

Abstract
Constitutive activation of the mammalian target of rapamycin complex 1 and S6 kinase (mTORC1→ S6K) attenuates insulin-stimulated Akt activity in certain tumors in part through “feedback” phosphorylation of the upstream insulin receptor substrate 1 (IRS1). However, the significance of this mechanism for regulating insulin sensitivity in normal tissue remains unclear. We investigated the function of Ser-302 in mouse IRS1, the major site of its phosphorylation by S6K in vitro, through genetic knock-in of a serine-to-alanine mutation (A302). Although insulin rapidly stimulated feedback phosphorylation of Ser-302 in mouse liver and muscle, homozygous A302 mice (A/A) and their knock-in controls (S/S) exhibited similar glucose homeostasis and muscle insulin signaling. Furthermore, both A302 and control primary hepatocytes from which Irs2 was deleted showed marked inhibition of insulin-stimulated IRS1 tyrosine phosphorylation and PI3K binding after emetine treatment to raise intracellular amino acids and activate mTORC1 → S6K signaling. To specifically activate mTORC1 in mouse tissue, we deleted hepatic Tsc1 using Cre adenovirus. Although it moderately decreased IRS1/PI3K association and Akt phosphorylation in liver, Tsc1 deletion failed to cause glucose intolerance or promote hyperinsulinemia in mixed background A/A or S/S mice. Moreover, Tsc1 deletion failed to stimulate phospho-Ser-302 or other putative S6K sites within IRS1, whereas ribosomal S6 protein was constitutively phosphorylated. Following acute Tsc1 deletion from hepatocytes, Akt phosphorylation, but not IRS1/PI3K association, was rapidly restored by treatment with the mTORC1 inhibitor rapamycin. Thus, within the hepatic compartment, mTORC1 → S6K signaling regulates Akt largely through IRS-independent means with little effect upon physiologic insulin sensitivity.
Keywords: insulin receptor substrate 1 (IRS-1), insulin resistance, mammalian target of rapamycin (mTOR), S6 kinase, tuberous sclerosis complex (TSC), amino acids, gene knock-in, insulin signaling, serine/threonine phosphorylation
Introduction
The membrane-proximal adaptor proteins IRS12 and IRS2 transmit the extracellular insulin signal to serine/threonine (Ser/Thr) kinases, such as Akt, which further diversify the signal to mediate the various actions of insulin (1). Upon binding insulin from the circulation, the insulin receptor tyrosine kinase (IR) autophosphorylates and is activated toward IRS. Phosphotyrosine (Tyr(P)) residues on IRS serve to recruit Src homology 2 domain proteins, including the dimeric class I-A PI 3-kinase. Although multiple adaptor and catalytic subunits of PI3K are found in most tissues, the p110α catalytic subunit appears to specifically mediate metabolic actions of insulin (2) as demonstrated recently in murine liver (3). By producing membrane phosphatidylinositol 3,4,5-trisphosphate, IRS-associated p110α stimulates PDPK1 (also known as PDK1, phosphoinositide-dependent kinase) to phosphorylate Akt on Thr-308 within its activation loop (4). Thus, mice having liver-specific deletions of genes encoding IR, IRS1 and IRS2, or p110α exhibit severely diminished phosphorylation of Akt Thr-308 (3, 5, 6). In contrast, Ser-473 within the Akt hydrophobic motif (HM) is phosphorylated in a partly IRS-independent fashion by the mTOR- and RICTOR-containing mTORC2 protein complex (6, 7).
A key target of activated Akt is the mTORC1 → S6 kinase cascade, which promotes protein synthesis following insulin stimulation or feeding. Akt indirectly activates mTORC1 by phosphorylating tuberous sclerosis complex (TSC) subunit TSC2 (8), which, with its obligate partner TSC1, negatively regulates RHEB, a small GTPase activator of mTORC1. The IRS proteins not only cooperate in activation of the mTORC1 → S6K and other insulin-stimulated kinase pathways but receive “feedback” from these in the form of multisite Ser/Thr phosphorylation. Through mutational studies in cultured cells, particular phospho-Ser/Thr (Ser(P)/Thr(P)) residues on IRS1 have been shown to impair, or in some cases enhance, its insulin-stimulated tyrosine phosphorylation and signaling (for a review, see Ref. 9). Regardless, the aggregate phosphorylation of IRS1 Ser/Thr residues in cells chronically exposed to insulin is associated with its diminished tyrosine phosphorylation and protein concentration (10). An integrative view of these data is that, in addition to (in specific cases) promoting IRS1 signaling, the phospho-Ser/Thr residues that accumulate on IRS1 during insulin signaling are permissive for its subcellular redistribution, inactivation, and proteasome-mediated degradation (11–13). Inhibitor studies support that the PI3K → Akt → mTORC1 cascade mediates insulin-promoted down-regulation of IRS1 in cultured cells (10, 14), although the involvement of mTORC1 appears to be cell type-specific (15).
Much attention has been given to the idea that non-insulin circulating factors associated with obesity can co-opt the Ser/Thr phosphorylation of IRS1 to mediate insulin resistance (16). For example, it has long been appreciated that amino acids, which independently stimulate the mTORC1 → S6K pathway, can diminish insulin-stimulated IRS1 Tyr(P) and/or glucose uptake when applied to cells (17) or infused into rodents or humans (18, 19). Collectively, these studies implicate at least Ser(P)-307, Ser(P)-632, and Ser(P)-1097 (mouse amino acid numbering used throughout) in the negative regulation of insulin sensitivity by mTORC1 → S6K. In support of such a mechanism, constitutive activity of mTORC1 → S6K in mouse embryonic fibroblasts lacking TSC1 or TSC2 (Tsc−/− MEFs) stimulates these and additional IRS1 Ser(P)/Thr(P) residues within canonical S6K motifs (RXRXX(pS/pT), where pS is phosphoserine and pT is phosphothreonine) as well as the marked degradation of IRS proteins (20–22). Nevertheless, Tsc2del3/del3 MEFs, which express a hypomorphic TSC2 protein, exhibit normal IRS1 protein concentration but retain impaired downstream Akt phosphorylation on par with that in Tsc−/− MEFs (23). To our knowledge, Ser/Thr phosphorylation of IRS1 or IRS2 has not been extensively investigated in these cells.
Among the IRS1 Ser/Thr residues phosphorylated in Tsc−/− MEFs, Ser-302 (RSRTEpS302; corresponding to Ser-307 in human IRS1) is the clearest target of S6K1 in vitro (22). Phosphorylation of this site is also diminished in S6k1 knock-out mice (24). However, earlier work in CHO cells indicated that the phosphorylation of Ser-302, apparently via mTORC1, is necessary for full insulin-stimulated IRS1 tyrosine phosphorylation and signaling to the downstream mTORC1 → S6K pathway (25). Compatible with this feed-forward role, studies in C2C12 and 3T3 Glut4myc cells showed that Ser-302 phosphorylation is an early insulin-stimulated event that, when mimicked chronically by mutation of Ser-302 to glutamic acid, enhances both Akt activity and glucose uptake (26). Rapid insulin-stimulated phosphorylation has also been noted for the Ser-302-homologous site (Ser-307) in studies of primary human adipocytes (27, 28).
Given such results, it is reasonable to question what effect physiologic mTORC1 → S6K activity, such as that promoted by dietary amino acids, has upon IRS1 function and insulin sensitivity in animal tissues. Indeed, extant studies of Tsc1 deletion in murine muscle and liver tissues make clear that hyperglycemia and hyperinsulinemia are not among the more remarkable consequences of this genetic manipulation (Refs. 29 and 30; see the supplement to the latter). Regardless, moderate glucose intolerance, apparently dependent upon genetic background, has been noted in mice following the acute deletion of hepatic Tsc1 with Cre adenovirus (31). Additionally, although IRS1 concentrations were not investigated, or were not diminished, in mouse liver and muscle lacking TSC1 (29–31), muscle-specific RAPTOR deficiency (which abolishes mTORC1 activity) reportedly increases IRS1 protein concentration by unknown mechanisms (32).
To our knowledge, there has not been a thorough description of mTORC1 → S6K-mediated IRS phosphorylation in Tsc-deficient mouse tissues, although it was noted that mutation of IRS1 Ser-307 to alanine does not beneficially affect glucose tolerance following hepatic Tsc1 deletion (31). It is additionally quite likely that TSC complex deficiency regulates tissue insulin sensitivity by IRS-independent mechanisms, including 1) phosphorylation and stabilization by mTORC1 of GRB10 (33) and 2) the inhibitory phosphorylation by S6K of the RICTOR subunit of Akt HM kinase mTORC2 (34). Thus, caution is warranted in generalizing a physiologic role of mTORC1 → S6K feedback to IRS1 based upon observations in Tsc−/− MEFs.
Here, we investigated the function of IRS1 Ser-302 in mouse tissues using genetic knock-in of a Ser-302-to-alanine mutation (A302). Consistent with several cell-based studies (25, 26), we found that Ser-302 is one of several rapidly phosphorylated IRS1 Ser/Thr residues in tissues of healthy, insulin-infused mice. Regardless, A302 knock-in mice exhibited glucose homeostasis indistinguishable from control mice with or without complementation in the liver by endogenous IRS2.
Surprisingly, we found that phosphorylation of Ser-302 is not required for the desensitization of IRS1 Tyr(P) and p110α binding in cells and tissues wherein S6K is highly active. Indeed, we failed to observe excessive phosphorylation of Ser-302, or of any other IRS1 Ser/Thr residues within canonical S6 kinase motifs, in Tsc1-deficient mouse liver despite demonstrably elevated mTORC1 → S6K activity. Regardless, we found substantially diminished Akt phosphorylation in Tsc1-deleted liver and primary hepatocytes from both A302 and control mice. Aggregate Ser/Thr phosphorylation of IRS1 in Tsc1-deficient tissue and cells may have contributed to this diminution by impairing IRS1 Tyr(P) and PI 3-kinase recruitment. However, we conclude from the evidence herein and elsewhere that IRS-independent pathways, such as the phosphorylation of RICTOR by S6 kinase, might be more relevant for mTORC1 → S6K regulation of insulin signaling in normal liver tissue.
Experimental Procedures
Luminex Assays
The IRS1 capture antibody (rabbit monoclonal antibody (mAb) 58-10C-31, Millipore catalog number 05-784R) was coupled to magnetic carboxylated microspheres (Luminex Magplex-C beads) using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride purchased from Sigma-Aldrich (catalog number 74104) (see Schlottmann et al. (35); a complete protocol is available upon request.) In brief, 3.75 × 106 Luminex beads were washed twice and resuspended in 450 μl of coupling buffer (50 mm monobasic sodium phosphate, pH 5.0) and then activated for 20 min at room temperature by addition of 50 μl of 50 mg/ml 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride dissolved in coupling buffer. The activated beads were washed three times in 500 μl of coupling buffer before addition of the antibody (5–10 μg) in 0.5–1.0 ml of phosphate-buffered saline (PBS). After vortexing and brief sonication using a bath sonicator, coupling was continued for 2 h at room temperature in the dark with end-over-end rotation. The coupled beads were washed three times with PBS-TBN (PBS containing 0.02% Tween 20, 0.1% bovine serum albumin (BSA), and 0.05% sodium azide) before being resuspended in storage buffer (PBS containing 1% BSA and 0.02% sodium azide). The antibody-coupled beads were counted using a hemocytometer, and antibody labeling was confirmed by detection of beads with a phycoerythrin-labeled anti-species (anti-rabbit) antibody. Antibodies used for the detection of captured IRS1 (and associated p110α) were biotinylated using reagents from Pierce/Thermo Fisher (EZ-Link NHS-PEG4-Biotin kit) following the manufacturer's guidelines and keeping the antibody concentrations between 1.5 and 2.0 mg/ml. Antibodies for detection of Ser/Thr phosphorylated IRS1 were described previously (36). Antibodies used for detection of total IRS1, Tyr(P) on IRS1, and p110α were the same as those used for immunoblotting (below); the anti-p110α antibody was rabbit mAb clone C73F8 (Cell Signaling Technology).
For Luminex assays, CHO cell lysates (10 μg) or mouse tissue lysates (80 μg) were diluted with IRS1 capture beads (5000 beads/well) into a total volume of 50 μl of Milliplex Assay Buffer 2 (Millipore) and then incubated overnight in 96-well round bottom plates (Costar catalog number 3789, white). During capture, the plates were maintained at 4 °C under constant agitation using an electromagnetic microplate shaker (Union Scientific); subsequent incubation steps were conducted at room temperature, protecting the IRS1 capture beads from light. Buffer exchange was facilitated by a magnetic plate (LifeSep 96F). Following IRS1 capture, the magnetic beads were washed twice with 100 μl of Assay Buffer 2 and then incubated with 25 μl of detection antibody for 1 h on a rotary plate shaker (80 rpm). After removal of the biotinylated detection antibody, the beads were incubated with shaking in 25 μl of 1 μg/ml streptavidin-phycoerythrin (Prozyme) for 15 min. All solutions were then removed, and beads were suspended in PBS-BN (Sigma) for analysis in a Luminex FlexMap 3D instrument. The IRS1 in each sample in Luminex assays was captured in triplicate wells for detection with a given detector antibody. Median fluorescence intensities (MFIs) were reported by the instrument. For IRS1 Ser(P)/Thr(P) assays, the IRS1 capture bead was multiplexed with a magnetic bead of different spectral address labeled with antibody to green fluorescent protein, which served as a control for background signal in addition to wells containing no cell or tissue lysate. The mean of triplicate measures of total (non-phospho-) IRS1 was used as the normalizing denominator for each IRS1 Ser(P)/Thr(P), Tyr(P), and p110α signal in each sample. For the Luminex assay of insulin-stimulated signaling pathways in hepatocyte lysates (18 μg/well) in Figs. 7 and 8, a kit from Millipore (catalog number 48-611MAG) supplemented by an in-house assay for IRS1-associated p110α was used.
FIGURE 7.

Comparison of insulin signaling in acutely Tsc1-deleted A/A and S/S hepatocytes. Primary hepatocytes were infected with control (lacZ) or Cre adenovirus and treated after 3 days with insulin (Ins.; 10 min) in concentrations spanning the portal physiologic range. A, D, F, and H, immunoblot analysis of insulin signaling to Akt (AL, activation loop) and downstream substrates. Numbers beneath blots of mTORC1 and S6 kinase targets (phospho-GRB10 (pGrb10), phospho-S6 (pS6), and phospho-RICTOR (pRictor)) show photometric quantitation of chemiluminescence signals (average, two experiments). B, C, E, G, I, and J, parallel Luminex-based quantitation of insulin-stimulated signals. Samples used in Luminex assays are the same as, or biologic replicates of, those immunoblotted at left; graphed points and error bars represent average MFIs ±S.E. from two experiments measured in triplicate. B and C, remnant TSC2 phosphorylation and S6 hyperphosphorylation; E and G, Akt activation loop and HM phosphorylation; I, IR autophosphorylation; J, IRS1/PI 3-kinase (p110α) association. Luminex signals (other than in J) are normalized by sample tubulin concentration. The immunoblot and Luminex signals, each expressed relative to the maximum insulin-stimulated value in adeno-lacZ-infected cells of the same genotype (=1.0), illustrate the similar, mild impairment of insulin signaling by mTORC1 → S6K activation in A/A and S/S cells.
FIGURE 8.

Effects of rapamycin upon signaling pathways regulating Akt phosphorylation in acutely Tsc1-deleted hepatocytes. A/A:Tsc1lox/lox PHCs were treated as in Fig. 7 but with (+) or without (−) addition of rapamycin (Rapa.) 30 min prior to insulin (Ins.) to inhibit mTORC1 → S6K signaling. A, D, F, and H, immunoblotting-based demonstration of mTORC1 → S6 kinase activation in Tsc1-deleted PHCs and inhibition by rapamycin. Numbers beneath the blots of mTORC1 and S6K substrates (GRB10, S6, and RICTOR) show photometric quantitation of chemiluminescence signals. B, C, E, G, I, and J, Luminex-based quantitation of insulin signaling in the immunoblotted lysates; graphed points and error bars represent average MFIs ±S.E. SC2 phosphorylation (pTsc2) (B), inhibition of S6 hyperphosphorylation (pS6) by rapamycin (C), restoration of insulin-stimulated Akt phosphorylation (pAkt) by rapamycin (E and G), normal IR autophosphorylation (pIR) (I), and persistently impaired IRS1/p110α association (J) in rapamycin-treated PHCs lacking Tsc1 are illustrated. K and L, Luminex quantitation of IRS1 Ser(P)/Thr(P) residues strongly affected or up-regulated in Tsc1-deficient mouse liver, illustrating their dissimilar phosphorylation in acutely Tsc1-deleted hepatocytes.
Immunoblotting Antibodies
All antibodies used in immunoprecipitation and immunoblotting (Western blotting) experiments were from Cell Signaling Technology with the following exceptions. A mouse mAb available from Millipore (catalog number 05-1085) was used to immunoprecipitate and/or detect IRS1. IRS2 was detected using a protein G-purified rabbit polyclonal antiserum raised against full-length mouse IRS2. Tyr(P) on IR and IRS1 was detected using mouse mAb 4G10, purified as described from hybridoma cell culture supernatant (36). The p85 subunit of PI3K and total (non-phosphorylated) GRB10 were detected with antibodies from Millipore (catalog numbers 06-195 and 07-2182, respectively).
Luminex Versus Western Blotting Comparison
CHO cells were cultured, serum-starved (fasted), and then stimulated or not with insulin for 30 min as described earlier (36). Luminex results represent the average of two experiments in which IRS1 Ser(P)/Thr(P) residues were determined from triplicate captures of IRS1 with Ser(P)/Thr(P) signals normalized by the sample total IRS1 determined in the same manner. Western blotting-based estimation of IRS1 Ser(P)/Thr(P) residues used 6–11 (average, 10) immunoblots on which each IRS1 Ser(P)/Thr(P) was determined by photometric quantitation of duplicate lanes. Total IRS1 was determined in the same manner from each fasted and insulin-stimulated CHO cell lysate (11 blots; duplicate fasted and stimulated lanes). All signals from individual immunoblot lanes were rescaled by dividing by the grand median. Signals quantitated from duplicate lanes on immunoblots were averaged to estimate the within-lysate signals for fasted and insulin-stimulated states for each Ser(P)/Thr(P) and total IRS1. Finally, each Ser(P)/Thr(P) signal was normalized by the total IRS1 within the same lysate and state, and grand averages (n = 6–11) were calculated for each IRS1-normalized Ser(P)/Thr(P).
Mice
Knock-in mice bearing the Irs1 S302A allele (“A”) were generated as described (37) except for the sequences of double-stranded DNA oligonucleotides used to introduce the S302A mutation and associated changes in the Irs1 targeting construct. Oligo pairs 1 and 2 introduced a translationally silent BamHI restriction enzyme recognition site at codon 317 of the Irs1 sequence, followed by the S302A mutation, respectively. Oligo pair 3 introduced a translationally silent EcoRI site within codon 259 of the Irs1 sequence. The sense strands of these oligonucleotide pairs with respect to the Irs1 genomic sequence were as follows: 1, 5′-CCTGCCAGTATGGTGGGTGGGAAACCAGGATCCTTCCGG-3′; 2, 5′-CCACCCAGCCAGGTAGGCCTGACTCGGAGATCTCGAACTGAGGCCATCACTGCCACC-3′; and 3, 5′-GGCCATGAGCGATGAATTCCGCCCTCGCAGC-3′. The S302A knock-in mice, produced by knock-in in R1 ES cells (Fig. 1A), were crossed with control knock-in mice of inbred (C57BL6) background and genotyped as described (37). The control knock-in mice bearing the Irs1 S302S allele (“S”) were described earlier (37). Liver-specific insulin receptor knock-out (LIRKO) mice lacking hepatic Insr expression (38) were provided by Dr. Sudha Biddinger. Liver Irs1/Irs2 double knock-out (LDKO) mice and floxed Tsc1 mice (Tsc1lox/lox) were genotyped as described earlier (6, 39). To maximize sensitivity of detecting impaired glucose homeostasis associated with diminished or absent IRS protein function, we used male mice. Mice were group-housed with littermates irrespective of genotype and were maintained on a 12/12-h light/dark cycle.
FIGURE 1.

Correlation of IRS1 Ser/Thr phosphorylation revealed by Western blotting and Luminex assays. CHO cells engineered to express insulin receptor and rat IRS1 were serum-starved or serum-starved and treated with insulin for 30 min prior to lysis. A, profiles of the average Ser(P)/Thr(P) signals in starved and insulin-stimulated cells determined by photometric quantitation of Western blots and by Luminex assays. Note that IRS1-normalized Ser(P)/Thr(P) signals lay naturally between 0 and 2 for both profiles; for details, see “Experimental Procedures.” B, linear fit of Western blot and Luminex data in A, showing measurements of IRS1 Ser(P)/Thr(P) residues in starved (basal) and insulin-stimulated CHO cells. r (Pearson) = 0.66.
Mouse Physiologic Assays and Adenovirus Injection
For glucose tolerance tests, mice were fasted for 16 h beginning 1 h before the end of the light cycle after which blood was drawn for measurement of fasting glucose and plasma insulin and mice were injected intraperitoneally with 2 g/kg glucose. For insulin tolerance tests, mice were fasted for 4 h beginning 3 h after the end of the dark cycle and then injected intraperitoneally with 1 unit/kg insulin (Humulin R, Lilly) diluted in sterile saline. Injection volumes of glucose and insulin were 10 μl/g of mouse body weight. Blood glucose was monitored using a hand-held glucometer (Contour, Bayer). Plasma insulin was measured using an ELISA (Crystal Chem catalog number 90080). Purified adenovirus stocks (Vector Biolabs) were thawed and diluted to a dose of 5.0 × 1010 (plaque-forming units (pfu)/kg of mouse body weight in 100 μl of sterile saline.
Adenovirus Infection of Hepatocytes
Primary hepatocytes (PHCs) were isolated as described (37) and plated in 6-well plates (∼5 × 105/well). The PHCs were allowed to attach for 3 h and then infected with adenoviruses (multiplicity of infection, 20) diluted in 1 ml of serum-free hepatocyte growth medium (Williams E supplemented with penicillin/streptomycin and l-glutamine only) for 1 h. The concentration of fetal bovine serum (FBS) in the medium was then adjusted to 10% by addition of 1.5 ml of growth medium containing excess FBS ( by volume; 16.66%), and the cells were incubated for 36 h. At this time (∼42 h after infection), the growth medium was replaced, and the PHCs were incubated another 8 h and then washed once with PBS and once with fasting medium before serum starvation overnight (16 h) in 2 ml/well fasting medium. Cells were then stimulated by addition of 0.5 ml of prewarmed serum-free medium containing 5× insulin (multiple concentrations used). After 10 min, the PHCs were placed on ice, rinsed once with ice-cold PBS, and then lysed in 1% Triton lysis buffer. Total time from infection to cell lysis was ∼72 h.
Results
Insulin-stimulated IRS1 Phosphorylation in Mouse Tissues
We previously generated and tested in CHO cells a library of monoclonal antibodies to phospho-Ser/Thr residues in IRS1 (αpS/TIrs1 mAb) (36). Insulin rapidly stimulates most IRS1 Ser(P)/Thr(P) residues in these cells, and nearly all of these are suppressible by small molecule inhibitors of PI 3-kinase and Akt (36). Thus, insulin sensitivity may be an important determinant of feedback phosphorylation of IRS1 in animal tissues. To simplify and improve quantitation of IRS1 Ser(P)/Thr(P) residues, we developed a Luminex bead-based assay for its immunocapture and detection using our αpS/TIrs1 mAb library in 96-well plates. Unlike immunoblotting, this assay probes the phosphorylation of native (non-denatured) IRS1 protein; regardless, the IRS1 Ser(P)/Thr(P) profile revealed by Luminex in insulin-stimulated versus serum-starved CHO cells was quite similar to that obtained through repeated photometric quantitation of Western blots (Fig. 1A). Inclusive of basal Ser(P)/Thr(P) measurements in serum-starved cells, the results of the two methods were significantly correlated (Pearson r = 0.66, p < 0.0001) (Fig. 1B); considering only the insulin-stimulated Ser(P)/Thr(P) signals, correlation of the two methods was even greater (r = 0.75, p < 0.0001).
To investigate insulin-stimulated feedback to IRS1 in tissues, we applied our Luminex-based assay to the study of 10-week-old wild-type C57BL6/J mice. Mice fed a standard chow diet were fasted for 6 h and then infused intravenously with saline or insulin (150 milliunits/g) for 5 min (n = 5 or 7) prior to sacrifice and tissue collection. The IRS1 Ser(P)/Thr(P) residues and Tyr(P) in individual liver, skeletal muscle, and heart samples were quantitated in triplicate wells and normalized by total sample IRS1 (determined in separate wells by detection with total IRS1 antibody). In insulin-stimulated liver and skeletal muscle samples, we noted an apparent diminution of total IRS1 protein (up to ∼10% less versus saline; not shown); this was assumed to have a basis in altered subcellular distribution of IRS1 after insulin stimulation, or other biologic mechanism, as the “total” IRS1 detector antibody was extensively qualified as phosphoinsensitive (36). Within a given mouse tissue, such as liver (Fig. 2A), the 25 Ser(P)/Thr(P) residues on IRS1 exhibited quite variable MFIs of the IRS1 capture bead as read with the library of αpS/TIrs1 mAb detectors. However, this was expected because of the unique characteristics (binding capacity or differential affinity) of each αpS/TIrs1 antibody, and there was not a marked correlation between higher absolute MFI and success of a given antibody to measure a stimulatory effect of insulin. Indeed, “non-informative” Ser(P)/Thr(P) assays, defined arbitrarily by average insulin-stimulated sample coefficient of variation greater than 20%, were few in number and unique within the three tissues assayed, compatible with tissue-specific differential phosphorylation as the basis. As a group, the remaining “informative” Ser(P)/Thr(P) assays in liver, skeletal muscle, and heart tissue (n = 21, 24, and 25 assays, respectively) yielded average sample coefficients of variation (±S.E.) of 6.6 ± 1, 6.5 ± 1, and 9.4 ± 1% from insulin-stimulated samples.
FIGURE 2.

Luminex-based quantitation of insulin-stimulated feedback to IRS1 within mouse tissues. Bars and error bars represent mean values ±S.E. A and B, tissues from C57BL6/J mice fasted for 6 h were collected beginning 5 min after intravenous infusion with saline (−) or insulin (+) (n = 5 and 7). Each Ser(P)/Thr(P) (or Tyr(P)) on IRS1 and total IRS1 protein concentration were determined in triplicate for each tissue sample. A, example of raw IRS1-normalized Ser(P)/Thr(P) signals in tissues of saline- and insulin-infused mice. B, raw insulin-stimulated MFIs in liver (see A) and other tissues were divided by the average MFIs measured in tissues of saline-infused mice (=1.0). Note that the average raw Tyr(P)/IRS1 MFI was similar among the three tissues, so unequal y axis scaling is due to lower saline values in skeletal muscle and heart tissue assays. Colored bars indicate nominally significant stimulation (green) or reduction (red) by insulin infusion (Student's t test, p < 0.05). The hash mark (#) indicates significance after Bonferroni correction (p < 0.05/26 comparisons). White bars (saline or insulin) indicate non-informative assays, defined arbitrarily by average sample coefficient of variation greater than or equal to 20%. C and D, survey of IRS1 Ser/Thr phosphorylation repeated in tissues of control (floxed) and LIRKO mice (n = 8 or 6 mice). C, tissues of saline-infused floxed and LIRKO mice exhibit indistinguishable basal phosphorylation of IRS1. D, liver, but not muscle tissue, from LIRKO mice shows appropriate loss of insulin-stimulated feedback to IRS1. n.s., not significant.
The composite insulin-stimulated Ser(P)/Thr(P) patterns in the liver and other tissues were analyzed through appropriate rescaling (such that saline = 1 for each Ser(P)/Thr(P) assay) (Fig. 2B). By comparison with CHO cells (36), the mouse tissues showed a more limited effect of insulin to stimulate IRS1 phosphorylation; however, a subset of Ser(P)/Thr(P) residues, including Ser(P)-265, Ser(P)-302, and Ser(P)-307, plus Tyr(P) on IRS1 were clearly stimulated by insulin in all three tissues (Bonferroni, p < 0.05/26; -fold insulin/saline, >1.5). Additionally, no Ser(P)/Thr(P) residues were significantly decreased by insulin across all three tissues (Bonferroni p values). To confirm the role of insulin-stimulated kinases (i.e. feedback) in these results, we replicated our survey of IRS1 phosphorylation in tissues of control (floxed) and LIRKO mice (38) on the same C57BL6 background (n = 8 and 6). Unexpectedly, the “basal” phosphorylation of IRS1 measured in tissues of saline-treated floxed and LIRKO mice was quite similar despite the profound hyperinsulinemia of LIRKO mice (5, 38) (Fig. 2C). However, basal IRS1 Tyr(P) was lower in liver and higher in muscle of LIRKO mice than in controls. As expected, insulin infusion failed to stimulate Ser(P)-265, Ser(P)-302, or Ser(P)-307 in liver from LIRKO mice, whereas the phosphorylation of all three sites was retained in LIRKO muscle samples (Fig. 2D). Of note, Ser(P)-307 was barely stimulated in LIRKO muscle, although this was nominally significant (Student's t test, p < 0.05). In general, we observed that Ser(P)-307 is more variable after short term insulin infusion than either Ser(P)-265 or Ser(P)-302. This is compatible with previous reports and our experience that Ser(P)-307 peaks relatively later than Ser(P)-302 following insulin stimulation in immortalized and primary human cells (26, 28).
Phenotyping of Irs1 S302A Knock-in Mice
Our survey of IRS1 Ser/Thr phosphorylation in mouse tissues supported that feedback to Ser-302, within the canonical S6 kinase (RXRXX(pS/pT)) motif, is a normal feature of early insulin signaling (25, 26, 28). To assess the physiologic role of Ser-302 phosphorylation by insulin and other stimuli, we generated knock-in mice in which this residue was substituted by alanine (Fig. 3A). To control for effects of genetic knock-in, we intercrossed the A302 mice with previously described control knock-in mice having a wild-type Irs1 gene sequence (S302) (37). Compound heterozygous mice (A302/S302) were then intercrossed to produce mice homozygous for either knock-in allele, referred to hereafter as A/A or S/S.
FIGURE 3.

Normal glucose homeostasis and insulin signaling in A302 Irs1 knock-in mice. A, construction of the A302 Irs1 knock-in allele by homologous recombination in R1 ES cells. Restriction enzymes are abbreviated as follows: K, KpnI; B, BamHI; N; NotI; X, XbaI. Black bars, Southern blot probes for KpnI or BamHI digestion of genomic DNA; s.d. and s.a., intron splice donor and acceptor, respectively. B–E show body weights (B), intraperitoneal glucose tolerance (C), fasting plasma insulin concentration (D), and response to intraperitoneally injected insulin (insulin tolerance) (E) in homozygous control (S/S) and A302 Irs1 knock-in mice (A/A) within cohort 1 described in the text (n = 12–13 mice/genotype). F and G, indistinguishable body weights and intraperitoneal glucose tolerance in a second set (cohort 2) of S/S and A/A mice (n = 9 and 9 mice). H, indistinguishable insulin-stimulated binding of PI 3-kinase (p85/p110α dimer) to Ala-302 and Ser-302 IRS1 proteins in skeletal muscles of cohort 1 mice. Each lane represents skeletal muscle protein from one mouse. IRS1 immunoprecipitates (IP) were analyzed by photometric quantitation of enhanced chemiluminescence signals on immunoblots. Graphed data and error bars represent mean ± S.E. AUC, areas under curves; AOC, areas over curves; n.s., not significant; *, p < 0.05 (Student's t test).
To compare glucose homeostasis in A/A and S/S mice, a cohort of males was produced and aged to 16 weeks (cohort 1; Fig. 3, B–E). The A/A mice within this cohort achieved significantly greater body weight (∼10%) and were significantly, if only moderately, glucose-intolerant versus their less massive S/S controls (Fig. 3, B and C). Nevertheless, fasting insulinemia and responses to injected insulin were statistically indistinguishable between A/A and S/S mice in cohort 1 (Fig. 3, D and E). Given these results, an insulin secretory defect in A/A mice was considered possible; however, a repeat of the above experiments in a second cohort of A/A and S/S males failed to confirm the previous observations of increased body weight or glucose intolerance in the A/A group (cohort 2; Fig. 3, F and G). Thus, it appeared that the influence of other factors, such as non-random distribution of modifier genes in recently intercrossed A/A and S/S mice, was sufficient to outweigh any real alteration of glucose homeostasis caused by the A302 mutation. Regardless, to investigate the signaling potential of the mutant IRS1 protein, we isolated skeletal muscle samples from A/A and S/S mice in cohort 1 that were fasted overnight and injected intravenously with saline or insulin. Immunoprecipitation of IRS1 from these muscles revealed equivalent insulin-stimulated Tyr(P) and binding of PI 3-kinase subunits (p85 and p110α) to Ala-302 and Ser-302 IRS1 proteins (Fig. 3H). Thus, a combination of physiologic and IRS1-specific signaling assays did not demonstrate a consistent difference in the function of Ala-302 and Ser-302 IRS1 in adult mice and tissues.
IRS2 Does Not Mask Ala-302 IRS1 Protein Function in the Liver
Because the influence of the A302 mutation might be masked by IRS2, we compared glucose homeostasis in A302 and S302 mice in which floxed Irs2 alleles (Irs2lox) could be deleted from the liver. We previously showed that mice lacking both Irs1 and Irs2 in the liver (LDKO mice) are profoundly glucose-intolerant, whereas those lacking only Irs2 (LKO2) are nearly normal (6). Moreover, we have shown that glucose homeostasis in LKO2 mice is sensitive to hepatic Irs1 allelic dose and function (37). We therefore used albumin-Cre to simultaneously delete one Irs1 allele (Irs1lox) along with hepatic Irs2, producing males that retained a single A302 or S302 Irs1 allele in the hepatocyte compartment (A/−:LKO2 and S/−:LKO2 mice, respectively). As controls, we produced a small number of Irs1lox/loxIrs2lox/lox males that lacked the albumin-Cre transgene (Cre−) as well as several LDKO males. Intraperitoneal glucose injection of these 16-week-old mice revealed equivalent blood glucose excursion in A/−:LKO2 and S/−:LKO2 mice, which, although far less than that in LDKO mice, was somewhat greater than in Cre− controls (Fig. 4A). Intraperitoneal insulin injection similarly produced nearly equivalent hypoglycemic responses in A/−:LKO2 and S/−:LKO2 mice (Fig. 4B). Fasting blood glucose and plasma insulin concentrations, although again higher than in Cre− controls, were not significantly different in A/−:LKO2 and S/−:LKO2 mice (Fig. 4C). Thus, the uncoupling of physiologic feedback to Ser-302 neither exacerbated nor improved the impairment of systemic glucose homeostasis in LKO2 mice.
FIGURE 4.
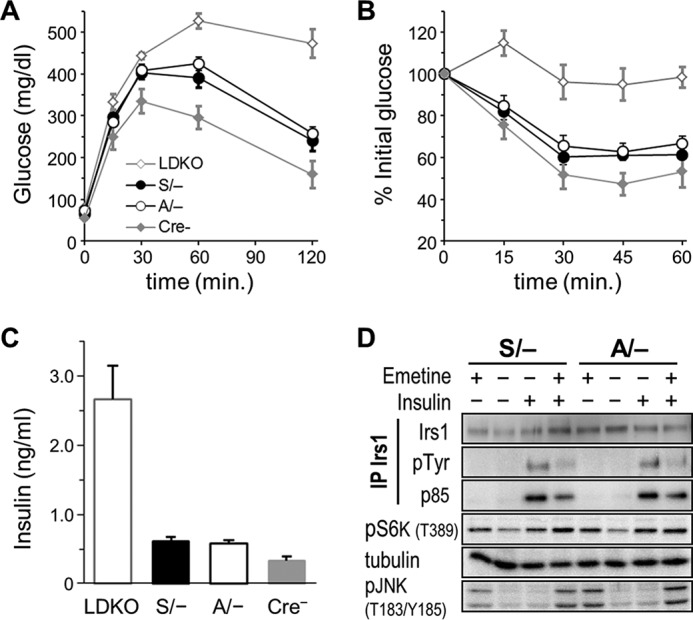
Equivalent physiologic and signaling function of Ala-302 and Ser-302 IRS1 proteins in mouse liver lacking IRS2. Livers of experimental mice had Cre-dependent knock-out of Irs2 as well as hemizygous knock-out of Irs1 and thus retained only a single A302 Irs1 allele (A/−) or S302 Irs1 allele (S/−) (n = 14 and 21). The livers of additional control mouse groups either completely lacked Irs1 and Irs2 (LDKO) (n = 5) or retained a full complement of floxed Irs1 and Irs2 alleles (Cre−). A–C show intraperitoneal glucose tolerance (A), responses to intraperitoneally injected insulin (B), and fasting plasma insulin concentrations (C) in the experimental and control mice. No significant differences were observed between A/− and S/− groups. Graphed data and error bars represent mean ± S.E. D, primary hepatocytes from A/− and S/− mice were treated (+) or not (−) with emetine (20 ng/ml) for 90 min to activate S6 kinase and then with insulin (10 nm) for 30 min to activate IRS1 Tyr(P) and PI 3-kinase (p85) binding. The presence of alanine at Ser-302 of IRS1 (A/−) did not appreciably alter the desensitization of IRS1/PI3K association by emetine. pJNK, phospho-JNK; IP, immunoprecipitate.
Ser-302 Is Not Required for Desensitization of Insulin Signaling Associated with S6K Activation
Despite the above findings, we reasoned that supraphysiologic S6 kinase activity might differentially impair insulin signaling in A/−:LKO2 and S/−:LKO2 tissues. We therefore treated PHCs from these mice with (or without) emetine, an inhibitor of protein synthesis, to raise intracellular amino acid concentrations and activate S6K (40). The cells were then treated (or not) with insulin to stimulate IRS1 phosphorylation. Emetine treatment was, by itself, highly effective at activating S6K (Thr-389 phosphorylation), and this was further augmented by insulin (Fig. 4D). Although we were unable to detect emetine stimulation of Ser(P)-302, treatment with emetine dramatically reduced insulin-stimulated IRS1 Tyr(P) and PI3K (p85) binding in both S/−:LKO2 and A/−:LKO2 cells. This result allows that other Ser/Thr residues targeted by S6K (but not Ser-302) mediated the desensitization of IRS1 signaling by emetine. However, as the protein translation block caused by such agents activates additional non-S6 kinases (e.g. JNK; Fig. 4D), the exact role of S6 kinase activity could not be determined in emetine-treated PHCs.
Assessment of Ala-302 IRS1 Protein Function in Tsc1-deficient Mouse Liver
To more specifically activate mTORC1 → S6K feedback to IRS1 within mouse tissue, we produced A/A and S/S mice in which Tsc1 could be deleted with Cre recombinase (i.e. A/A:Tsc1lox/lox and S/S:Tsc1lox/lox mice). To acutely activate mTORC1 in the hepatocyte compartment, we transduced these mice with a Cre-expressing adenovirus (adeno-Cre) or control lacZ adenovirus (adeno-lacZ). Two cohorts of males were characterized prior to virus injection by measurement of body weights, fasting insulinemia, and intraperitoneal glucose tolerance and then followed up 9 or 18 days after injection with the same assays and collection of liver tissues. Similar results were achieved after 9- or 18-day follow-up, and only the latter data are presented. The mice in this experiment were 12 weeks of age at time of injection and randomly assigned to adeno-lacZ or adeno-Cre virus (n = 10–12 per group). Baseline body weights, fasting plasma insulin concentrations, and glucose tolerance assayed 4 days before virus injection were indistinguishable in A/A:Tsc1lox/lox and S/S:Tsc1lox/lox mice, confirming the similar function of A302 and S302 Irs1 alleles after several generations of intercrossing and maintenance (Fig. 5, A–D; see also cohort 2 in Fig. 3, F and G). During the follow-up period, two adeno-lacZ-injected mice became lethargic and were removed from the study; however, average body weights of the remaining mice (n = 9–12 per group) continued to increase in a manner typical for age (Fig. 5, compare A and E).
FIGURE 5.

Minimal effect of hepatic Tsc1 deletion upon glucose homeostasis in A/A or S/S mice. Twelve-week-old A/A and S/S mice homozygous for floxed Tsc1 alleles (Tsc1lox/lox) were compared 4 days prior to injection with adenoviruses expressing Cre recombinase or control (lacZ) and then followed up after 18 days using the same assays (n = 10–12 mice/genotype per virus). A–D, baseline measurements of mouse body weights (A), fasting plasma insulin concentration (B), and intraperitoneal glucose tolerance (C) showed indistinguishable glucose homeostasis in A/A:Tsc1lox/lox and S/S:Tsc1lox/lox mice. D, areas under glucose curves (AUC) in C. E–H, assays identical to those in A–D repeated 18 days after adenoviral infection. I, liver enlargement due to mTORC1 → S6K activation in adeno-Cre- but not adeno-lacZ-infected mice (tissue collected 21 days postinfection). Graphed data and error bars represent mean ± S.E. The results in E, F, H, and I were analyzed by two-way analysis of variance; *, significant main effect of virus only (no interaction with genotype).
Earlier studies of mice with hepatic Tsc1 deletion have either failed to note any effect upon glucose homeostasis (30) or showed only moderate glucose intolerance dependent upon genetic background (31). Compatible with these studies, S/S:Tsc1lox/lox mice injected with adeno-Cre virus exhibited fasting insulinemia and glucose tolerance equivalent to those in adeno-lacZ-injected controls (Fig. 5, F and G). In fact, by reference to the baseline data (Fig. 5, B and D), there was a general tendency of decreased insulinemia and improved glucose tolerance in all virus-infected mice (Fig. 5, F and G) save adeno-Cre-infected A/A:Tsc1lox/lox mice in which glucose tolerance was somewhat less improved (Fig. 5G). Data from the virus-injected mice were analyzed by two-way analysis of variance with genotype and virus as the independent variables; glucose tolerance data were summarized as areas under the curves (Fig. 5H). Only for glucose tolerance was there a significant effect of genotype or virus (main effect of virus, p < 0.05; no interaction with genotype). As this was mostly due to less improved glucose tolerance in the adeno-Cre-infected A/A:Tsc1lox/lox group, deletion of hepatic Tsc1 did not have a clear effect in our mixed background mice to impair systemic glucose homeostasis. Neither did it reveal differential function of Ala-302 versus Ser-302 IRS1. Given the observed weak effect of adeno-Cre infection upon glucose homeostasis, we questioned whether Tsc1 was sufficiently deleted. However, as reported by others (30), infection of Tsc1lox/lox mice with adeno-Cre, but not adeno-lacZ virus, caused significant liver enlargement (main effect of virus, p < 0.05; no interaction with genotype) (Fig. 5I).
Insulin Signaling and Irs1 Ser/Thr Phosphorylation in Tsc1-deleted Liver
To directly assay Tsc1 deletion and its effects upon hepatic insulin signaling, we isolated liver tissues approximately 3 weeks after adenovirus injection. The virus-infected mice were fasted overnight and then injected intravenously with saline or insulin, and liver tissues were completely excised after 5 min. Correlating with liver enlargement (Fig. 5I), all liver samples from adeno-Cre-infected mice exhibited near total loss of TSC1 protein as well as its obligate partner protein, TSC2 (Fig. 6A). As expected, these samples showed constitutive phosphorylation of mTOR (Ser-2448) and S6K (Thr-389) that was greater than in insulin-stimulated control (adeno-lacZ) samples. To a lesser degree, phosphorylation of RICTOR on Ser-1135, presumably because of S6K activation (34), was also constitutively elevated in Tsc1-deleted liver. IRS2 protein concentration was apparently unchanged by adeno-Cre infection, whereas IRS1 concentration was mildly decreased in line with the degree of TSC1 loss in both A/A and S/S samples. In immunoprecipitates of IRS1, binding of p110α was decreased by Tsc1 deletion, although this was more remarkable in blots of A/A:Tsc1lox/lox than of S/S:Tsc1lox/lox livers. Downstream of the IRS proteins, phosphorylation of Akt on Thr-308 appeared to be more reduced by deletion of Tsc1 than was phosphorylation of Ser-473; however, this was not the case in further experiments conducted in Tsc1-deleted hepatocytes (below; Figs. 7 and 8).
FIGURE 6.

Analysis of insulin signaling and IRS1 Ser/Thr phosphorylation in Tsc1-deleted liver. Mice were stimulated for 5 min by intravenous injection of insulin (+) or control saline (−) before removal of liver tissue. A, top, immunoblots showing deletion of hepatic Tsc1, degradation of TSC2 protein, and constitutive (insulin-independent) activation of mTORC → S6K toward RICTOR in adeno-Cre-infected liver; bottom, immunoblots illustrating moderate effects of mTORC1 → S6K activation upon IRS- and insulin-stimulated Akt phosphorylation (pAkt). B and C, Luminex-based comparison of IRS1 Ser(P)/Thr(P) profiles in control and Tsc1-deleted liver. Samples are those immunoblotted in the first panel; bars show the average IRS1-normalized MFIs (±S.E.) expressed relative to the value in adeno-lacZ-infected S/S:Tsc1lox/lox mice treated with saline (=1); error bars represent S.E. B, total IRS1 protein and insulin-responsive hepatic IRS1 Ser(P)/Thr(P) residues (defined in wild-type liver; Fig. 2B). C, remaining insulin-non-responsive Ser(P)/Thr(P) residues addressed by the αpS/TIrs1 mAb library; insulin (ins.) bars (−/+) are superposed over the average of the two treatments for reference. *, p < 0.05 versus saline; #, p < 0.05 versus same condition in control (adeno-lacZ-infected) liver. IP, immunoprecipitate.
Given the indistinguishable phenotypes and hepatic insulin signaling of adeno-Cre-infected A/A:Tsc1lox/lox and S/S:Tsc1lox/lox mice, we sought to identify commonalities in the Ser/Thr phosphorylation of Ala-302 and Ser-302 IRS1 proteins that might explain the impairment of IRS1/p110α association and downstream Akt phosphorylation. For this experiment, we relied upon Luminex bead assays for capture of IRS1 and detection with our αpS/TIrs1 mAb library plus anti-phosphotyrosine (4G10) and total IRS1 antibodies (Fig. 6, B and C). The study groups were saline- and insulin-treated adeno-lacZ-infected A/A and S/S mice (n = 2 and 3) and adeno-Cre-infected A/A and S/S mice (n = 3 and 4). The various IRS1 Ser(P)/Thr(P) and Tyr(P) MFIs determined by Luminex were normalized by their respective per-sample total IRS1. As suggested by Western blotting of the same lysates (Fig. 6A), the IRS1 protein concentrations quantitated by Luminex were somewhat decreased by adeno-Cre (by ∼22% versus adeno-lacZ, pooling A/A:Tsc1lox/lox and S/S:Tsc1lox/lox mice; p < 0.05) (Fig. 6B, inset).
Overall, the IRS1 Ser(P)/Thr(P) patterns observed in S/S and A/A liver were highly similar other than at Ser-302. The Ser(P)-302 signal in A/A liver measured by Luminex was not zero (presumably due to a poorly corrected background signal) but was consistently ∼40% of that in saline-treated S/S liver and not stimulated by insulin (Fig. 6B). Because of the small sample numbers available, we used simple Student's t tests to screen for apparent changes in IRS1 Ser/Thr phosphorylation caused by insulin stimulation and/or virus treatment. As in the livers of younger, wild-type mice (Fig. 1B), insulin clearly stimulated only IRS1 Tyr(P) plus Ser(P)-265, Ser(P)-302, and Ser(P)-307 in adeno-lacZ-infected S/S liver (Fig. 6B). The remaining “insulin-non-responsive” Ser(P)/Thr(P) residues (Fig. 6C) were nearly equivalent in adeno-lacZ-infected A/A:Tsc1lox/lox and S/S:Tsc1lox/lox livers, and so we focused upon changes in their phosphorylation in adeno-Cre-infected samples. For consistent reference, all of the Luminex data in Fig. 6, B and C, are expressed relative to the average value in adeno-lacZ-infected S/S:Tsc1lox/lox liver treated with saline.
Surprisingly, the deletion of Tsc1 from mouse liver did not stimulate phosphorylation of canonical S6K sites within IRS1 (RXRXX(pS/pT)) defined in vitro and in cultured cells, such as Ser-265 (41) or Ser-302 and Ser-522 (22) (Fig. 6B). On the contrary, Tsc1 deletion diminished the insulin-stimulated phosphorylation of Ser-265 in both A/A and S/S liver samples. Reduced insulin-stimulated phosphorylation of IRS1 Ser/Thr residues (i.e. feedback) is suggestive of insulin resistance; however, in keeping with the minimal phenotype of hepatic Tsc1 deletion, insulin-stimulated IRS1 Tyr(P) was diminished only in A/A:Tsc1lox/lox samples (versus adeno-lacZ) and then quite mildly (Fig. 6B). In contrast with RXRXX(pS/pT) sites in IRS1, Ser(P)-307 (lying within a “proline-directed” p(S/T)P motif) was constitutively elevated by Tsc1 deletion; the Ser(P)-307 signals in adeno-Cre-infected A/A and S/S livers exceeded by 4–6 times the insulin-stimulated signals in adeno-lacZ-infected S/S liver (Fig. 6B). Of note, the pronounced phosphorylation of Ser-307 within adeno-Cre-infected A/A liver indicates insensitivity of the Ser(P)-307 antibody to the state of nearby Ser-302; thus, the relatively weak insulin stimulation of Ser(P)-307 in adeno-lacZ-infected A/A samples was most likely an effect of small sample size or of unintended variation in the short insulin stimulation.
Besides Ser(P)-307, only Ser(P)-632 and Ser(P)-498, the latter sharing a sequence highly similar to that around Ser-307, showed marked stimulation by Tsc1 deletion (i.e. >2-fold versus the average value in liver of adeno-lacZ-infected S/S:Tsc1lox/lox mice that received saline) (Fig. 6C). Four other insulin-non-responsive Ser(P)/Thr(P) residues, Ser(P)-408, Ser(P)-416, Ser(P)-857, and Ser(P)-899, were conversely reduced ∼50% in Tsc1-deleted A/A and S/S samples (Fig. 6C). Although we were able to survey only the 25 Ser/Thr residues addressed by our αpS/TIrs1 mAb library on the Luminex platform, these results appear incompatible with the hyperphosphorylation of IRS1 expected from studies of Tsc1−/− MEF cells. Moreover, it was reported previously that IRS1 Ser-307-to-alanine knock-in mutation does not improve glucose tolerance in C57BL6 mice with hepatic Tsc1 deletion (31). Thus, the diminished Akt phosphorylation that we observed in Tsc1-deleted liver might have been due to concerted effects of multiple Ser(P)/Thr(P) residues or additional factors, such as inhibitory phosphorylation of RICTOR by S6 kinase (see Fig. 6A).
Signaling in Acutely Tsc1-deleted Primary Hepatocytes
To better quantitate the effects of mTORC1 → S6K upon insulin signaling, we used adeno-Cre virus to acutely delete Tsc1 in primary hepatocytes from A/A:Tsc1lox/lox and S/S:Tsc1lox/lox mice. PHCs infected with adeno-Cre (or adeno-lacZ) virus were cultured for a total of 72 h, ending in 16 h of serum starvation after which they were treated with varying insulin concentrations spanning the rodent portal physiologic range (∼1–6 nm) (42). Quantitation by immunoblotting and/or Luminex assays revealed similar down-regulation of insulin-stimulated signals in adeno-Cre-infected A/A and S/S cells (Fig. 7). The degree of Tsc1 deletion after 72 h (Fig. 7A) was similar to that observed in mouse liver 3 weeks after adeno-Cre infection (Fig. 6A); however, whereas TSC2 was almost undetectable in adeno-Cre-infected liver, it was incompletely degraded after 3 days in adeno-Cre-infected PHCs (Fig. 7A). From Luminex quantitation of Ser(P)-939 on TSC2, adeno-Cre-infected PHCs may have retained up to 30% of normal TSC2 phosphoregulation by Akt (Fig. 7B). Nevertheless, these cells exhibited constitutive phosphorylation of GRB10 by mTORC1 (Fig. 7A) and of ribosomal S6 protein by S6K (Fig. 7, A and C, Luminex), in each case exceeding that in control PHCs treated with supraphysiologic insulin. Thus, although the mTORC1 → S6K activity in Tsc1-deleted PHCs was perhaps less than that in Tsc1−/− MEFs (20, 21), it was likely well in excess of physiologic activity in normal liver tissue. Similar to Tsc2del3/del3 MEFs, which express a truncated hypomorphic TSC2 protein (23), adeno-Cre-infected A/A and S/S PHCs failed to show marked degradation of IRS1 or IRS2 (Fig. 7D).
In hepatocyte preparations from different A/A:Tsc1lox/lox and S/S:Tsc1lox/lox donors, we observed substantial intrinsic variation in the phosphorylation of Akt and its substrates. However, any differences in the ability of adeno-Cre to diminish insulin-stimulated phosphorylation of Akt on its activation loop (Thr-308) or HM (Ser-473) were minor. In paired experiments, Tsc1 deletion reduced Akt Thr-308 phosphorylation (at the maximum insulin concentration) by ∼12% in A/A cells and by ∼6% in S/S cells (Fig. 7, D and E, Luminex). Regardless, Thr(P)-308 was more strongly diminished in PHCs from other mice infected with the same lot of adeno-Cre virus (see Fig. 8E). (The cause of this variability is unknown but does not seem to be differential deletion of Tsc1 based upon inspection of remnant TSC1 and TSC2 proteins.) In comparison with the Thr-308 site, phosphorylation of Akt on Ser-473 was more routinely diminished in adeno-Cre-infected cells (by ∼30% at maximal insulin concentration) (Fig. 7, F and G, Luminex; see also Fig. 8G). Compatible with undisturbed glucose homeostasis in adeno-Cre-infected mice (Fig. 5), these alterations in Akt activation loop and HM site phosphorylation did not appreciably affect the insulin-stimulated phosphorylation of diverse Akt substrates (blots in Fig. 7H; see also Fig. 8H).
Although likely influenced by other factors, insulin-stimulated phosphorylation of Akt Thr-308 in mouse liver requires insulin receptor signaling through the IRS and PI 3-kinase (p110α) (3, 5, 6). The GRB10 protein, when stabilized through phosphorylation by mTORC1, is hypothesized to compete with IRS1 and IRS2 for productive binding to the IR (33, 43); however, we were unable to observe an increase in GRB10 protein concentration in Tsc1-deleted PHCs (Fig. 7A; see also Fig. 8A). Autophosphorylation by the IR appeared normal in immunoblots of Tsc1-deleted cells (Fig. 7D) but was diminished by around 20–30% at the highest insulin concentration in several experiments quantitated by Luminex (Fig. 7I; see also Fig. 8I). In contrast, we observed diminished IRS1/p110α association at all insulin concentrations in adeno-Cre-infected PHCs regardless of the state of downstream Akt Thr-308 phosphorylation (Fig. 7J; see also Fig. 8J). The effects of adeno-Cre infection upon IR autophosphorylation and IRS1/p110α association were also quantitatively similar in A/A and S/S cells (Fig. 7, I and J).
In contrast with Thr-308, phosphorylation of the Akt HM site (Ser-473) depends upon the activity of the RICTOR/mTOR-containing kinase mTORC2 (7). In our experiments, inhibitory phosphorylation of RICTOR on Ser-1135 was consistently and strongly stimulated by adeno-Cre infection of both A/A:Tsc1lox/lox and control S/S:Tsc1lox/lox PHCs (Figs. 7F and 8F), potentially explaining the regular diminution of insulin-stimulated Ser(P)-473 versus Thr(P)-308 (Fig. 7G; see also Fig. 8G).
Effect of Rapamycin on the Regulation of Akt Phosphorylation in Tsc1-deleted Hepatocytes
To better gauge the mechanisms by which moderately elevated mTORC1 activity regulates hepatic Akt, we next examined the effect of the mTORC1 inhibitor rapamycin in adeno-Cre-infected A/A:Tsc1lox/lox PHCs (Fig. 8). The results of this experiment confirm that Ser-302 on IRS1 is not needed for the desensitization of insulin signaling by mTORC1 → S6K activity as phosphorylation of both Thr-308 and Ser-473 of Akt was strongly decreased by adeno-Cre (Fig. 8, E and G). As in prior experiments, adeno-Cre infection strongly stimulated the phosphorylation of mTORC1 and S6K substrates GRB10 and S6 (Fig. 8, A and C) as well as the inhibitory phosphorylation of RICTOR on Thr-1135 (Fig. 8F). As expected, short term rapamycin treatment did not restore the concentration or phosphorylation of TSC2 (Fig. 8, A and B) but completely inhibited the phosphorylation of S6K (Thr-389) and S6 (Ser-235/236) in adeno-Cre-infected cells (Fig. 8, A and C). The phosphorylation of mTORC2 subunit RICTOR was similarly strongly reduced by rapamycin, although this could still be somewhat stimulated by insulin (Fig. 8F). Consistent with an inhibitory effect of RICTOR phosphorylation on mTORC2 activity in Tsc1-deleted cells, rapamycin treatment restored insulin stimulation of Ser(P)-473 as well as Thr(P)-308 on Akt to levels exceeding those in adeno-lacZ-infected cells (Fig. 8, G and E).
Neither the IRS1 nor IRS2 protein concentration was markedly affected by inhibition of the mTORC1 → S6K pathway in adeno-Cre-infected PHCs. However, rapamycin noticeably decreased the apparent molecular weight (mobility) of IRS2 on SDS-PAGE gels under insulin-free conditions, compatible with rapamycin-activated phosphatase activity (44) (Fig. 8D). Although autophosphorylation of the IR was comparable within control and Tsc1-deleted cells (Fig. 8I), insulin-stimulated IRS1/p110α association was markedly reduced by Tsc1 deletion and could not be restored by rapamycin treatment (Fig. 8J). We thus queried the status of IRS1 Ser(P)/Thr(P) residues highlighted in our studies of Tsc1-deficient liver (Fig. 6, B and C). As in the liver, adeno-Cre infection of PHCs reduced both basal and insulin-stimulated phosphorylation of IRS1 on Ser-265 (Fig. 8K). However, Ser(P)-307, the most strongly up-regulated Ser(P)/Thr(P) in Tsc1-deleted liver, was not increased in adeno-Cre-infected PHCs despite being somewhat reduced by rapamycin treatment (Fig. 8L). Phosphorylation of Ser-498 was (as expected from the liver results) not stimulated by insulin but was also unaffected in any way by adeno-Cre infection or rapamycin treatment (data not shown). Thus, diminished Akt phosphorylation in Tsc1-deleted liver and in PHCs was not accompanied by a consistent pattern of IRS1 Ser/Thr phosphorylation. Although we cannot exclude that aggregate phosphorylation of IRS1 contributes to its diminished interaction with p110α in Tsc1-deleted tissues and cells, our data best support that excessive mTORC1 → S6K activity regulates Akt phosphorylation through predominantly IRS-independent means.
Discussion
Consistent with earlier studies in transfected cells and primary human adipocytes (25–28), we found that Ser-302 is among several IRS1 Ser/Thr residues rapidly and significantly phosphorylated in response to intravenous insulin in rodent liver and muscle tissues. This result, obtained with a library of anti-Ser(P)/Thr(P) antibodies in fluorescent bead-based assays, is somewhat at odds with the expectation of broader IRS1 phosphorylation developed from transfected cell studies. However, our methodological comparison (using IR and IRS1-transfected CHO cell lysates; Fig. 1) supports that Luminex assays produce results similar to much more laborious and inherently less quantitative Western blotting. Surprisingly, Luminex-based interrogation of muscle tissue from hyperinsulinemic LIRKO mice does not identify significant up-regulation of basal IRS1 Ser/Thr phosphorylation even at apparent insulin-sensitive sites. The seemingly normal insulin stimulation of Ser(P)/Thr(P) residues in LIRKO muscle further suggests that this tissue is not insulin-resistant. Although additional, more directed studies are required to prove this point, our data do not suggest a model whereby hyperinsulinemia promotes insulin resistance through IRS phosphorylation.
The paucity of robustly stimulated IRS1 Ser(P)/Thr(P) residues in mouse tissues would appear to be auspicious for confirmation of their function(s) by genetic knock-in. However, contrary to earlier studies in cultured cells, we found little evidence that the phosphorylation of Ser-302 is required for normal insulin signaling or sensitivity in mice (see Figs. 3, D–H, and 4, A–C, and baseline physiologic data in Fig. 5, A–D). Just as clearly, knock-in mutation of Ser-302 in IRS1 to alanine fails to protect against the desensitization of insulin signaling associated with chemical activation of S6K (Fig. 4D) or with specific activation of mTORC1 → S6K in liver and hepatocytes (Figs. 6A, 7, and 8). In the first instance, it seems possible that physiologic assays are simply insufficient to detect a positive effect of Ser-302 phosphorylation even within the sensitizing milieu of hepatic Irs2 deletion (Fig. 4, A–C). Conversely, our results appear to definitively rule out a role of Ser(P)-302 (or sequence-similar Ser(P)/Thr(P) residues) in feedback inhibition of IRS1 by the mTORC1 → S6K pathway.
The deletion of hepatic Tsc1 using Cre adenovirus provides a clean system for investigating the effects of excess mTORC1 → S6K activity free from complications associated with high fat feeding or genetic obesity. Consistent with earlier work in this system (30), we found that Tsc1 deletion causes liver enlargement but does not increase insulinemia. This result confirms that, despite other metabolic abnormalities, liver-specific Tsc1 knock-out mice do not have need of compensatory insulin secretion to overcome insulin resistance, such as occurs in high fat-fed mice. Consistent with this conclusion, we found that glucose tolerance in mixed genetic background mice is minimally altered by hepatic Tsc1 deletion, being generally equivalent to or improved versus that prior to Tsc1 knock-out (Fig. 5, cf. C and G). Despite these findings, we observed decreased insulin-stimulated Akt phosphorylation in Tsc1-deleted mouse liver (Fig. 6A). A potential explanation for this “disconnect” is that intravenous insulin infusion maximally stimulates tissue Akt, revealing a ceiling imposed by mTORC1 → S6K activation, whereas the stimulation of Akt by physiologic insulin (or intraperitoneal glucose) peaks below such a ceiling. Additionally, we note that the stimulation of Tsc1-deleted hepatocytes with physiologic insulin concentrations (equivalent to portal insulinemia in mice) does not reveal a sensitive connection between the phosphorylation of Akt and that of its downstream substrates (e.g. Fig. 7H).
We find it unlikely that the phenotype of mice with Cre-mediated Tsc1 deletion reflects developmental or other compensation for chronic mTORC1 → S6K activity as we infected adult mice with Cre adenovirus and observed similar results in mice followed for up for just 9 days (half the time of those in Fig. 5). Moreover, we observed similar moderate impairment of insulin signaling to Akt in liver and hepatocytes collected 3 weeks or 3 days after Tsc1 deletion, respectively. We conclude that activity of the mTORC1 → S6K pathway in normal liver, which is probably far less than in Tsc1-deleted liver or hepatocytes, is unlikely to substantially dysregulate systemic glucose homeostasis.
In contrast with Tsc−/− MEF cells, the acutely Tsc1-deleted mouse liver fails to augment IRS1 phosphorylation at canonical S6K sites, including Ser-302 and Ser-265 (Fig. 6, B and C), despite demonstrably elevated mTORC1 → S6K activity (Fig. 6). These data, however, are compatible with the pattern of insulin-stimulated IRS1 phosphorylation seen in CHO cells (36) in which only Ser(P)-302 (among 21 Ser(P)/Thr(P) residues) appeared somewhat reduced by S6 kinase inhibition. There is also some doubt whether S6 kinase mediates insulin-stimulated phosphorylation of the Ser-302-equivalent site in human tissue (45). In contrast, mTORC1 has been implicated previously in phosphorylation of IRS1 on Ser-307 in diverse cultured cells (13, 36, 46), and we found this site to be excessively phosphorylated in Tsc1-deficient liver (Fig. 6B) albeit not hepatocytes (Fig. 8L). Our experiments do not suggest a basis for this differential phosphorylation (apart from the shorter 3-day follow-up in hepatocytes versus Tsc1-deleted liver) but further support that Ser-307 by itself is dispensable for the effects of hepatic Tsc1 deletion in inbred mice (31). We suggest that the insulin-stimulated phosphorylation of Ser-265 in mouse liver and hepatocytes might be mediated by Akt (rather than S6K) as Akt and S6K phosphorylate Ser/Thr residues within the same consensus sequence. In support of this notion, insulin-stimulated Ser(P)-265 in CHO cells is reduced by Akt inhibition (36), and we found deceased Ser(P)-265 in both liver and hepatocytes lacking Tsc1 (Figs. 6B and 8K) in conjunction with decreased phosphorylation of Akt upon Thr-308 and Ser-473.
By recruiting PI 3-kinase, tyrosine phosphorylated IRS1 promotes the activity of PDPK1 toward Thr-308 of Akt. However, it appears unlikely that the Ser/Thr phosphorylation of IRS1 at non-S6K sites significantly impairs Akt phosphorylation during Tsc1 deficiency. First, inconsistent IRS1 phosphorylation within Tsc1-deficient liver and hepatocytes (e.g. at Ser-307) calls into question the connection with mTORC1 → S6K activity. Second, although we do not exclude that such phosphorylation (in aggregate) diminishes IRS1/p110α association, reduced IR activity in some of our experiments might have directly impaired IRS1 tyrosine phosphorylation, particularly at higher insulin concentrations. Third, the observation of diminished IRS1/p110α interaction does not explain impaired Akt Thr-308 phosphorylation in Tsc1-deleted hepatocytes as rapamycin treatment is without effect upon IRS1/p110α interaction but rapidly and fully restores Thr(P)-308. Regarding impaired Akt Ser-473 phosphorylation, our data are fully compatible with S6K-dependent phosphorylation of the mTORC2 component RICTOR as the mechanism (34) as we observed constitutive phosphorylation of RICTOR on Thr-1135 in Tsc1-deficient cells that is both rapamycin-sensitive and inversely correlated with that of Akt Ser-473. Potentially, the restoration of this “priming” phosphorylation could be a factor supporting the full phosphorylation of Akt Thr-308 in rapamycin-treated cells.
Phosphorylation of IRS1 upon Ser/Thr residues has routinely been invoked as a mechanism regulating insulin signaling and/or insulin sensitivity in mammalian tissues. Moreover, such a role is supported by diverse experiments and phenomena observable within cultured cells. Among these phenomena, the mTORC1 → S6K-dependent phosphorylation and degradation of IRS1 in Tsc−/− MEFs is foundational for interpreting the role of IRS1 Ser/Thr phosphorylation. Our observations of Irs1 knock-in mice, including S307A knock-in mice previously (37), question the significance of this feedback loop within animal tissue and caution, more generally, against the extrapolation of physiologic function from signaling in cultured cells.
Author Contributions
The study was conceived and overseen by M. F. W. W. Q. purified monoclonal antibodies to IRS1. N. J. H. contributed and K. D. C. analyzed the data on IRS1 Ser/Thr phosphorylation in CHO cells in Fig. 1. K. D. C. and N. J. H. designed and performed experiments on IRS1 Ser/Thr phosphorylation in mouse tissues in Figs. 2 and 6, B and C. K. D. C. designed and performed all other experiments and analyses, prepared figures, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Dr. Sudha Biddinger for providing LIRKO mice.
This work was supported by National Institutes of Health Grants DK38712, DK55326, DK098655, and GM021700 (to M. F. W.) with further support by an award from the Juvenile Diabetes Foundation International (to M. F. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IRS
- insulin receptor substrate
- IR
- insulin receptor tyrosine kinase
- mTOR
- mammalian target of rapamycin
- mTORC
- mammalian target of rapamycin complex
- S6K
- S6 kinase
- PI
- phosphatidylinositol
- HM
- hydrophobic motif
- TSC
- tuberous sclerosis complex
- MEF
- mouse embryonic fibroblast
- MFI
- median fluorescence intensity
- LIRKO
- liver-specific insulin receptor knock-out
- LDKO
- liver Irs1/Irs2 double knock-out
- PHC
- primary hepatocyte.
References
- 1. White M. F., Copps K. D., Ozcan U., and Tseng Y. D. (2010) The molecular basis of insulin action, in Endocrinology (Jameson J. L., and DeGroot L. J., eds) pp. 636–659, 6th Ed., Elsevier, Philadelphia [Google Scholar]
- 2. Foukas L. C., Claret M., Pearce W., Okkenhaug K., Meek S., Peskett E., Sancho S., Smith A. J., Withers D. J., and Vanhaesebroeck B. (2006) Critical role for the p110α phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441, 366–370 [DOI] [PubMed] [Google Scholar]
- 3. Sopasakis V. R., Liu P., Suzuki R., Kondo T., Winnay J., Tran T. T., Asano T., Smyth G., Sajan M. P., Farese R. V., Kahn C. R., and Zhao J. J. (2010) Specific roles of the p110α isoform of phosphatidylinositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 11, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pearce L. R., Komander D., and Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22 [DOI] [PubMed] [Google Scholar]
- 5. Haas J. T., Miao J., Chanda D., Wang Y., Zhao E., Haas M. E., Hirschey M., Vaitheesvaran B., Farese R. V. Jr., Kurland I. J., Graham M., Crooke R., Foufelle F., and Biddinger S. B. (2012) Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 15, 873–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong X. C., Copps K. D., Guo S., Li Y., Kollipara R., DePinho R. A., and White M. F. (2008) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 8. Manning B. D., Tee A. R., Logsdon M. N., Blenis J., and Cantley L. C. (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 10, 151–162 [DOI] [PubMed] [Google Scholar]
- 9. Copps K. D., and White M. F. (2012) Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55, 2565–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haruta T., Uno T., Kawahara J., Takano A., Egawa K., Sharma P. M., Olefsky J. M., and Kobayashi M. (2000) A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 14, 783–794 [DOI] [PubMed] [Google Scholar]
- 11. Heller-Harrison R. A., Morin M., and Czech M. (1995) Insulin regulation of membrane-associated insulin receptor substrate 1. J. Biol. Chem. 270, 24442–24450 [DOI] [PubMed] [Google Scholar]
- 12. Takano A., Usui I., Haruta T., Kawahara J., Uno T., Iwata M., and Kobayashi M. (2001) Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol. Cell. Biol. 21, 5050–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carlson C. J., White M. F., and Rondinone C. M. (2004) Mammalian target of rapamycin regulates IRS-1 serine 307 phosphorylation. Biochem. Biophys. Res. Commun. 316, 533–539 [DOI] [PubMed] [Google Scholar]
- 14. Pederson T. M., Kramer D. L., and Rondinone C. M. (2001) Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes 50, 24–31 [DOI] [PubMed] [Google Scholar]
- 15. Zhande R., Mitchell J. J., Wu J., and Sun X. J. (2002) Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol. Cell. Biol. 22, 1016–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanti J. F., and Jager J. (2009) Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr. Opin. Pharmacol. 9, 753–762 [DOI] [PubMed] [Google Scholar]
- 17. Patti M. E., Brambilla E., Luzi L., Landaker E. J., and Kahn C. R. (1998) Bidirectional modulation of insulin action by amino acids. J. Clin. Investig. 101, 1519–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tremblay F., and Marette A. (2001) Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem. 276, 38052–38060 [DOI] [PubMed] [Google Scholar]
- 19. Krebs M., Brunmair B., Brehm A., Artwohl M., Szendroedi J., Nowotny P., Roth E., Fürnsinn C., Promintzer M., Anderwald C., Bischof M., and Roden M. (2007) The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes 56, 1600–1607 [DOI] [PubMed] [Google Scholar]
- 20. Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R., Gout I., Downes C. P., and Lamb R. F. (2004) The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah O. J., Wang Z., and Hunter T. (2004) Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14, 1650–1656 [DOI] [PubMed] [Google Scholar]
- 22. Shah O. J., and Hunter T. (2006) Turnover of the active fraction of IRS1 involves raptor-mTOR- and S6K1-dependent serine phosphorylation in cell culture models of tuberous sclerosis. Mol. Cell. Biol. 26, 6425–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pollizzi K., Malinowska-Kolodziej I., Doughty C., Betz C., Ma J., Goto J., and Kwiatkowski D. J. (2009) A hypomorphic allele of Tsc2 highlights the role of TSC1/TSC2 in signaling to AKT and models mild human TSC2 alleles. Hum. Mol. Genet. 18, 2378–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Um S. H., Frigerio F., Watanabe M., Picard F., Joaquin M., Sticker M., Fumagalli S., Allegrini P. R., Kozma S. C., Auwerx J., and Thomas G. (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205 [DOI] [PubMed] [Google Scholar]
- 25. Giraud J., Leshan R., Lee Y. H., and White M. F. (2004) Nutrient-dependent and insulin-stimulated phosphorylation of insulin receptor substrate-1 on serine 302 correlates with increased insulin signaling. J. Biol. Chem. 279, 3447–3454 [DOI] [PubMed] [Google Scholar]
- 26. Weigert C., Kron M., Kalbacher H., Pohl A. K., Runge H., Häring H. U., Schleicher E., and Lehmann R. (2008) Interplay and effects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin action in skeletal muscle cells. Mol. Endocrinol. 22, 2729–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Danielsson A., Ost A., Nystrom F. H., and Strålfors P. (2005) Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J. Biol. Chem. 280, 34389–34392 [DOI] [PubMed] [Google Scholar]
- 28. Danielsson A., Nystrom F. H., and Strålfors P. (2006) Phosphorylation of IRS1 at serine 307 and serine 312 in response to insulin in human adipocytes. Biochem. Biophys. Res. Commun. 342, 1183–1187 [DOI] [PubMed] [Google Scholar]
- 29. Castets P., Lin S., Rion N., Di Fulvio S., Romanino K., Guridi M., Frank S., Tintignac L. A., Sinnreich M., and Rüegg M. A. (2013) Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 17, 731–744 [DOI] [PubMed] [Google Scholar]
- 30. Sengupta S., Peterson T. R., Laplante M., Oh S., and Sabatini D. M. (2010) mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104 [DOI] [PubMed] [Google Scholar]
- 31. Herrema H., Lee J., Zhou Y., Copps K. D., White M. F., and Ozcan U. (2014) IRS1(Ser307) phosphorylation does not mediate mTORC1-induced insulin resistance. Biochem. Biophys. Res. Commun. 443, 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bentzinger C. F., Romanino K., Cloëtta D., Lin S., Mascarenhas J. B., Oliveri F., Xia J., Casanova E., Costa C. F., Brink M., Zorzato F., Hall M. N., and Rüegg M. A. (2008) Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8, 411–424 [DOI] [PubMed] [Google Scholar]
- 33. Hsu P. P., Kang S. A., Rameseder J., Zhang Y., Ottina K. A., Lim D., Peterson T. R., Choi Y., Gray N. S., Yaffe M. B., Marto J. A., and Sabatini D. M. (2011) The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dibble C. C., Asara J. M., and Manning B. D. (2009) Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 29, 5657–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schlottmann S. A., Jain N., Chirmule N., and Esser M. T. (2006) A novel chemistry for conjugating pneumococcal polysaccharides to Luminex microspheres. J. Immunol. Methods 309, 75–85 [DOI] [PubMed] [Google Scholar]
- 36. Hançer N. J., Qiu W., Cherella C., Li Y., Copps K. D., and White M. F. (2014) Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation. J. Biol. Chem. 289, 12467–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Copps K. D., Hancer N. J., Opare-Ado L., Qiu W., Walsh C., and White M. F. (2010) Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 11, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Michael M. D., Kulkarni R. N., Postic C., Previs S. F., Shulman G. I., Magnuson M. A., and Kahn C. R. (2000) Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 6, 87–97 [PubMed] [Google Scholar]
- 39. Kwiatkowski D. J., Zhang H., Bandura J. L., Heiberger K. M., Glogauer M., el-Hashemite N., and Onda H. (2002) A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 11, 525–534 [DOI] [PubMed] [Google Scholar]
- 40. Beugnet A., Tee A. R., Taylor P. M., and Proud C. G. (2003) Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem. J. 372, 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang J., Gao Z., Yin J., Quon M. J., and Ye J. (2008) S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 283, 35375–35382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bergsten P., Westerlund J., Liss P., and Carlsson P. O. (2002) Primary in vivo oscillations of metabolism in the pancreas. Diabetes 51, 699–703 [DOI] [PubMed] [Google Scholar]
- 43. Wick K. R., Werner E. D., Langlais P., Ramos F. J., Dong L. Q., Shoelson S. E., and Liu F. (2003) Grb10 inhibits insulin-stimulated insulin receptor substrate (IRS)-phosphatidylinositol 3-kinase/Akt signaling pathway by disrupting the association of IRS-1/IRS-2 with the insulin receptor. J. Biol. Chem. 278, 8460–8467 [DOI] [PubMed] [Google Scholar]
- 44. Hartley D., and Cooper G. M. (2002) Role of mTOR in the degradation of IRS-1: regulation of PP2A activity. J. Cell. Biochem. 85, 304–314 [DOI] [PubMed] [Google Scholar]
- 45. Rajan M. R., Fagerholm S., Jönsson C., Kjølhede P., Turkina M. V., and Strålfors P. (2013) Phosphorylation of IRS1 at serine 307 in response to insulin in human adipocytes is not likely to be catalyzed by p70 ribosomal S6 kinase. PLoS One 8, e59725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greene M. W., Sakaue H., Wang L., Alessi D. R., and Roth R. A. (2003) Modulation of insulin-stimulated degradation of human insulin receptor substrate-1 by serine 312 phosphorylation. J. Biol. Chem. 278, 8199–8211 [DOI] [PubMed] [Google Scholar]