum fluoride, which mimics the transition state of GTP, and the two catalytic residues (Arg343 and Gln378) are shown for the Rab-TBC complex. TBC1D7 lacks these two critical residues and has a very different structure to other TBC domains.
Both TSC1 and TSC2 form large oligomers or aggregates, and gel filtration studies indicate that the TSC complex is a large hetero-oligomer (26, 42, 43). TBC1D7 may promote TSC1-TSC2 interaction in two ways: by directly interacting with both TSC1 and TSC2, or, by affecting the conformation or oligomerization state of TSC1. Although the TBC1D7- and TSC2-binding sites on TSC1 are adjacent to each other, there is so far no evidence that TBC1D7 directly interacts with TSC2 in the TSC complex. Indeed our co-IP results suggest that this is unlikely. Future structural and biochemical analysis will be needed to examine this possibility.
Regarding the second possibility, at least two TSC1 regions are involved in oligomerization: the N-terminal domain (34) and the coiled-coil domain (42). As a result, the full-length TSC1 and truncated TSC1 isoforms containing both domains (especially the coiled-coil domain) form large aggregates (42). In our hands, the purified TSC1 coiled-coil domain (residues 725–994) forms large oligomers (data not shown), whereas some shorter fragments (e.g. residues 871–992) mostly form dimers (Fig. 1c). It is interesting that the TBC1D7-binding domain of TSC1 (residues 939–992) is monomeric (Fig. 1c), but forms a dimer upon TBC1D7 binding. This region is not predicted to form a continuous coiled-coil by the COILS server, due to a weakened hydrophobic “spine” (Leu941, Ala944, Tyr948, Gln951, Thr955, Phe958, Ile962, Leu965, and Leu969; with less hydrophobic residues in italics). In the absence of TBC1D7, it remains unclear what kind of structure the TSC1 TBC1D7-binding domain may have in the context of full-length TSC1 or the TSC complex. Nevertheless, it is plausible that one structural consequence of TBC1D7 binding is the stabilization, or induced parallel dimerization, of the otherwise potentially floppy C-terminal end of the TSC1 coiled-coil domain. Coiled-coil domains may adopt multiple conformations or oligomerization states in different functional states (e.g. HIV gp41 and proteins involved in membrane fusion) (44–46). One interesting possibility is that TBC1D7 tethers the C-terminal ends of two TSC1 coiled-coil domain, thereby affecting the overall oligomerization equilibrium of TSC1 and TSC2 in the TSC complex.
One puzzling issue regarding the TSC1-TBC1D7 interaction is the role of this interaction in lung cancer (24). In the canonical mTORC1 pathway, TSC1 and TBC1D7 function as tumor suppressors. How do they function as oncoproteins in lung cancer? One possibility is that the TSC1-TBC1D7 interaction regulates pathways other than mTORC1. For example, it has recently been shown that TSC1 interacts with SMAD2/3 to activate TGF-β signaling (22). Alternatively, elevated levels of TBC1D7 might affect TSC1 stability and change the equilibrium of the oligomerization/functional states of TSC complexes. In summary, our work provides a solid foundation for understanding the structural basis of the TBC1D7-TSC1 interaction, and allows for specific testing of the role of this interaction in various biological and pathological processes.
Author Contributions
J. Q., W. G., M. N., and W. X. designed the study and wrote the paper. J. Q. and Z. W. purified and crystallized the protein complex and determined its x-ray structure. J. Q. and G. S. performed the in vitro biochemical analysis. M. H. and M. N. performed the co-IP assay. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to the staff at ALS beamlines BL 8.2.1 and 8.2.2 for assistance with synchrotron data collection. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02-05CH11231.
This work was supported by CAS Grant XDB08010303, and the Department of Defense Grant W81XWH-13–1-0059 (TS120073) (to W. X.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5EJC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TSC
- tuberous sclerosis complex
- TBC1D7
- Tre2-Bub2-Cdc16 (TBC) 1 domain family member 7
- mTORC1
- mechanistic target of rapamycin complex 1
- co-IP
- co-immunoprecipitation
- ITC
- isothermal titration calorimetry
- AUC
- analytical ultracentrifugation assay
- SEC-MALS
- size exclusion chromatography-coupled multi-angle light scattering
- r.m.s.
- root mean square
- GAP
- GTPase-activating protein.
References
- 1. Curatolo P., Verdecchia M., and Bombardieri R. (2002) Tuberous sclerosis complex: a review of neurological aspects. Eur. J. Paediatr. Neurol. 6, 15–23 [DOI] [PubMed] [Google Scholar]
- 2. Crino P. B., Nathanson K. L., and Henske E. P. (2006) The tuberous sclerosis complex. N. Engl. J. Med. 355, 1345–1356 [DOI] [PubMed] [Google Scholar]
- 3. de Vries P. J., and Howe C. J. (2007) The tuberous sclerosis complex proteins: a GRIPP on cognition and neurodevelopment. Trends Mol. Med. 13, 319–326 [DOI] [PubMed] [Google Scholar]
- 4. van Slegtenhorst M., Nellist M., Nagelkerken B., Cheadle J., Snell R., van den Ouweland A., Reuser A., Sampson J., Halley D., and van der Sluijs P. (1998) Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 7, 1053–1057 [DOI] [PubMed] [Google Scholar]
- 5. Kwiatkowski D. J., and Manning B. D. (2005) Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 14, R251–258 [DOI] [PubMed] [Google Scholar]
- 6. Inoki K., Corradetti M. N., and Guan K. L. (2005) Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 37, 19–24 [DOI] [PubMed] [Google Scholar]
- 7. Tee A. R., and Blenis J. (2005) mTOR, translational control and human disease. Semin. Cell Dev. Biol. 16, 29–37 [DOI] [PubMed] [Google Scholar]
- 8. Han J. M., and Sahin M. (2011) TSC1/TSC2 signaling in the CNS. FEBS Lett. 585, 973–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tomasoni R., and Mondino A. (2011) The tuberous sclerosis complex: balancing proliferation and survival. Biochem. Soc. Trans. 39, 466–471 [DOI] [PubMed] [Google Scholar]
- 10. Khwaja O. S., and Sahin M. (2011) Translational research: Rett syndrome and tuberous sclerosis complex. Curr. Opin. Pediatr. 23, 633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sahin M. (2012) Targeted treatment trials for tuberous sclerosis and autism: no longer a dream. Curr. Opin. Neurobiol. 22, 895–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chong-Kopera H., Inoki K., Li Y., Zhu T., Garcia-Gonzalo F. R., Rosa J. L., and Guan K. L. (2006) TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J. Biol. Chem. 281, 8313–8316 [DOI] [PubMed] [Google Scholar]
- 13. Nellist M., Sancak O., Goedbloed M. A., Rohe C., van Netten D., Mayer K., Tucker-Williams A., van den Ouweland A. M., and Halley D. J. (2005) Distinct effects of single amino-acid changes to tuberin on the function of the tuberin-hamartin complex. Eur. J. Hum. Genet. 13, 59–68 [DOI] [PubMed] [Google Scholar]
- 14. Zhang Y., Gao X., Saucedo L. J., Ru B., Edgar B. A., and Pan D. (2003) Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 5, 578–581 [DOI] [PubMed] [Google Scholar]
- 15. Garami A., Zwartkruis F. J., Nobukuni T., Joaquin M., Roccio M., Stocker H., Kozma S. C., Hafen E., Bos J. L., and Thomas G. (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466 [DOI] [PubMed] [Google Scholar]
- 16. Tee A. R., Manning B. D., Roux P. P., Cantley L. C., and Blenis J. (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13, 1259–1268 [DOI] [PubMed] [Google Scholar]
- 17. Castro A. F., Rebhun J. F., Clark G. J., and Quilliam L. A. (2003) Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 278, 32493–32496 [DOI] [PubMed] [Google Scholar]
- 18. Inoki K., Li Y., Xu T., and Guan K. L. (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 17, 1829–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu J., Zacharek S., He Y. J., Lee H., Shumway S., Duronio R. J., and Xiong Y. (2008) WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev. 22, 866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosner M., Hanneder M., Siegel N., Valli A., and Hengstschläger M. (2008) The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat. Res. 658, 234–246 [DOI] [PubMed] [Google Scholar]
- 21. Neuman N. A., and Henske E. P. (2011) Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol. Med. 3, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thien A., Prentzell M. T., Holzwarth B., Kläsener K., Kuper I., Boehlke C., Sonntag A. G., Ruf S., Maerz L., Nitschke R., Grellscheid S. N., Reth M., Walz G., Baumeister R., Neumann-Haefelin E., and Thedieck K. (2015) TSC1 activates TGF-β-Smad2/3 signaling in growth arrest and epithelial-to-mesenchymal transition. Dev. Cell 32, 617–630 [DOI] [PubMed] [Google Scholar]
- 23. Nakashima A., Yoshino K., Miyamoto T., Eguchi S., Oshiro N., Kikkawa U., and Yonezawa K. (2007) Identification of TBC7 having TBC domain as a novel binding protein to TSC1-TSC2 complex. Biochem. Biophys. Res. Commun. 361, 218–223 [DOI] [PubMed] [Google Scholar]
- 24. Sato N., Koinuma J., Ito T., Tsuchiya E., Kondo S., Nakamura Y., and Daigo Y. (2010) Activation of an oncogenic TBC1D7 (TBC1 domain family, member 7) protein in pulmonary carcinogenesis. Genes Chromosomes Cancer 49, 353–367 [DOI] [PubMed] [Google Scholar]
- 25. Dibble C. C., Elis W., Menon S., Qin W., Klekota J., Asara J. M., Finan P. M., Kwiatkowski D. J., Murphy L. O., and Manning B. D. (2012) TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. cell 47, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Menon S., Dibble C. C., Talbott G., Hoxhaj G., Valvezan A. J., Takahashi H., Cantley L. C., and Manning B. D. (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Capo-Chichi J. M., Tcherkezian J., Hamdan F. F., Décarie J. C., Dobrzeniecka S., Patry L., Nadon M. A., Mucha B. E., Major P., Shevell M., Bencheikh B. O., Joober R., Samuels M. E., Rouleau G. A., Roux P. P., and Michaud J. L. (2013) Disruption of TBC1D7, a subunit of the TSC1-TSC2 protein complex, in intellectual disability and megalencephaly. J. Med. Genet. 50, 740–744 [DOI] [PubMed] [Google Scholar]
- 28. Anttila V., Winsvold B. S., Gormley P., Kurth T., Bettella F., McMahon G., Kallela M., Malik R., de Vries B., Terwindt G., Medland S. E., Todt U., McArdle W. L., Quaye L., Koiranen M., et al. (2013) Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 45, 912–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alfaiz A. A., Micale L., Mandriani B., Augello B., Pellico M. T., Chrast J., Xenarios I., Zelante L., Merla G., and Reymond A. (2014) TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum. Mutat. 35, 447–451 [DOI] [PubMed] [Google Scholar]
- 30. Pan X., Eathiraj S., Munson M., and Lambright D. G. (2006) TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature 442, 303–306 [DOI] [PubMed] [Google Scholar]
- 31. Frasa M. A., Koessmeier K. T., Ahmadian M. R., and Braga V. M. (2012) Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat. Rev. Mol. Cell Biol. 13, 67–73 [DOI] [PubMed] [Google Scholar]
- 32. Yoshimura S., Egerer J., Fuchs E., Haas A. K., and Barr F. A. (2007) Functional dissection of Rab GTPases involved in primary cilium formation. J. Cell Biol. 178, 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mozaffari M., Hoogeveen-Westerveld M., Kwiatkowski D., Sampson J., Ekong R., Povey S., den Dunnen J. T., van den Ouweland A., Halley D., and Nellist M. (2009) Identification of a region required for TSC1 stability by functional analysis of TSC1 missense mutations found in individuals with tuberous sclerosis complex. BMC Med. Genet. 10, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun W., Zhu Y. J., Wang Z., Zhong Q., Gao F., Lou J., Gong W., and Xu W. (2013) Crystal structure of the yeast TSC1 core domain and implications for tuberous sclerosis pathological mutations. Nat. Commun. 4, 2135. [DOI] [PubMed] [Google Scholar]
- 35. Santiago Lima A. J., Hoogeveen-Westerveld M., Nakashima A., Maat-Kievit A., van den Ouweland A., Halley D., Kikkawa U., and Nellist M. (2014) Identification of regions critical for the integrity of the TSC1-TSC2-TBC1D7 complex. PloS One 9, e93940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Otwinowski Z., and Minor W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collaborative Computational Project, Number 4. (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeLano W. L., and Brünger A. T. (1994) Helix packing in proteins: prediction and energetic analysis of dimeric, trimeric, and tetrameric GCN4 coiled-coil structures. Proteins 20, 105–123 [DOI] [PubMed] [Google Scholar]
- 41. Ishida T., and Kinoshita K. (2008) Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 24, 1344–1348 [DOI] [PubMed] [Google Scholar]
- 42. Hoogeveen-Westerveld M., Exalto C., Maat-Kievit A., van den Ouweland A., Halley D., and Nellist M. (2010) Analysis of TSC1 truncations defines regions involved in TSC1 stability, aggregation and interaction. Biochim. Biophys. Acta 1802, 774–781 [DOI] [PubMed] [Google Scholar]
- 43. Hoogeveen-Westerveld M., van Unen L., van den Ouweland A., Halley D., Hoogeveen A., and Nellist M. (2012) The TSC1-TSC2 complex consists of multiple TSC1 and TSC2 subunits. BMC Biochem. 13, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gillingham A. K., and Munro S. (2003) Long coiled-coil proteins and membrane traffic. Biochim. Biophys. Acta 1641, 71–85 [DOI] [PubMed] [Google Scholar]
- 45. Apostolovic B., Danial M., and Klok H. A. (2010) Coiled coils: attractive protein folding motifs for the fabrication of self-assembled, responsive and bioactive materials. Chem. Soc. Rev. 39, 3541–3575 [DOI] [PubMed] [Google Scholar]
- 46. Woolfson D. N., Bartlett G. J., Bruning M., and Thomson A. R. (2012) New currency for old rope: from coiled-coil assemblies to α-helical barrels. Curr. Opin. Struct. Biol. 22, 432–441 slcompose,+ zbc-bc zbc-bc zbc01716 zbc4163–16a$sl [DOI] [PubMed] [Google Scholar]