

Abstract
The endothelium is exposed to various flow patterns such as vasoprotective unidirectional laminar shear stress (LSS) and atherogenic oscillatory shear stress (OSS). A software-controlled, valve-operated OsciFlow device with parallel chambers was used to apply LSS and OSS to endothelial cells. Although LSS inhibited superoxide over time, OSS time-dependently increased superoxide production from endothelial cells. Immunocytochemical staining revealed that, at resting state, p47phox colocalizes with NOX2, whereas NOXO1 colocalizes with NOX1. RNAi of p47phox had no effects on superoxide or NO production in response to OSS but significantly reduced NO production in LSS, implicating a p47phox-bound NADPH oxidase (NOX) in mediating basal NO production. Indeed, RNAi of p47phox inhibited endothelial nitric oxide synthase (eNOS) serine 1179 phosphorylation, whereas PEG-catalase scavenging of intracellular hydrogen peroxide or RNAi of NOX2 produced similar results, indicating a role of NOX2/p47phox-derived hydrogen peroxide in mediating the basal activity of NO production from eNOS. In contrast, RNAi of NOXO1 resulted in no significant changes in NO and superoxide levels in response to LSS but significantly reduced superoxide while increasing NO in response to OSS. Furthermore, we identified, for the first time, that OSS uncouples eNOS, which was corrected by RNAi of NOXO1. In summary, LSS activates the NOX2-p47phox complex to activate eNOS phosphorylation and NO production. OSS instead activates the NOX1-NOXO1 complex to uncouple eNOS. These results demonstrate differential roles of NOXs in modulating the redox state in response to different shear stresses, which may promote the development of novel therapeutic agents to mimic the protective effects of LSS while inhibiting the injurious effects of OSS.
Keywords: NADPH oxidase, nitric oxide, nitric oxide synthase, oxidative stress, shear stress, signal transduction
Introduction
The endothelium is exposed to various flow patterns, such as laminar shear stress (LSS),2 which is the steady tangential force applied at the flat area of the vasculature, and oscillatory shear stress (OSS), generated by forward-reverse flow cycles observed at the bifurcations or branching points of blood vessels (1). A large body of evidence has shown that increased LSS, such as during exercise training, promotes beneficial responses in the vasculature, such as increased NO bioavailability (2, 3) and a reduction in oxidative stress (4). In contrast, OSS has been implicated in vascular pathogenic conditions, at least partially, by increasing reactive oxygen species (ROS) production (5, 6). ROS that are generated under different flow conditions have been shown to play a key role in the signaling of various vascular responses to shear stress. LSS has been shown to reduce excessive ROS levels (7, 8), whereas OSS increases them (5, 6). ROS produced by different shear stresses can have many downstream effects. For example, reduced ROS and increased NO from LSS have been shown to activate anti-inflammatory and anti-atherogenic genes (9).
NADPH oxidase (NOX) is a major source of ROS in the vascular wall. The activity of this enzyme requires the assembly of NOX catalytic subunits with its regulatory subunits (10, 11). The major NOX isoforms in the vasculature are NOX1, 2, and 4 (10). NOX proteins are important in a variety of physiological processes, such as cell proliferation (12, 13) and angiogenesis (14), when producing low levels of ROS at baseline (10). NOXs have been implicated in many pathological conditions, such as diabetes (15) and atherosclerosis (16). These data establish the importance of NOXs in both physiology and pathophysiology.
NOXs have also been implicated in the modulation of differential production of ROS from different types of shear stresses. For example, NOX2 mRNA and protein expression have both been reported to decrease in LSS-treated human endothelial cells (17). In contrast, OSS is known to stimulate the release of ROS from NOX1 (18). Although these studies showed that different NOX isoforms are involved in different shear stress responses, further regulatory mechanisms and downstream details about the activation of these enzymes have yet to be elucidated.
The aim of the current study is to define the specific roles and mechanisms of NOX isoforms and their regulatory subunits in the endothelial cell responses to shear stress. We used a flow chamber to simulate the conditions of LSS and OSS. We found that LSS activates the NOX2-p47phox complex, which, in turn, activates eNOS to increase NO production and reduce superoxide levels, mediated by low levels of intracellular H2O2. In the case of OSS, the NOX1-NOXO1 complex is activated, leading to uncoupling of eNOS, which lowers NO production and increases superoxide. The results here reveal important details by which different NOX isoforms and their regulatory subunits regulate the response of endothelial cells to different shear stress environments.
Experimental Procedures
Material and Reagents
Chemicals of the highest purity were obtained from Sigma unless otherwise specified.
Cell Culture
Bovine aortic endothelial cells (Cell Systems, Kirkland, WA) were cultured in medium 199 (Invitrogen Life Technologies) containing 10% FCS (Hyclone Laboratories, Logan, UT) as described previously (19). Cells were starved with media containing 5% FCS for 16 h before shear experiments.
Shear Stress Device
A software-controlled, valve-operated device (OsciFlow, Flexcell International Corp.) was used to generate LSS or OSS. The bovine aortic endothelial cells were grown on glass slides until 90% confluence, and the slides were inserted into the streamer for exposure to LSS or OSS. During the experiments, 15 dynes/cm2 of either LSS or OSS was applied to cells for 2 h. Cells were harvested after 18–24 h. A schematic of this is shown in Fig. 1A.
FIGURE 1.

A, schematic of the shear stress protocol. B, LSS and OSS regulation of superoxide production. Superoxide production was measured using electron spin resonance at different time points after exposure of bovine aortic endothelial cells to LSS or OSS (15 dynes/cm2). Data shown are mean ± S.E. from four to seven individual experiments. *, p < 0.05 compared with the static condition; #, p < 0.05 compared with corresponding LSS.
Immunoblotting
20 μg of protein from cellular lysates was separated in 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were probed with a specific antibody against NOX1 (Santa Cruz Biotechnology), NOX2 (BD Biosciences), p47phox (BD Biosciences), eNOS (BD Biosciences), or β-actin (Sigma).
Immunostaining
Endothelial cells were fixed in 4% paraformaldehyde for 1 h after the application of shear stress. After fixation, the cell membrane was permeabilized by 0.1% Triton. Cell monolayers were blocked with 5% milk and incubated with specific primary antibodies against NOXO1 (Abcam), NOX2 (BD Biosciences), NOX1 (Santa Cruz Biotechnology), and p47phox (BD Biosciences) for 1 h in the dark. After washing with PBST (0.1% PBS-Tween), they were incubated with Alexa Fluor secondary antibodies. The fluorescence was captured and analyzed with a Zeiss confocal microscope.
Electron Spin Resonance Detection of Nitric Oxide and Superoxide Radicals
NO in endothelial cells after shearing was detected by using ESR as described previously (15, 19, 20), with small modifications to adapt the use of slides in the shear stress setup. All five slides from one shear stress experiment were used for the measurement. Slides were washed with ice-cold Krebs-Henseleit Buffer (KHB) and then moved to a small box to minimize oxidation of the spin trap. NO was trapped by incubation with spin probe Fe2+(DETC)2 colloid (0.5 mmol/liter) for 1 h. Cell pellets were gently collected and snap-frozen in liquid nitrogen. The frozen column of cell pellets was loaded into a finger Dewar for analysis of NO production using an ESR spectrometer (eScan, Bruker). Cells for superoxide measurements were resuspended in spin trap solution and loaded into a glass capillary (Fisher Scientific), where superoxide production was monitored as superoxide dismutase-inhibited accumulation over 10 min. Superoxide production was monitored kinetically, and the rate was calculated over time (minutes).
RNAi Transfection
Proliferating endothelial cells were transfected with control siRNA (25 nmol/liter), p47phox siRNA (25 nmol/liter, CCAAAGAUGGCAAGAACAA), and NOXO1 siRNA (25 nmol/liter, GCAGAAGCUGGGAGGACUU) using Oligofectamine (Invitrogen) in 5% FBS. Twenty-four hours later, cells were subjected to shear stress as described above. Then they were either harvested for Western blotting analysis of NOX isoform expression or ESR analysis of NO or superoxide production 18–24 h after shearing.
DCF-DA Measurement of Intracellular H2O
Endothelial cells were harvested and resuspended in Krebs-Ringer buffer (145 mm NaCl, 5.7 mm Na2HPO4, 4.86 mm KCl, 5.5 mm d-glucose, 0.54 mm CaCl2, and 1.22 mm MgSO4). Samples were incubated with DCF-DA solution (20 μm DCF-DA, final concentration) for 30 min in the dark at 37 °C. H2O2 standards were concurrently incubated. Samples were then measured using a Bio-Tek plate reader at 420 nm excitation and 520 nm emission. H2O2 levels were calculated from the standard curve for each run. The protein concentration from each sample was then measured using a protein assay kit (Bio-Rad) and used for normalization.
Statistical Analysis
One-way analysis of variance was used to test for statistical significance when comparing multiple groups, followed by a Newman-Keuls post hoc test. Student's t test was used for comparison between two groups, also with a significance level of p < 0.05. All statistical analyses were done using the GraphPad Prism software package. All grouped data shown in the figures are presented as mean ± S.E.
Results
LSS and OSS Regulation of Superoxide Production
Superoxide production, determined by ESR, was increased by OSS at both 1 and 24 h but decreased by LSS at 24 h despite a transient increase at 1 h that was consistent with previous observations (7, 17, 18, 21). Therefore, chronic OSS stimulates superoxide production, whereas LSS suppresses it (Fig. 1B, n = 6 each).
Colocalization and Assembly of NOX Catalytic Subunits and Their Regulatory Binding Partners
Before analysis of roles of different NOX isoforms and their regulatory subunits in shear regulation of endothelial NO and ROS signaling, the expression of each NOX isoform and its colocalization with different regulatory subunits was examined by immunocytochemistry. Confocal images indicate p47phox co-localization with NOX2 (Fig. 2F), whereas NOXO1 co-localizes with NOX1 (Fig. 2I). These proteins seem to aggregate in membrane microdomains, likely lipid rafts, and this shares similarities with reports by Hilenski et al. (22). Of note, it is known that the different NOX isoforms localize in different subcellular compartments in different cell types. For example, NOX1 has been found in the caveolae on the plasma membranes of vascular smooth muscle cells (VSMCs) (22). NOX2 has been found on the lamellipodia of phagosomes (23).
FIGURE 2.

Colocalization of NOX catalytic and regulatory subunits in endothelial cells at baseline. Bovine aortic endothelial cells were double-labeled with NOXs and p47phox (A–F) or NOXs and NOXO1 (G–L) to study colocalization of different membrane catalytic subunits with p47phox-NOXO1. Colocalization of NOX1 with p47phox (C) or NOXO1 (I) is demonstrated by overlap of immunofluorescent images of NOX1 (A or G, green) with p47phox (B, red) or NOXO1 (H, red). Colocalization of NOX2 with p47phox (F) or NOXO1 (L) is demonstrated by overlap of immunofluorescent images of NOX2 (D or J, green) with p47phox (E, red) or NOXO1 (K, red).
LSS and OSS Regulation of NOX and eNOS Expression
As described above, under basal conditions, NOX2 colocalizes with p47phox, whereas NOX1 colocalizes with NOXO1. Next we explored whether and how LSS and OSS regulate NOX and eNOS protein expression. Although NOX2 expression was significantly lowered by both LSS and OSS, NOX1 was unaffected (Fig. 3, n = 5 each). The expression of eNOS was up-regulated by both LSS and OSS, as documented by our previous work and that of others (2, 21, 24–27).
FIGURE 3.
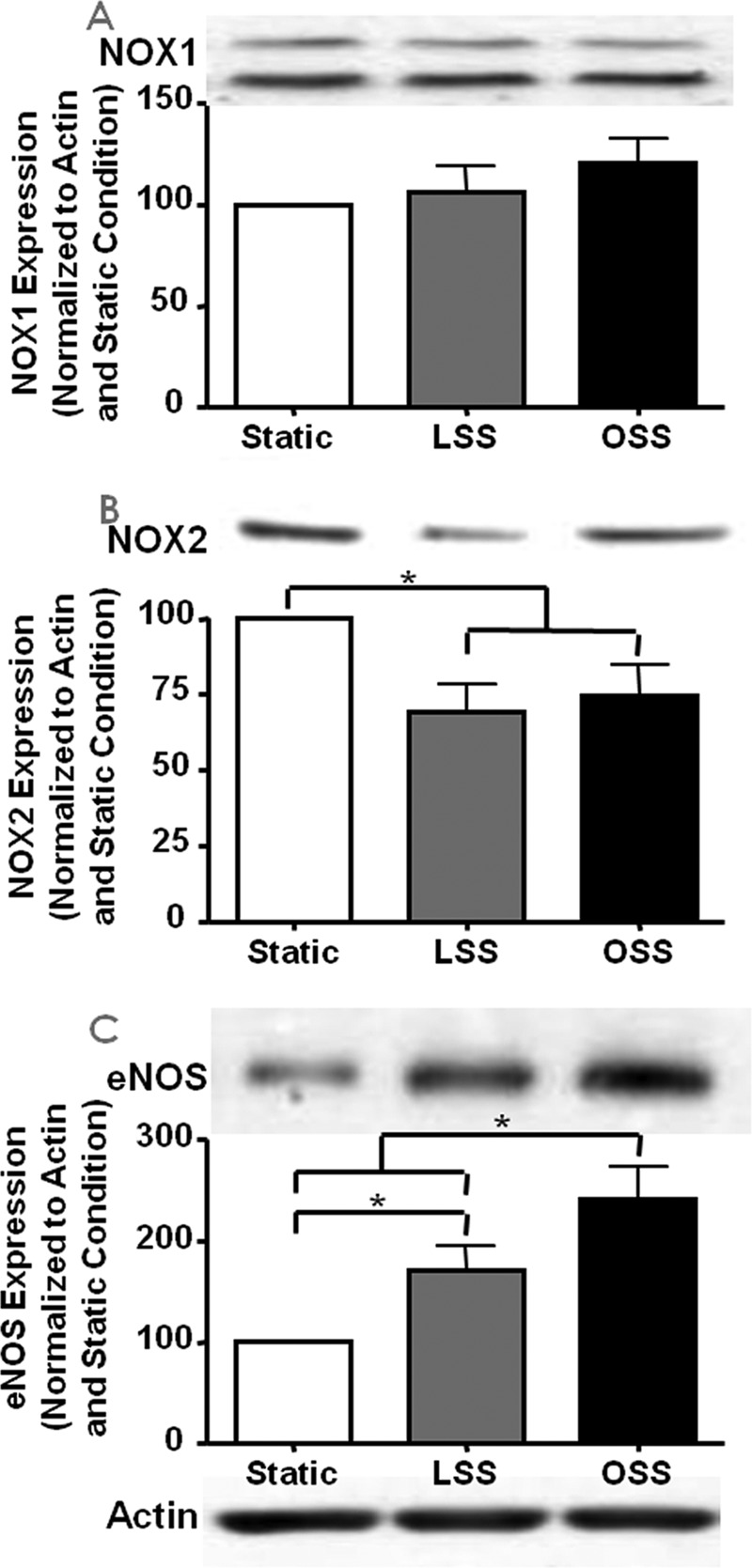
Shear stress regulation of NOX isoforms and eNOS expression. Expression of the NOX isoforms and eNOS was determined by Western blotting analyses from bovine aortic endothelial cells harvested 24 h after exposure to different shear stresses. The representative Western blotting analyses are shown at the top of each panel, and the grouped densitometric data are shown at the bottom (n = 5). The blots are of NOX1 (A), NOX2 (B), and eNOS (C); *, p ≤ 0.05.
Role of p47phox in LSS/OSS Regulation of NO and Superoxide Production
We next examined the role of p47phox in superoxide and NO production in response to different shear stresses through the use of siRNA knockdown. We first examined the expression of p47phox in response to LSS and OSS with and without treatment with p47phox siRNA. Fig. 4A shows the representative Western blotting analysis and grouped densitometric data of p47phox (n = 4 each) protein expression in control or p47phox siRNA-treated cells that were also subjected to LSS and OSS stimulation. Of note, p47phox protein levels were increased by both shear types in control siRNA-treated cells, and these responses were completely attenuated by p47phox siRNA, showing the efficacy of the siRNA treatment.
FIGURE 4.

The role of p47phox in LSS/OSS regulation of NO and superoxide production. siRNA was used to inhibit the NOX regulatory subunit p47phox to examine its role in the endothelial responses to LSS and OSS. A, expression of p47phox in response to LSS/OSS with and without siRNA treatment (n = 4). B, superoxide measured from control or p47phox siRNA-treated cells under static, LSS, or OSS conditions (n = 3). C, NO measured from control or p47phox siRNA-treated cells under static, LSS, or OSS conditions (n = 3); *, p ≤ 0.05.
We then examined the role of p47phox in endothelial cell production of superoxide and NO in response to both LSS and OSS. Of note, RNAi inhibition of p47phox had no effect on either superoxide (Fig. 4B, n = 3) or NO (Fig. 4C, n = 3) production compared with control siRNA-treated cells that were subjected to OSS. In LSS-treated cells, however, superoxide production was further decreased by the inhibition of p47phox, whereas the increase in LSS-induced NO was attenuated. This is actually due to the fact that the p47phox subunit is required for NOX2-dependent hydrogen peroxide production and hydrogen peroxide-dependent eNOS phosphorylation and NO production in response to LSS (see below). But, clearly, p47phox is not involved in the responses of OSS, as shown by these observations.
Role of NOXO1 in LSS/OSS Regulation of NO and Superoxide Production
Similar experiments were performed with NOXO1 siRNA to examine the role of NOXO1 in endothelial cell production of superoxide and NO in response to the two types of shear. We first examined the expression of the NOXO1 protein in endothelial cells subjected to shear stress with control or NOXO1 siRNA treatment. The results (Fig. 5A, n = 4 each) showed that NOXO1 siRNA was effective in the inhibition of NOXO1. Furthermore, although both LSS and OSS induced a significant increase in NOXO1 expression, OSS induced a significantly higher level of NOXO1 expression than LSS.
FIGURE 5.

The role of NOXO1 in LSS/OSS regulation of NO and superoxide production. siRNA was used to inhibit the NOX regulatory subunit NOXO1 to examine its role in the endothelial responses to LSS and OSS. A, expression of NOXO1 in response to LSS/OSS with and without siRNA treatment (n = 4). B, superoxide measured from control or NOXO1-siRNA treated cells under static, LSS, or OSS conditions (n = 3). C, NO measured from control or NOXO1 siRNA-treated cells under static, LSS, or OSS conditions (n = 3); *, p ≤ 0.05.
As shown in Fig. 5, B (n = 3) and C (n = 3), the inhibition of NOXO1 by siRNA treatment did not significantly alter LSS-induced superoxide or NO response compared with control siRNA-treated cells. However, the OSS-induced superoxide increase was significantly attenuated by NOXO1 RNAi, whereas NO deficiency was restored.
Mechanism of p47phox-mediated Response to LSS
The above data suggested an important regulatory role of p47phox in LSS production of NO and superoxide. We further explored the molecular mechanisms leading to this response.
Inhibition of p47phox Attenuates LSS-induced eNOS Activation
The above data showed that p47phox was involved in LSS-induced production of NO. Here we examined whether this increase is related to the activation of eNOSs1179 phosphorylation, which is known to activate eNOS (28). Endothelial cells transfected with control or p47phox siRNA were subjected to static or LSS and probed for total and phosphorylated eNOSs1179 using Western blotting (n = 4 each). The results show that, although the LSS-induced increase in total eNOS was not affected by p47phox inhibition, its increase in phosphorylation was completely attenuated (Fig. 6A).
FIGURE 6.

LSS activates eNOS via p47phox-dependent H2O2 production. siRNA against p47phox was used to examine its role in LSS-stimulated NO production. A, Western blotting data of total and phosphorylated eNOS in bovine aortic endothelial cells treated with p47phox siRNA and LSS (n = 4; *, p < 0.01). B, H2O2 levels in LSS- and OSS-stimulated cells indicating a modest increase with LSS and a marked increase with OSS (n = 6–7; *, p < 0.05; #, p < 0.01). C, Western blotting data of total and phosphorylated eNOS in LSS and PEG-catalase (PEG-CAT)-treated endothelial cells (n = 3; *, p < 0.01; #, p < 0.01 versus all others); *, p < = 0.05.
LSS Results in a Modest Increase in Intracellular H2O2 Production
Previous studies have shown that eNOS can be activated by H2O2 (29–32). Therefore, intracellular H2O2 was measured by DCF fluorescence (n = 6–7 each). The results, shown in Fig. 6B, indicate that both LSS and OSS significantly increase intracellular H2O2 but at substantially different thresholds. It is logical to speculate that this difference in hydrogen peroxide levels stimulated by LSS versus OSS is one of the major factors determining differential downstream signaling of the two flow forces.
H2O2 Mediates LSS-induced eNOS Activation
To examine whether H2O2 mediates LSS-induced activation of eNOSs1179, endothelial cells were treated with the H2O2 scavenger PEG-catalase prior to being subjected to static or LSS. The results, shown in Fig. 6C, indicate that, in PEG-catalase-treated cells where intracellular H2O2 was reduced, the increase in p-eNOSs1179 was blunted (n = 3 each). The results here show that the increase in eNOS activation from LSS is mediated by intracellular H2O2 derived from a p47phox-dependent mechanism.
Role of NOX2 in the Endothelial Response to LSS
As shown in Fig. 2F, p47phox colocalizes with NOX2 at baseline. Here we examined whether NOX2 also participates in the response of endothelial cells to LSS.
Inhibition of NOX2 Attenuates LSS-induced eNOS Activation
As described above, LSS activates eNOS via phosphorylation at serine 1179. Here we used siRNA knockdown of NOX2 to examine whether NOX2 also plays a role in this response. Endothelial cells were treated with control or NOX2 siRNA prior to being subjected to LSS and then probed for NOX2 and total and phosphorylated eNOSs1179 by Western blotting analysis (n = 4). Fig. 7A, top panel, shows the efficacy of the NOX2 siRNA treatment in inhibiting the expression of NOX2. Fig. 7A, center and bottom panels, demonstrate total and phosphorylated eNOSs1179, respectively. The results indicate that, although total eNOS expression was not affected by NOX2 siRNA treatment compared with control siRNA, NOX2 siRNA attenuated the LSS-induced increase in eNOSs1179 phosphorylation.
FIGURE 7.

NOX2 regulates LSS production of NO via eNOS phosphorylation. siRNA against NOX2 was used to examine whether NOX2 plays a role in LSS activation of eNOS. A, Western blotting data of total and phosphorylated eNOS in bovine aortic endothelial cells treated with NOX2 siRNA and LSS (n = 4; *, p < 0.001; #, p < 0.001 versus all others). Of note, NOX2 siRNA inhibited LSS activation of eNOS. B, superoxide production in cells treated with NOX2 siRNA and LSS (n = 4; *, p < 0.001 versus static; #, p < 0.05 versus static control siRNA; &, p < 0.05 versus LSS control siRNA). C, NO production in cells treated with NOX2 siRNA and LSS (n = 4; #, p < 0.01 versus all others). Consistent with eNOS activation data, NOX2 siRNA attenuated NO production in response to LSS.
Inhibition of NOX2 Reduces the Superoxide and LSS-induced NO Increase
Superoxide and NO were directly measured from control and NOX2 siRNA-treated cells subjected to LSS to examine the involvement of NOX2 in the endothelial response to LSS. The results in Fig. 7B show that NOX2 siRNA reduced superoxide production under both static and LSS conditions (n = 4 each), indicating a housekeeping role under the static condition that shares some similarity with previous reports (33). They also indicate a role of NOX2 in activating p47phox-dependent superoxide production. In addition, NOX2 inhibition attenuated the LSS-induced increase in NO production (n = 4 each). Taken together, these data show that the p47phox-NOX2 complex is responsible for the increase in NO production from LSS via H2O2-dependent phosphorylation of eNOS.
NOXO1/NOX1 Mediates OSS Uncoupling of eNOS
The data above showed that NOXO1 inhibition attenuated OSS induced superoxide increase and NO inhibition. One possible way that this can occur is through the uncoupling of eNOS. We and others have shown previously that indeed, NOX1 lies upstream of uncoupled eNOS under different conditions, such as diabetes (15) and hypertension (34) and in response to angiotensin II treatment (35). To examine the coupling state of eNOS, we compared superoxide production from measurements with and without the NOS inhibitor L-NAME. If eNOS is coupled and producing NO, then its inhibition with L-NAME will increase total superoxide production because of the decrease in NO-mediated superoxide buffering. However, if eNOS is uncoupled and producing superoxide, then its inhibition with L-NAME will decrease superoxide production. The results shown in Fig. 8 indicate that, with control siRNA treatment, OSS induces eNOS uncoupling, as shown by the decrease in superoxide production in L-NAME samples (n = 3). However, inhibition of NOXO1 restored the coupling state of eNOS. Taken together with the data from Fig. 2I, which showed that NOXO1 and NOX1 colocalize, these data suggest that, under OSS, the NOXO1-NOX1 complex is responsible for increased superoxide production and reduced NO bioavailability via the mechanism of uncoupling eNOS.
FIGURE 8.
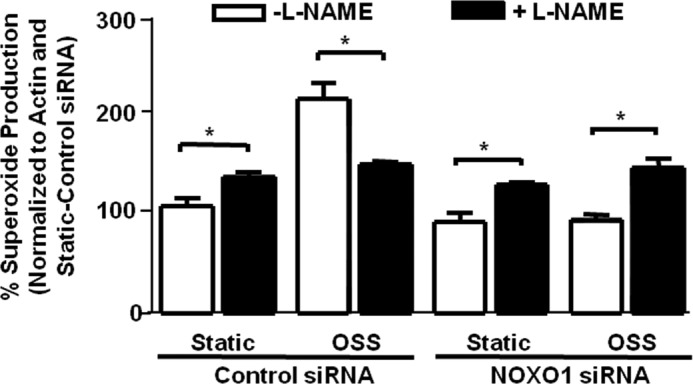
Inhibition of NOXO1 restores eNOS activity uncoupled by OSS. siRNA against NOXO1 was used to examine whether OSS-induced changes in superoxide and NO production are related to the uncoupling of eNOS. Bovine aortic endothelial cells were treated with control or NOXO1 siRNA prior to being subjected to static or OSS conditions. Superoxide was measured from these cells in the presence or absence of L-NAME, a NOS inhibitor, to assess the coupling state of eNOS. Obviously, L-NAME inhibited superoxide production under OSS conditions, implicating uncoupling of eNOS. This response was fully reversed by the treatment with NOXO1 siRNA (n = 3); *, p ≤ 0.05.
Discussion
In this study, we demonstrated that LSS and OSS differentially regulate NOX and eNOS pathways to affect the redox status of endothelial cells. For LSS, the p47phox-NOX2 complex is activated to produce NO, which is mediated by H2O2-dependent eNOS phosphorylation. In response to OSS, NOXO1 is substantially up-regulated under both static and LSS conditions and complexes with NOX1. The resulting NOXO1-NOX1 complex increases superoxide production and reduces NO, which is mediated by uncoupling of eNOS. These results elucidate detailed molecular roles of the specific NOX isoforms and their regulatory subunits as well as their differential regulations of eNOS in controlling the redox status of endothelial cells subjected to different types of shear stress. These pathways are summarized in a schematic in Fig. 9.
FIGURE 9.
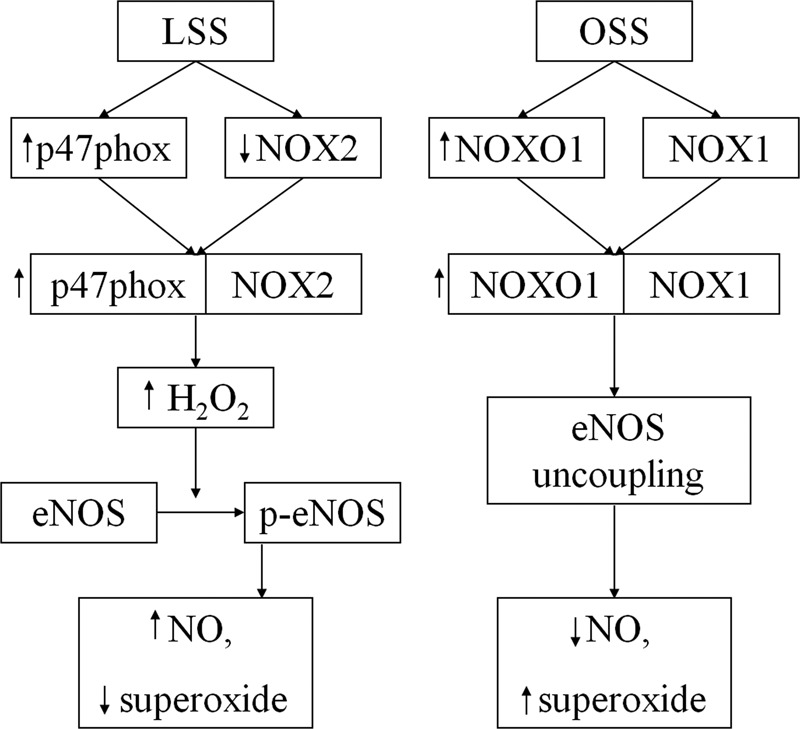
Schematic of the pathways elucidated in this work. Under LSS, p47phox and NOX2 complex, causing a modest increase in H2O2, which results in activation of eNOS, a consequent increase in NO production, and a decline in superoxide levels. Under OSS, NOX1 and NOXO1 complex, resulting in uncoupling of eNOS. This leads to increased production of superoxide and a decrease in NO bioavailability.
NOXs have been shown to be involved in the pathophysiology of many cardiovascular diseases. For example, NOX1 is activated in diabetes, resulting in endothelial dysfunction (15). NOXs, specifically NOX1 and NOX2, and their subunits have also been implicated in hypertension in many studies (for reviews, see Refs. 36, 37). In atherosclerosis, the role of NOX1 as a major contributing factor has been established (16). In contrast, the role of NOX2 is more controversial, with some studies showing a pathological effect (38), whereas others show no effect (39). In the heart, NOX4 has been shown to play in a role in ischemic damage (40) and heart failure (41). Taken together, these data show that NOXs are important mediators of cardiovascular pathologies.
Past observations have shown that atherosclerotic lesions tend to preferentially localize at curved or branched regions of the arterial tree (42). At these regions, local blood flow becomes disturbed, leading to flow conditions that are irregular or oscillating, subjecting endothelial cells to OSS (1). In contrast, atherosclerotic lesions are less frequent in continuous, non-branched parts of the artery, which are characterized by regular flow conditions, subjecting endothelial cells to LSS. These different flow patterns ultimately lead to signaling cascades that result in the activation of anti-atherogenic or pro-atherogenic genes (1).
Accumulating evidence has implicated an important signaling role of ROS in the response of cells to different flow conditions. Studies have shown that irregular flow or OSS conditions have decreased NO production and increased superoxide production compared with steady or LSS conditions (5, 6, 43, 44). Our observations match these earlier findings well in that, under LSS conditions, we detected an increase in NO production and decrease in superoxide, whereas, under OSS conditions, we observed the opposite. These changes in ROS under different shear stress conditions have important signaling and downstream effects. For example, OSS stimulates NOX-dependent ROS that increase ICAM-1 expression and subsequent monocyte adhesion, events that are pro-atherogenic (18). Superoxide from NOXs has also been shown to oxidize LDL (45), which enhances macrophage infiltration through the expression of adhesion molecules, leading to inflammation (46). On the other hand, LSS has been shown to increase NO production from eNOS, which is a potent vasodilator and anti-inflammatory mediator (28). Furthermore, this NO can lead to decreased MCP-1 expression (47), which will decrease monocyte adhesion. Our results here show the details of this ROS regulation, with LSS inducing the formation of the p47phox-NOX2 complex to activate eNOS/NO in an H2O2-dependent fashion and OSS inducing the formation of the NOXO1-NOX1 complex to uncouple eNOS and enhance superoxide production.
In a previous study, LSS was found to down-regulate the expression of NOX2 (17). In this study, we observed a similar trend in both LSS- and OSS-treated endothelial cells. Furthermore, we identified a NOX2-p47phox-dependent activation of eNOS mediated by a modest increase in intracellular H2O2. These results agree with previous studies that have also shown that H2O2 can lead to the activation of eNOS (29, 30, 48). Furthermore, although we observed that both LSS and OSS increased H2O2 extracellularly, its level inside the cells was further increased when exposed to OSS. Interestingly, this is coupled to an uncoupling of eNOS. Taken together, these data indicate that, although a modest increase in H2O2 may improve eNOS activity via the NOX2-p47phox complex, a larger increase will lead to the uncoupling of eNOS and endothelial dysfunction.
A previous study have shown that OSS stimulates ROS from NOX1 in a BMP4-dependent manner (18). Other studies have also shown that NOX1-derived ROS is at least partly responsible for the formation of lesions in a murine model of atherosclerosis (15, 16, 49). In this study, we expanded upon these earlier findings by showing that NOXO1, a specific regulatory protein for NOX1, is greatly up-regulated by OSS and is responsible for the observed increase in ROS. In addition, we identified that activation of the NOX1-NOXO1 complex via OSS leads to uncoupling of eNOS, which would further exacerbate oxidative stress by increasing superoxide and reducing NO production. The uncoupling of eNOS has been observed in the vasculature of several diseases, such as hypercholesterolemia (50), diabetes (15, 51), hypertension (19, 52, 53), and atherosclerosis (54). Taken together, these observations again show the importance of the NOX-eNOS uncoupling pathway in vascular diseases, specifically under conditions of irregular hemodynamic flow.
In this study, we did not examine the effects of NOX4. From our data of NOX2 knockdown in LSS and NOXO1 knockdown in OSS, both were sufficient in restoring ROS levels back to baseline. Therefore, it is unlikely that NOX4 would further contribute to the endothelial cell response to the different shear stresses. In one study using long term LSS (continuously for 24 h), NOX4 expression was decreased in endothelial cells (55). NOX4 has been shown in other studies to be involved in chronic conditions such as angiogenesis (14, 56) and heart failure (41). It is possible that NOX4 activity is involved more in the long-term regulation of shear stress (beyond 24 h), which was not examined in this study. Nonetheless, its expression and regulation have been found to be much more relevant to cardiac biology rather than vascular biology (41, 57).
In conclusion, our study identifies the specific roles that NOX proteins and its regulatory subunits play in the response of endothelial cells to different shear stresses. Under regular flow or LSS conditions, NOX2 complexes with the regulatory subunit p47phox, enhancing H2O2 production, which, in turn, activates eNOS to increase NO and reduce superoxide. Under irregular flow or OSS conditions, the NOX1-NOXO1 complex is activated, which, in turn, leads to uncoupling of eNOS, resulting in increased superoxide production and a reduction in NO. These results not only elucidate key details of shear stress regulation of endothelial redox status but also identify possible therapeutic targets that can be exploited in the treatment of atherosclerosis that are highly prevalent in vasculature exposed to oscillatory flow.
Author Contributions
K. L. S. performed data collection, data analysis, and manuscript preparation. L. G. performed data collection and analysis. H. C. designed the research and performed data analysis and manuscript preparation.
This study was supported by NHLBI/National Institutes of Health Grants HL077440 (to H. C.), HL088975 (to H. C.), HL108701 (to H. C. and D. G. H.), and HL119968 (to H. C.) and American Heart Association Established Investigator Award 12EIA8990025 (to H. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LSS
- laminar shear stress
- OSS
- oscillatory shear stress
- ROS
- reactive oxygen species
- NOX
- NADPH oxidase
- eNOS
- endothelial nitric oxide synthase
- l-NAME
- l-NG-nitroarginine methyl ester
- DCF-FA
- 2′,7′-dichlorofluorescein diacetate.
References
- 1. Chiu J. J., and Chien S. (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev. 91, 327–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davis M. E., Cai H., Drummond G. R., and Harrison D. G. (2001) Shear stress regulates endothelial nitric oxide synthase expression through c-Src by divergent signaling pathways. Circ. Res. 89, 1073–1080 [DOI] [PubMed] [Google Scholar]
- 3. Wang J., Wolin M. S., and Hintze T. H. (1993) Chronic exercise enhances endothelium-mediated dilation of epicardial coronary artery in conscious dogs. Circ. Res. 73, 829–838 [DOI] [PubMed] [Google Scholar]
- 4. Fukai T., Siegfried M. R., Ushio-Fukai M., Cheng Y., Kojda G., and Harrison D. G. (2000) Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J. Clin. Invest. 105, 1631–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hwang J., Saha A., Boo Y. C., Sorescu G. P., McNally J. S., Holland S. M., Dikalov S., Giddens D. P., Griendling K. K., Harrison D. G., and Jo H. (2003) Oscillatory shear stress stimulates endothelial production of O2˙̄ from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J. Biol. Chem. 278, 47291–47298 [DOI] [PubMed] [Google Scholar]
- 6. De Keulenaer G. W., Chappell D. C., Ishizaka N., Nerem R. M., Alexander R. W., and Griendling K. K. (1998) Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. Circ. Res. 82, 1094–1101 [DOI] [PubMed] [Google Scholar]
- 7. Goettsch C., Goettsch W., Brux M., Haschke C., Brunssen C., Muller G., Bornstein S. R., Duerrschmidt N., Wagner A. H., and Morawietz H. (2011) Arterial flow reduces oxidative stress via an antioxidant response element and Oct-1 binding site within the NADPH oxidase 4 promoter in endothelial cells. Basic Res. Cardiol. 106, 551–561 [DOI] [PubMed] [Google Scholar]
- 8. White S. J., Hayes E. M., Lehoux S., Jeremy J. Y., Horrevoets A. J., and Newby A. C. (2011) Characterization of the differential response of endothelial cells exposed to normal and elevated laminar shear stress. J. Cell Physiol. 226, 2841–2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lehoux S. (2006) Redox signalling in vascular responses to shear and stretch. Cardiovasc. Res. 71, 269–279 [DOI] [PubMed] [Google Scholar]
- 10. Lassègue B., San Martín A., and Griendling K. K. (2012) Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 110, 1364–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai H., Griendling K. K., and Harrison D. G. (2003) The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 24, 471–478 [DOI] [PubMed] [Google Scholar]
- 12. Lee M. Y., San Martin A., Mehta P. K., Dikalova A. E., Garrido A. M., Datla S. R., Lyons E., Krause K. H., Banfi B., Lambeth J. D., Lassègue B., and Griendling K. K. (2009) Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 29, 480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suh Y. A., Arnold R. S., Lassegue B., Shi J., Xu X., Sorescu D., Chung A. B., Griendling K. K., and Lambeth J. D. (1999) Cell transformation by the superoxide-generating oxidase Mox1. Nature 401, 79–82 [DOI] [PubMed] [Google Scholar]
- 14. Craige S. M., Chen K., Pei Y., Li C., Huang X., Chen C., Shibata R., Sato K., Walsh K., and Keaney J. F. Jr. (2011) NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 124, 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Youn J. Y., Gao L., and Cai H. (2012) The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 55, 2069–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheehan A. L., Carrell S., Johnson B., Stanic B., Banfi B., and Miller F. J. Jr. (2011) Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 216, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duerrschmidt N., Stielow C., Muller G., Pagano P. J., and Morawietz H. (2006) NO-mediated regulation of NAD(P)H oxidase by laminar shear stress in human endothelial cells. J. Physiol. 576, 557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sorescu G. P., Song H., Tressel S. L., Hwang J., Dikalov S., Smith D. A., Boyd N. L., Platt M. O., Lassègue B., Griendling K. K., and Jo H. (2004) Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ. Res. 95, 773–779 [DOI] [PubMed] [Google Scholar]
- 19. Chalupsky K., and Cai H. (2005) Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 102, 9056–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nguyen A., and Cai H. (2006) Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 6530–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai H., McNally J. S., Weber M., and Harrison D. G. (2004) Oscillatory shear stress upregulation of endothelial nitric oxide synthase requires intracellular hydrogen peroxide and CaMKII. J. Mol. Cell Cardiol. 37, 121–125 [DOI] [PubMed] [Google Scholar]
- 22. Hilenski L. L., Clempus R. E., Quinn M. T., Lambeth J. D., and Griendling K. K. (2004) Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 24, 677–683 [DOI] [PubMed] [Google Scholar]
- 23. Ushio-Fukai M. (2006) Localizing NADPH oxidase-derived ROS. Sci. STKE 2006, re8. [DOI] [PubMed] [Google Scholar]
- 24. Harrison D. G., Cai H., Landmesser U., and Griendling K. K. (2003) Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 4, 51–61 [DOI] [PubMed] [Google Scholar]
- 25. Davis M. E., Cai H., McCann L., Fukai T., and Harrison D. G. (2003) Role of c-Src in regulation of endothelial nitric oxide synthase expression during exercise training. Am. J. Physiol. Heart Circ. Physiol. 284, H1449–1453 [DOI] [PubMed] [Google Scholar]
- 26. Woodman C. R., Muller J. M., Laughlin M. H., and Price E. M. (1997) Induction of nitric oxide synthase mRNA in coronary resistance arteries isolated from exercise-trained pigs. Am. J. Physiol. 273, H2575–2579 [DOI] [PubMed] [Google Scholar]
- 27. Sessa W. C., Pritchard K., Seyedi N., Wang J., and Hintze T. H. (1994) Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ. Res. 74, 349–353 [DOI] [PubMed] [Google Scholar]
- 28. Dimmeler S., Fleming I., Fisslthaler B., Hermann C., Busse R., and Zeiher A. M. (1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605 [DOI] [PubMed] [Google Scholar]
- 29. Cai H., Davis M. E., Drummond G. R., and Harrison D. G. (2001) Induction of endothelial NO synthase by hydrogen peroxide via a Ca2+/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 21, 1571–1576 [DOI] [PubMed] [Google Scholar]
- 30. Drummond G. R., Cai H., Davis M. E., Ramasamy S., and Harrison D. G. (2000) Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ. Res. 86, 347–354 [DOI] [PubMed] [Google Scholar]
- 31. Sartoretto J. L., Kalwa H., Pluth M. D., Lippard S. J., and Michel T. (2011) Hydrogen peroxide differentially modulates cardiac myocyte nitric oxide synthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 15792–15797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lauer N., Suvorava T., Rüther U., Jacob R., Meyer W., Harrison D. G., and Kojda G. (2005) Critical involvement of hydrogen peroxide in exercise-induced up-regulation of endothelial NO synthase. Cardiovasc. Res. 65, 254–262 [DOI] [PubMed] [Google Scholar]
- 33. Peshavariya H., Dusting G. J., Jiang F., Halmos L. R., Sobey C. G., Drummond G. R., and Selemidis S. (2009) NADPH oxidase isoform selective regulation of endothelial cell proliferation and survival. Naunyn Schmiedebergs Arch. Pharmacol. 380, 193–204 [DOI] [PubMed] [Google Scholar]
- 34. Li H., Witte K., August M., Brausch I., Gödtel-Armbrust U., Habermeier A., Closs E. I., Oelze M., Münzel T., and Förstermann U. (2006) Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 47, 2536–2544 [DOI] [PubMed] [Google Scholar]
- 35. Dikalova A. E., Góngora M. C., Harrison D. G., Lambeth J. D., Dikalov S., and Griendling K. K. (2010) Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 299, H673–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lassègue B., and Griendling K. K. (2010) NADPH oxidases: functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 30, 653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Montezano A. C., and Touyz R. M. (2014) Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid. Redox. Signal 20, 164–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Judkins C. P., Diep H., Broughton B. R., Mast A. E., Hooker E. U., Miller A. A., Selemidis S., Dusting G. J., Sobey C. G., and Drummond G. R. (2010) Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol. Heart Circ. Physiol. 298, H24–32 [DOI] [PubMed] [Google Scholar]
- 39. Kirk E. A., Dinauer M. C., Rosen H., Chait A., Heinecke J. W., and LeBoeuf R. C. (2000) Impaired superoxide production due to a deficiency in phagocyte NADPH oxidase fails to inhibit atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 20, 1529–1535 [DOI] [PubMed] [Google Scholar]
- 40. Siu K. L., Lotz C., Ping P., and Cai H. (2015) Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J. Mol. Cell Cardiol. 78, 174–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M. D., and Sadoshima J. (2010) NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 107, 15565–15570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. VanderLaan P. A., Reardon C. A., and Getz G. S. (2004) Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler. Thromb. Vasc. Biol. 24, 12–22 [DOI] [PubMed] [Google Scholar]
- 43. Lu X., and Kassab G. S. (2004) Nitric oxide is significantly reduced in ex vivo porcine arteries during reverse flow because of increased superoxide production. J. Physiol. 561, 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hsiai T. K., Hwang J., Barr M. L., Correa A., Hamilton R., Alavi M., Rouhanizadeh M., Cadenas E., and Hazen S. L. (2007) Hemodynamics influences vascular peroxynitrite formation: implication for low-density lipoprotein apo-B-100 nitration. Free Radic. Biol. Med. 42, 519–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hwang J., Ing M. H., Salazar A., Lassègue B., Griendling K., Navab M., Sevanian A., and Hsiai T. K. (2003) Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ. Res. 93, 1225–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Berliner J. A., Territo M. C., Sevanian A., Ramin S., Kim J. A., Bamshad B., Esterson M., and Fogelman A. M. (1990) Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J. Clin. Invest. 85, 1260–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chien S. (2007) Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am. J. Physiol. Heart Circ. Physiol. 292, H1209–1224 [DOI] [PubMed] [Google Scholar]
- 48. Laude K., Cai H., Fink B., Hoch N., Weber D. S., McCann L., Kojda G., Fukai T., Schmidt H. H., Dikalov S., Ramasamy S., Gamez G., Griendling K. K., and Harrison D. G. (2005) Hemodynamic and biochemical adaptations to vascular smooth muscle overexpression of p22phox in mice. Am. J. Physiol. Heart Circ. Physiol. 288, H7–12 [DOI] [PubMed] [Google Scholar]
- 49. Gray S. P., Di Marco E., Okabe J., Szyndralewiez C., Heitz F., Montezano A. C., de Haan J. B., Koulis C., El-Osta A., Andrews K. L., Chin-Dusting J. P., Touyz R. M., Wingler K., Cooper M. E., Schmidt H. H., and Jandeleit-Dahm K. A. (2013) NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127, 1888–1902 [DOI] [PubMed] [Google Scholar]
- 50. Stroes E., Kastelein J., Cosentino F., Erkelens W., Wever R., Koomans H., Lüscher T., and Rabelink T. (1997) Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J. Clin. Invest. 99, 41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heitzer T., Krohn K., Albers S., and Meinertz T. (2000) Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with type II diabetes mellitus. Diabetologia 43, 1435–1438 [DOI] [PubMed] [Google Scholar]
- 52. Gao L., Chalupsky K., Stefani E., and Cai H. (2009) Mechanistic insights into folic acid-dependent vascular protection: dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice. A Novel HPLC-based Fluorescent Assay for DHFR Activity. J. Mol. Cell. Cardiol. 47, 752–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Higashi Y., Sasaki S., Nakagawa K., Fukuda Y., Matsuura H., Oshima T., and Chayama K. (2002) Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am. J. Hypertens. 15, 326–332 [DOI] [PubMed] [Google Scholar]
- 54. Laursen J. B., Somers M., Kurz S., McCann L., Warnholtz A., Freeman B. A., Tarpey M., Fukai T., and Harrison D. G. (2001) Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103, 1282–1288 [DOI] [PubMed] [Google Scholar]
- 55. Goettsch C., Goettsch W., Brux M., Haschke C., Brunssen C., Muller G., Bornstein S. R., Duerrschmidt N., Wagner A. H., and Morawietz H. (2011) Arterial flow reduces oxidative stress via an antioxidant response element and Oct-1 binding site within the NADPH oxidase 4 promoter in endothelial cells. Basic Res. Cardiol. 106, 551–561 [DOI] [PubMed] [Google Scholar]
- 56. Schröder K., Zhang M., Benkhoff S., Mieth A., Pliquett R., Kosowski J., Kruse C., Luedike P., Michaelis U. R., Weissmann N., Dimmeler S., Shah A. M., and Brandes R. P. (2012) Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 110, 1217–1225 [DOI] [PubMed] [Google Scholar]
- 57. Altenhöfer S., Kleikers P. W., Radermacher K. A., Scheurer P., Rob Hermans J. J., Schiffers P., Ho H., Wingler K., and Schmidt H. H. (2012) The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci. 69, 2327–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]