

Abstract
Genome-wide association studies as well as murine models have shown that the interleukin 23 receptor (IL23R) pathway plays a pivotal role in chronic inflammatory diseases such as Crohn disease (CD), ulcerative colitis, psoriasis, and type 1 diabetes. Genome-wide association studies and targeted re-sequencing studies have revealed the presence of multiple potentially causal variants of the IL23R. Specifically the G149R, V362I, and R381Q IL23Rα chain variants are linked to protection against the development of Crohn disease and ulcerative colitis in humans. Moreover, the exact mechanism of action of these receptor variants has not been elucidated. We show that all three of these IL23Rα variants cause a reduction in IL23 receptor activation-mediated phosphorylation of the signal-transducing activator of transcription 3 (STAT3) and phosphorylation of signal transducing activator of transcription 4 (STAT4). The reduction in signaling is due to lower levels of cell surface receptor expression. For G149R, the receptor retention in the endoplasmic reticulum is due to an impairment of receptor maturation, whereas the R381Q and V362I variants have reduced protein stability. Finally, we demonstrate that the endogenous expression of IL23Rα protein from V362I and R381Q variants in human lymphoblastoid cell lines exhibited lower expression levels relative to susceptibility alleles. Our results suggest a convergent cause of IL23Rα variant protection against chronic inflammatory disease.
Keywords: genetic polymorphism, glycosylation, inflammatory bowel disease (IBD), protein trafficking (Golgi), receptor, interleukin 23 receptor
Introduction
Interleukin 23 receptors (IL23Rs) have been implicated in chronic inflammatory diseases through their role in initiating the differentiation of helper T cells (Th17). The Th17 pathway is important to control acute microbial infections (1–3), and deregulation of the pathway results in inflammatory bowel disease (IBD)2 (4–6). IL23R is expressed in specific subsets of T cells as well as other immune cells (7). Cells that express the IL23R polypeptide chain do not express IL12Rβ2, which is also a partner for IL12Rβ1 forming the interleukin 12 receptor (IL12R) (7). Recently, evidence has been presented that IL23R in type 3 innate lymphoid cells is involved in development of colitis through IL23 signaling (8). It is thus essential to understand how IL23Rα-protective variants affect IL23 signaling as evidence to support development of therapeutic strategies for IBD. To avoid confusion due to the nomenclature that denotes the IL23R heterodimer receptor from the individual chains, in this report the chain distinct to IL23R will be denoted as IL23Rα and the heterodimer denoted as IL23R. IL23R signals through the interaction between two receptor chains, IL23Rα and IL12Rβ1, and together signals by binding IL23 cytokine (9, 10). IL23Rα constitutively interacts with Janus kinase 2 (JAK2). Binding of IL23 by IL23R results in activation of JAK2 and subsequent phosphorylation of the IL23R followed by recruitment of STAT3 and STAT4 and their phosphorylation (9). Phosphorylated STAT3 and STAT4 both form homodimers that translocate to the nucleus to transcribe proinflammatory cytokines. The increase in transcription of proinflammatory cytokines leads to differentiation of CD4+ T cells into Th17 cells resulting in inflammation (11, 12). Multiple evidence suggests that IL23R may play a pivotal role in chronic inflammation (13–16).
Genome-wide association studies have identified potential biological pathways that are responsible for disease establishment and pathogenesis (17–20), and the IL23R gene was reported to show strong association (susceptibility or protection) with IBD (18, 21–23). IL23R-protective coding variants such as R381Q were identified by genome-wide association studies (24), whereas G149R and V362I were identified by targeted re-sequencing (20). Four notable studies were performed on IL23R variants discovered in genome-wide association studies to gain insight on the role of these variants. One study reported that a risk variant for CD and ulcerative colitis located in the 3′-untranslated region lost its binding to microRNA, Let7f, and thereby its deregulation led to an increase in mRNA and protein levels of IL23Rα. This suggests that the deregulation of IL23Rα expression could lead to a hypersensitivity of the IL23R and resultant overstimulation of the IL23R-activated signaling pathway (25). In contrast, an IL23Rα variant that has been studied in some detail is R381Q and is predicted to be a protective variant. Three studies reported that this variant results in a loss of function because cells expressing this variant show reduced levels of phosphorylated STAT3 and consequently reduced production of proinflammatory cytokines with reduced Th17 effector functions (26–28).
In this study we sought to identify the biochemical mechanisms by which IL23R variants may provide protection against IBD. The IL23R variants are positioned within distinct domains of the folded receptor. R381Q is found in the C-terminal cytoplasmic portion, V362I is in the transmembrane region, and G149R is in the extracellular domain of the receptor, suggesting that these variants will have distinct biochemical properties. We show here that IL23Rα R381Q and V362I variants have lower protein stability leading to reduced expression levels, whereas the G149R variant is retained in the endoplasmic reticulum (ER) as unfolded polypeptides.
Experimental Procedures
Antibodies and Reagents
IL23 was purchased from R&D Systems, and IL23R antibody was purchased from Abcam. Antibody to IL12Rβ1 was purchased from Santa Cruz Biotechnology, and antibody toward the FLAG tag was purchased from Sigma. Antibodies to detect ER stress proteins, phosphorylated STAT3 (Tyr-705), STAT3, phosphorylated STAT4, STAT4, and α-tubulin, purchased from Cell Signaling Technology. Transfection reagents such as X-treme gene transfection reagent were purchased from Roche Applied Science, and the TransIT-HeLa MONSTER transfection kit was purchased from Mirus Bio. Chemical compounds such as cycloheximide, brefeldin A, and tunicamycin were purchased from Sigma. Reagents for glycosidase experiments such as Endo H, peptide N-glycosidase F, and neuraminidase were purchased from New England BioLabs. Nuc Blue live cell stain (Hoechst 33342), Cell light ER-RFP (BacMam 2.0), and MTT were purchased from Molecular Probes by Life Technologies. Alexa Fluor 647 mouse IgG was purchased from Molecular Probes by Life Technologies.
Plasmid Construction
cDNAs coding for IL23Rα and IL12Rβ1 were amplified from pCDNA3.1IL23Rα and pCDNA3.1IL12Rβ1 (the Dr. Rioux laboratory). To generate IL23Rα-protective variants, pCDNA3.1IL23Rα was used as template, and the QuikChange method was performed as described by the manufacturer of the QuikChange II site-directed mutagenesis kit (Agilent Technologies) to create pCDNA3.1IL23RαR381Q, -G149R, and -V362I. To account for transfection efficiency and to approximate the expression levels of the receptors, we decided to insert fluorescent proteins Venus YFP (vYFP) and mPlum (mRFP) downstream of an IRES sequence found in the pLpC binary vector as described previously (29). The pCDNA3.1 carrying the cDNA to IL23Rα and variants was used as the template to PCR-amplify and subclone into PacI/ClaI restriction sites found pLpC + Venus retroviral vector to generate pLpCIL23Rα+vYFP, pLpCIL23RαR381Q+vYFP, pLpCIL23RαG149R+vYFP, pLpCIL23RαV362I+vYFP. To construct pLpCIL12Rβ1+mPlum, the pCDNA3.1IL12Rβ1 was used as a template to PCR-amplify and subclone into PacI/ClaI restriction sites of pLpC +mPlum vector. These vectors were used to generate stable cell lines in HEK293.
To generate N-terminal tagging of IL23Rα with 3×FLAG tags, first, 5′-phosphorylated oligonucleotides that correspond to the signal peptide of IL23Rα was annealed followed by subcloning into the BamHI/PacI restriction site of pLpC+vYFP vector to create pLpCSP+vYFP. Next, the cDNA corresponding to IL23Rα and its variants were PCR-amplified from their corresponding pCDNA3.1 templates using primers specific for 3×FLAG and IL23Rα and subcloned into PacI/ClaI restriction sites found in pLpCSP+Venus vector.
For generation of constructs to be used in a Renilla luciferase Protein-fragment Complementation Assay (Rluc PCA), first, Rluc F[1] and F[2] fragments were PCR-amplified from pCDNA3.1Reg-F[1] and pCDNA3.1Cat-F[2] (30) using primers that contain the sequence to encode a 5-amino acid GGGGS amino acid sequence (5-aa linker) followed by the sequence of Rluc F[1] and F[2]. The resulting PCR fragments were subcloned into NotI/XbaI sites in pCDNA3.1 to create pCDNA3.1- 5aa-F[1] and pCDNA3.1-5aa-F[2], respectively. To construct 10- and 20-aa linker fusions to F[1] and F[2], first Rluc F[1] and Rluc F[2] fragments were PCR-amplified from the templates mentioned above and subcloned into XhoI/XbaI sites found in pCDNA3.1 to create pCDNA3.1Rluc-F[1] and pCDNA3.1Rluc-F[2]. After this, oligonucleotides that encode 2× GGGGS and 4× GGGGS sequences were 5′-phosphorylated and then annealed and ligated into NotI/XhoI sites found in pCDNA3.1Rluc-F[1] and pCDNA3.1Rluc-F[2] to create vectors that carry both F[1] and F[2] fragments with 10- and 20-amino acid linkers upstream to it. Finally, to create Rluc fusions to IL23Rα and IL12Rβ1, primers were designed to amplify the sequence coding for the N terminus of both receptors until the end of the transmembrane region using the pCDNA3.1IL23Rα and pCDNA3.1IL12Rβ1 as templates followed by subcloning into NheI/NotI site of pCDNA3.1-5aa-F[2] and pCDN3.1-5aa-F[1] to construct pCDNA3.1IL23Rαext-5aa-F[2] and pCDNA3.1IL12Rβ1ext-5aa-F[1]. The same strategy was used for construction of IL23R Rluc PCA reporter vectors containing 10- and 20-aa linkers. The cDNA for full-length of IL23Rα and IL12Rβ1 was PCR-amplified using the templates mentioned above and subcloned into NheI/NotI sites found in pCDNA3.1-10aa-F[2] and pCDNA3.1–10aa-F[1] to construct pCDNA3.1IL323Rα-F[2] and pCDN3.1IL12Rβ1-F[1]. The IL23Rα variant fusions to Rluc F[2] were constructed in a similar fashion.
For generation of C-terminal vYFP tagging of IL23Rα (pCDNA3.1IL23Rα-Venus) and its variants, first, oligonucleotides that encode the 2× GGGGS sequence were 5′-phosphorylated and annealed followed by ligation into NotI/XhoI restriction sites of pCDNA3.1 vector to create the pCDNA3.110aa construct. The coding sequence of IL23Rα common and protective variants were PCR-amplified followed by subcloning into NheI/NotI restriction sites of the pCDNA3.110aa construct to create the pcDNA3.1IL23Rα10aa and the variant derivative constructs. Finally, the pCDNA3.1IL23Rα and variants constructs were digested using XhoI/XbaI restriction enzymes followed by ligation of DNA sequence coding for Venus fluorescent protein that was PCR-amplified from pLpC+Venus construct.
Cell Culture
Stable cell lines were generated from HEK293 cells using pLpC retroviral vector as described elsewhere (31). Both HEK293 cell lines and HeLa cells were maintained in DMEM supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 units/ml), and for propagation of stable cell lines, puromycin at 2.5 μg/ml was added. Human lymphoblastoid cell lines were obtained from the NIDDK, National Institutes of Health Central Repository (www.niddkrepository.org) and had been generated by the NIDDK Inflammatory Bowel Disease Genetics Consortium (IBDGC) study. These lymphoblastoid cell lines were maintained in RPMI with 20% FBS, penicillin (100 units/ml), and streptomycin (100 units/ml). All cell lines were propagated at 37 °C and 5% CO2.
Rluc PCA
Rluc PCA was performed as described previously (30). HEK293 cells grown in 6-well plates were transfected with pCDNA3.1 constructs encoding IL23Rα Rluc F[2] fusion and IL12Rβ1 Rluc F[1] fusions. After 24 h of transfection, cells were harvested, washed with PBS, and resuspended in 500 μl of DMEM minus phenol red. Approximately 100,000 cells were transferred to 96-well white-walled plates (Corning), and after the addition of benzyl-coelenterazine (5 μm, Nanolight) to the cells; bioluminescence was monitored by using the LMaxII384 luminometer (Molecular Devices). For detection of receptor activation by IL23, the cells were treated with IL23 per 96-well for 10 min before the addition of benzyl-coelenterazine and measurement of luminescence.
STAT3 and STAT4 Activation Assays
To study the biological importance of IL23Rα variants, STAT3 phosphorylation experiments were performed as reported recently (32). Briefly, pLpCIL23Rα + vYFP or variants of IL23Rα and pLpCIL12Rβ1 + mPlum was transfected into HeLa cells at ∼70% confluence using Trans-IT HeLa Monster transfection reagent. The next day the cells were washed three times with PBS, followed by overnight growth in DMEM without (FBS) serum. After overnight serum starvation, IL23 was added to the cells for 30 min followed by preparation of cell lysate and Western blotting. Antibodies against pSTAT3 (1/1000 dilution), STAT3 (1/5000 dilution), IL23Rα (1/100 dilution), and IL12Rβ1 (1/2500 dilution) were used in Western blotting. To probe for phosphorylation of STAT4, STAT4 cDNA (pLSV-STAT4, the Dr. Rioux laboratory) was co-transfected along with IL23Rα and IL12Rβ1 into HeLa cells, and activation experiments were performed by the same procedure used to measure STAT3 activation. Antibodies against pSTAT4 (1/1000 dilution) and STAT4 (1/5000 dilution) were used in Western blotting.
Western Blotting of Cell Lysates
Generally, after cells were harvested, the cell pellet was lysed in 250 mm Tris-HCl, pH 6.8, 5% SDS containing protease inhibitors (Roche Applied Science). The cell lysate was boiled for 10 min and centrifuged to remove any traces of cell debris, and the supernatant was used to quantify total protein concentration by BCA (Pierce). Equal amounts of proteins were separated on SDS-PAGE after Western blotting using antibodies specific to the protein of interest. The membrane was blocked with 5% skim milk in TBS-T (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Tween 20) and probed with primary and the corresponding secondary antibodies (anti-mouse-HRP and anti rabbit-HRP) in 5% skim milk in TBS-T. In the case for STAT4 and pSTAT4 antibodies, both the primary and secondary antibodies were diluted in TBST with 5% BSA. After both primary antibody (1 h at room temperature or overnight at 4 °C) and secondary antibody incubation step (1 h at room temperature), the blots were washed three times with TBS-T followed by detection using the chemiluminescent detection reagent (Luminata Forte, Millipore). This procedure was followed for analysis of cell lysates from all cell lines used in this study (HEK293 and HeLa; NIDDK) unless otherwise stated. For stability experiments, 500 μm cycloheximide was added to HEK293 cells that stably expressed IL23Rα and its variants at ∼80% confluency for 0, 0.5, 1, 2, and 4 h. Cell lysates from the stability experiments were analyzed by Western blotting as described here using IL23Rα antibody at 1/1000 dilution. Similarly, brefeldin A (BFA; 1 μg/ml) or tunicamycin (1 μg/ml) treatment of HEK293 cells stably expressing IL23Rα and its variants for 5 h at 37 °C with 5% CO2 was followed by preparation of cell lysates and analyzed by Western blotting as described here. For analysis of the rate of IL23Rα synthesis, cycloheximide was added to cells at 500 μm as stated before followed by incubation at 37 °C with 5% CO2 for 4 h. After this, the cells were washed with PBS to remove cycloheximide, and then the cells were incubated for the indicated times at 37 °C with 5% CO2. After specified time points, (0, 30, 60, 120, and 240 min) the cells were harvested, and the cell lysates were subjected to SDS-PAGE and Western blotting for IL23Rα.
Glycosidase Digestion of IL23Rα
HEK293 cells stably expressing IL23Rα and its variants were lysed with Nonidet buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40) containing 0.5% SDS, 0.04 m DTT, and protease inhibitors as described elsewhere (33). Briefly, the lysate was boiled for 10 min, and the supernatant containing soluble proteins (40 μg of total protein) were digested with either Endo H (1000 units) or with peptide N-glycosidase F (1000 units) and neuraminidase (100 units) at 37 °C for 16 h. SDS-PAGE and Western blotting of the digested products were conducted as described above.
Immunofluorescence and Confocal Microscopy
For all microscopy, HEK293 cells were seeded onto 96-well black, clear glass-bottom plates (MGB101–1-2-LG, Matrical Bioscience) followed by transfection using X-treme GENE reagent. Plasmids constructs harboring N-terminally 3×FLAG-tagged IL23Rα and its variants (e.g. pLpCSP-3×FLAGIL23Rα+vYFP) were transfected, and the following day the cells were prepared for immunofluorescence as described elsewhere (34, 35). The cells were washed with ice-cold PBS followed by gentle fixation by 15 min of incubation on ice with 1.25% formaldehyde. After removal of the formaldehyde and washing with PBS, the cells were blocked with 5% goat serum in PBS for 30 min. After the blocking step, the cells were incubated with FLAG-tag antibody (mouse, 1/100 dilution) diluted in 3% skimmed milk for 2 h at room temperature or overnight at 4 °C. The cells were washed with PBS followed by incubation with Alexa Fluor 647 goat anti-mouse (1/100 dilution) diluted in 3% skimmed milk for 1 h at room temperature. The cells were then washed with PBS followed by confocal imaging using an INCell 6000 confocal microscope (GE Healthcare). Cells were imaged in PBS with NucBlue stain (2 drops/ml of PBS) for nuclear staining. For detection of the nucleus, cells were excited at 405 nm, and emission was measured at 455/50 nm; to detect membrane bound receptors, cells were excited at 642 nm, and emission was measured at 706.5/70 nm. To control for transfection and to normalize for expression level of IL23Rα and its variants, vYFP fluorescence was imaged at 488-nm excitation and 525/20-nm emission wavelengths. Images were processed using Metamorph software (version 7.5.3, MDS Analytical Technologies). Images were thresholded followed by creation of binary masks. The binary masks were used in integrated morphometry analysis to quantify the total average fluorescence intensity of the population of cells in the field of view.
For detection of IL23Rα-vYFP fusions, HEK293 cells were transfected with pCDNA3.1IL23Rα-vYFP or its variants, and 6 h after transfection cells were incubated with BacMam 2.0 ER-RFP viruses (Life Technologies), which encode the ER signal peptide fused to the red fluorescent protein (RFP) to measure co-localization between IL23Rα and ER. The cells were maintained in DMEM minus phenol red, NucBlue stain was added (2 drops/ml), and image acquisition was performed using INCell6000 (GE Healthcare) as stated above. To detect the RFP, cells were irradiated at 561-nm excitation and detected at 605/52-nm emission. Image analysis was performed using Metamorph software (version 7.5.3, MDS Analytical Technologies). To quantify the level of IL23Rα retained in the ER, the fluorescent images from ER localization was thresholded, and binary masks were generated to identify the ER, which were then used in integrated morphometry analysis to measure the fluorescent intensities from vYFP fluorescent proteins that are C-terminally tagged to the IL23Rα. The ratios between the total IL23Rα levels and ER retained were calculated.
Results
IL23R Quaternary Structure and Activation Is Similar to Erythropoietin Receptor (EpoR)
IL23R is grouped under cytokine class I receptor superfamily, which also includes EpoR. Both receptors contain the WSXWS motif in the extracellular N-terminal domain, a single transmembrane hydrophobic domain, and a cytoplasmic C-terminal domain. Binding of Epo was shown to induce a scissor-like conformational change at the interface of the EpoR dimer that bring together JAK2 kinases associated with the EpoR cytosolic domains to allow for cross-phosphorylation and activation (Fig. 1A) (36, 37). IL23R is composed of IL23Rα and IL12Rβ1. Heterodimerization of these two chains is essential to IL23R activation by IL23, and we hypothesize that the receptor is activated in the same manner as EpoR. To test this hypothesis, we used a Rluc PCA as a molecular ruler to measure heterodimer dynamics (Fig. 1A, middle and bottom) (36). Briefly, we fused cDNA coding for the IL23Rα and IL12Rβ1 chains to those coding for Rluc complementary N- and C-terminal fragments separated by sequences coding for 5, 10, or 20 amino acid (20 aa) peptide linkers composed of the 5-aa repeats of GGGGS (Fig. 1A). Our reasoning was that linkers of 5 aa would not be long enough for the Rluc fragments to associate and fold in the inactive form of the receptor dimer, where the C termini of the receptor chains are separated by ∼70 Å, but would be separated by ∼40 Å in the active dimer (Fig. 1A, middle; assuming the length of a peptide bond is ∼4 Å, two 5-aa peptides could traverse a maximum distance of ∼28 Å). Rluc PCA signal for the 5-aa linker was increased 4-fold after IL23 treatment but not with constructs with 10- or 20-aa linkers, consistent with what we observed with EpoR (Fig. 1B) (36). We also showed that IL23Rα and IL12Rβ1 interact with their cognate kinases JAK2 and TYK2, respectively (Fig. 1C). Finally and consistent with the EpoR activation model, when JAK2 and TYK2 were expressed as fusions to the Rluc PCA reporter fragments along with their respective IL23Rα and IL12Rβ1 binding partners, the Rluc signal increased in cells treated with IL23 (Fig. 1D) (36).
FIGURE 1.

Characterization of IL23R activation mechanism model. A, schematic representation of allosteric IL23R activation. IL23R exists as a heterodimer before interaction with IL23 cytokine similar to the EpoR receptor (36, 62). The extracellular domain exists in a conformation that keeps the intracellular domains and associated JAK2 and TYK2 separated from each other by ∼70 Å. Upon binding to IL23, the extracellular dimer is reorganized and brings the intracellular cytoplasmic domains within ∼ 40 Å of each other, allowing for interaction between and activation of their associated kinases. For identification of IL23R activation mechanism, the extracellular and transmembrane domains of IL23R and IL12Rβ1 are fused to Renilla luciferase (Rluc) complementary N- and C-terminal fragments, Rluc F[1] and F[2], via flexible linkers consisting of 1, 2, or 4 five-amino acid repeats to generate the following: IL23R and IL12Rβ1 fused to 5-, 10-, and 20-amino acid linkers that are in turn fused to the Rluc fragments. HEK293 cells transfected with these receptor fusions and PCA were detected by measuring the luminescence. When the receptors are fused via a 5-aa linker, folding of the Rluc reporter protein from the fragments only occurs for the IL23 hormone-bound active form of the receptor dimer. If receptors fused to Rluc fragments via 10- or 20-aa linkers, Rluc reporter protein can fold from the fragments in either inactive or active dimer conformations. The structural model of Renilla luciferase was rendered using information from 2pSj.pdb (63) using PyMOL 1.3. B, Rluc enzyme assay detected the IL23R activation in HEK293 cells. Cells were co-transfected with IL23Rα and IL12Rβ1 fused to Rluc PCA fragments via various linker lengths. The next day the cells were incubated with and without 100 ng of IL23 cytokine followed by measurement of Rluc enzyme activity using benzyl coelenterazine as the substrate and detection using a luminometer. The signal is expressed as relative light units (RLU). C, Rluc PCA reveals the interaction between IL23Rα and their subunits and their associated kinases (JAK2 and TYK2). HEK293T cells were transfected with pCDNA3.1 harboring the cDNA of full-length IL23R receptor subunits and the kinases JAK2 and TYK2 fused to Rluc fragments to detect the interaction as measured by PCA (RLU) of Rluc. D, JAK2 and TYK2 association in the presence of IL23 binding to IL23R was detected by Rluc PCA. Rluc fragments were fused to JAK2 and TYK2 and were co-transfected in HEK293T cells stably expressing IL23R subunits followed by measuring Rluc luminescence. Statistical significance denoted by the asterisk (*) was calculated by Student's t test, where p < 0.05. E, IL23Rα variants show the reduction in activation by IL23. IL23Rα, its, variants and IL12Rβ1 were fused to Rluc fragments to detect activation by IL23 (10 ng/ml), and luminescence was measured as in B. The change in RLU between cells not treated and treated with IL23 was calculated, and activation was thus observed as a function of change in RLU. Statistical significance denoted by the asterisk (*) was calculated by ANOVA, where p < 0.01. F, G, and H, IL23Rα variants retain their ability to interact with IL12Rβ1, JAK2, and TYK2. IL23Rα and variants Rluc fragment 2 (F[2]) fusions were co-transfected with either IL12Rβ1 fused to Rluc fragment 1 F[1], IL12Rβ1 F[1] (F), JAK2 F[1] (G), and TYK2 F[1] (H), and reconstitution of Rluc enzyme activity was measured to assess their interaction. Rluc enzyme activity (RLU) is expressed as percentage relative to IL23Rα. Data and S.D. are representative of three independent experiments.
IL23R Activation Is Reduced in Protective Variants
Having established that IL23R complex structure and activation is similar to that of EpoR, we now assessed the effects of sequence variants of IL23Rα on IL23-induced receptor conformation change. IL23Rα-protective variants are located at three distinct sites: the G149R change is found in the extracellular domain of the receptor, whereas the V362I change is located within the transmembrane domain, and the R381Q change is located at the C terminus of the receptor five amino acids from the transmembrane domain. We found that the IL23Rα variants retained their ability to interact with IL12Rβ1; however, the extent to which they are induced by IL23 was reduced compared with the common variant (Fig. 1E). Finally, interactions of IL23Rα to IL12Rβ1, JAK2, and to TYK2 were comparable for all variants (Fig. 1, F, G, and H).
Protective Variants of IL23Rα All Result in Reduced IL23R-activated STAT3 and STAT4 Phosphorylation
STAT3 is a primary effector of IL23R signaling, and to investigate IL23-dependent STAT3 phosphorylation, we co-transfected HeLa cells with a plasmid encoding the cDNA for IL23Rα or its variants together with a plasmid encoding the cDNA for IL12Rβ1 (32). After serum starvation, we measured IL23-induced STAT3 phosphorylation (pSTAT3). Cells expressing the protective R381Q, G149R, and V362I variants showed reduced pSTAT3 levels compared with cells expressing the common variant (Fig. 2, A and B). Transfection of HeLa cells with only IL12Rβ1 or together with IL23Rα, but without IL23, did not show significant STAT3 phosphorylation (Fig. 3A). Western blotting with antibodies specific to both IL23Rα and IL12Rβ1 confirmed expression of both receptor subunits (Fig. 3B). Furthermore, we investigated the effects of IL23R variants on IL23-mediated STAT4 signaling as several studies have demonstrated that IL23R subunits activate STAT4 in response to IL23 (9, 10, 38). To this end we co-transfected HeLa cells with cDNA of STAT4 with plasmid encoding the cDNA for IL23Rα or its variants together with a plasmid encoding the cDNA for IL12Rβ1. The same experimental setup was followed as stated above for STAT3 signaling. We found that the variants show reduced STAT4 phosphorylation levels compared with the common variant (Fig. 2, E and F). The reduction in signaling corroborates well with the reduced activation in protective variants by IL23 (Fig. 1E). We, therefore, turned to questions of receptor maturation and expression.
FIGURE 2.

IL23Rα protective variants R381Q, G149R, and V362I display reduction in IL23R signaling. A, HeLa cells were co-transfected with plasmids carrying IL23Rα or variants and IL12Rβ1. After washing with PBS to remove media and overnight growth in serum-free medium, transfected cells were incubated with or without IL23 (15 ng/ml) for 30 min. Cell lysates were subjected to SDS-PAGE followed by Western blotting with antibodies specific to phospho-STAT3 (pSTAT3) and STAT3. Representative blots from three independent experiments are shown here. B, corresponding densitometry of the levels of pSTAT3 in IL23Rα and IL23Rα variants obtained from Western blots above. The signal of pSTAT3 detected on Western blot was measured as pixels using ImageJ 1.46r (National Institutes of Health) and were further normalized to levels of total STAT3. Data and S.D. are representative of three independent experiments. Statistical significance calculated by ANOVA is denoted by the asterisk (*), where p < 0.01 is compared with IL23Rα; however, R381Q was significantly different from V362I, with a p < 0.05. C, HeLa cells were co-transfected with plasmids carrying STAT4, IL23Rα or variants, and IL12Rβ1. As stated for STAT3 experiments (A), the cell lysates were subjected to SDS-PAGE followed by Western blotting with antibodies specific to phospho-STAT4 (pSTAT4) and STAT4. D, corresponding densitometry of the levels of pSTAT4 in IL23Rα and IL23Rα variants obtained from Western blots of C. The signal of pSTAT4 detected on Western blot was measured as pixels using ImageJ 1.46r (National Institutes of Health) and were further normalized to levels of total STAT4. Data and S.D. are representative of three independent experiments. Statistical significance calculated by ANOVA is denoted by the asterisk (*), where p < 0.01 is compared with IL23Rα; in addition, R381Q was significantly different from G149R and V362I with a p < 0.01. EV denotes empty vector.
FIGURE 3.
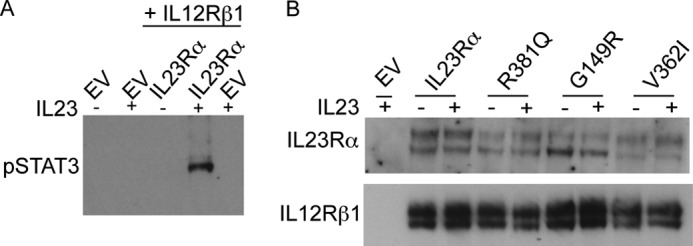
IL23-induced STAT3 phosphorylation is dependent on the presence of both IL23Rα and IL12Rβ1 and IL23 cytokine. A, HeLa cells were co-transfected with plasmids carrying either IL12Rβ1 or IL12Rβ1 together with IL23Rα. After washing with PBS to remove medium and overnight growth in serum free medium, transfected cells were incubated with or without IL23 (15 ng/ml) for 30 min. Cell lysates were subjected to SDS-PAGE followed by Western blotting with antibodies specific to phospho-STAT3 (pSTAT3) and tubulin. EV denotes empty vector. B, HeLa cells were co-transfected with plasmids carrying IL23Rα or variants and IL12Rβ1. After washing with PBS to remove medium and overnight growth in serum free medium, transfected cells were incubated with or without IL23 (15 ng/ml) for 30 min. Cell lysates were subjected to SDS-PAGE followed by Western blotting with antibodies specific to IL23Rα and IL12Rβ1.
IL23Rα, R381Q, and G149R Variants Perturb Receptor Maturation in HEK293 Cells
Both IL23Rα and IL12Rβ1 migrated as two bands (Fig. 3B), consistent with the persistent presence of early and mature glycosylated forms of these receptors (39, 40). One possible explanation for reduced activity of IL23R variants is that they could be defective in maturation. We thus examined the maturation of all IL23R variant polypeptides in HEK293 cells. We tested for mature versus early glycosylation marks of IL23Rα in stably transfected HEK293 cells. We found that the low molecular mass signal of IL23Rα was sensitive to Endo H, which cleaves N-linked glycopeptides, but the high molecular mass band was Endo H-resistant (Fig. 4A). However, treatment with peptide N-glycosidase and neuraminidase, glycosidases that together hydrolyze all types of glycosides, removed both the high and low molecular mass bands. These results suggested that both bands corresponded to glycosylated species of the IL23Rα, but the lower molecular mass form corresponded to immature receptor, and the high molecular mass corresponded to mature receptor.
FIGURE 4.

IL23Rα-protective variants display different receptor maturation. A, glycosylation states of IL23Rα stably expressed in HEK293 cells. Cell lysates were treated with Endo H or with peptide N-glycosidase and neuraminidase and resolved by SDS-PAGE and Western blot using antibodies against IL23Rα. Endo H-resistant receptors are mature in glycosylation, whereas Endo H-sensitive receptor corresponds to immature glycosylated receptor. B, Western blots of cell lysates from HEK293 transfected with IL23Rα common or protective variants. The amount of cDNA transfected is noted in parentheses. Mature (M IL23Rα) or immature IL23Rα (IM IL23Rα) receptors are indicated to the right. Molecular markers are displayed to the left of the blots in kDa. C, the ratio of mature IL23Rα (M) to immature IL23Rα (IM) was calculated. Data represented are the means ± S.D. calculated from three independent experiments. Statistical significance-calculated ANOVA is denoted by the asterisk (*), where p < 0.01.
To investigate whether there are any differences in maturation between the IL23Rα common and disease protective variants, we performed quantitative Western blotting of cell lysates from cells that transiently expressed each variant, and we determined the ratio of mature to immature receptors (Fig. 4, B and C). R381Q and G149R were reduced in their mature-to-immature receptor ratio to ∼50 and 30%, respectively. In contrast, the V362I variant showed a similar ratio to the common variant, although the protein level was much reduced (discussed further below), requiring that a higher amount of V362I cDNA had to be transfected to achieve a similar level of receptor expression as that of the common variant (Fig. 4B). Thus both R381Q and G149R displayed impairment in maturation.
IL23Rα R381Q and G149R Show Reduced Surface Expression and Impairment in ER to Golgi Trafficking
We next asked whether the impairment in maturation of R381Q and G149R variants could result in decreased cell surface expression. IL23Rα common and protective variant surface expression was quantified by immunofluorescence on HEK293 cells expressing FLAG-tag IL23Rα. The 3×FLAG sequence was fused to the N terminus of the IL23Rα receptor and its variants as there are no effective commercial antibodies available. Using antibody against the FLAG tag, surface- expressed IL23Rα were imaged by confocal microscopy and quantified by normalizing for total receptor expression (Fig. 5A). We found that relative to the common variant of IL23Rα, both R381Q and G149R were reduced in surface expression by ∼60 and 30%, respectively, consistent with the maturation data (Fig. 5B).
FIGURE 5.

Localization of IL23Rα common and protective variants in HEK293 cells. A, confocal microscopy images of cells expressing N-terminal FLAG-tagged receptors in HEK293 cells. Cells were imaged for vYFP fluorescent protein to show IL23Rα expression levels; Hoeschst (Nuc-Blue, Molecular Probes), to identify the nucleus and antibody toward FLAG tag, was used to localize IL23Rα. Overlay images (Merge) from Hoechst and anti-FLAG staining is shown to demonstrate IL23Rα localization. Scale bar, 10 μm. B, mean fluorescent intensities from the corresponding confocal images were used to quantify IL23Rα, which were normalized to the total expression of IL23Rα and its variants using levels of vYFP fluorescent protein as described under “Experimental Procedures.” Different fields of images were quantified, and S.E. values are n = 3. Statistical significance calculated by ANOVA is denoted by asterisk (*), where p < 0.01 compared with IL23Rα.R381Q was significantly different from G149R and V362I, and G149R is significantly different from V362I with a p < 0.01. C, localization of IL23Rα variant-vYFP fusions; green, IL23Rα; red, ER. Hoeschst staining was used to identify the nucleus, and cell light ER-RFP was used to identify ER. Scale bar, 10 μm. D, ratio of total fluorescence from v-YFP-tagged IL23Rα and ER-localized v-YFP-tagged IL23Rα is calculated as described under “Experimental Procedures.” On average, 10 cells were quantified for each variant, and the S.E. was calculated. Statistical significance calculated by ANOVA is denoted by the asterisk (*) compared with IL23Rα, where p < 0.05; in addition, R381Q was significantly different from G149R, with a p < 0.05, and G149R was significantly different from V362I, with a p < 0.01.
It is generally accepted that immaturely glycosylated forms of proteins are mostly retained in the ER/cis-Golgi, whereas mature proteins (both N- and O- glycosylated) are found in post-Golgi and other compartments (40). We tested for ER retention of IL23Rα common and protective variants fused to vYFP (41) (C terminus to receptor) and quantified expression in the ER using confocal microscopy. Co-localization with ER (ER retention signal-mRFP) with IL23Rα-vYFP revealed that a fraction of the receptors are retained within the ER (Fig. 5C), and quantification of ER-retained IL23Rα revealed that variants R381Q and G149R display higher retention in the ER than the common variant (Fig. 5D). These findings further confirmed that R381Q and G149R maturation is impaired in ER-Golgi trafficking.
The glycosidase tests strongly suggest that ER retention is reflected in failed completion of glycosylation. It is, however, also possible that the R381Q and G149R variants also had immature O-glycosylation. To test for differences in O-glycosylation, HEK293 cells expressing stable IL23Rα and its variants were treated with BFA, which fuses the ER and Golgi membranes, thereby allowing for the partial redistribution of enzymes such as O-GlcNAc transferase found in the Golgi membranes to the ER (42). BFA treatment resulted in intermediate molecular weight products that migrated between the mature and immature IL23Rα receptor, the novel species of signal named aberrantly glycosylated receptor (Fig. 6). The ability of all the IL23Rα variants showing this aberrantly glycosylation confirms that these variants are able to undergo O-glycosylation when they interact with the necessary enzymes. Alternatively, proteins are also retained in the ER when they do not pass through the quality check by the ER resident chaperones (43).
FIGURE 6.
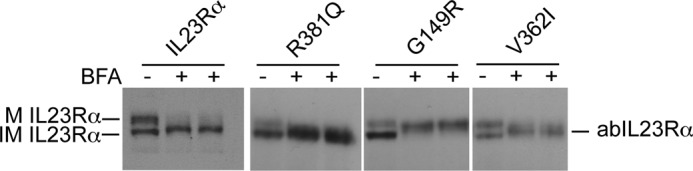
BFA treatment of HEK293 cells stably expressing IL23Rα and variants results in partial O-glycosylation. HEK293 cells expressing IL23Rα and its variants when exposed to BFA (1 μg/ml) for 5 h is shown. Cell lysates from two independent experiments were subjected to SDS-PAGE, and Western blotting with antibodies to IL23Rα show that the immature receptors in both wild type and variants are able to be partially O-glycosylated when the responsible enzymes found in Golgi are available to them. Arrows indicate the mature (M IL23Rα) and immature (IM IL23Rα) and the aberrant IL23Rα (abIL23Rα) that is partially O-glycosylated.
IL23Rα G149R Expressing HEK293 Cells Display an Increase in Unfolded Protein Response
To determine whether the IL23Rα-protective variants cause disturbances to the homeostasis of the ER, we probed the cell lysates from HEK293 cell lines stably expressing IL23Rα and its variants for accumulation of proteins involved in the unfolded protein response (43). Expression levels of Ero1Lα (endoplasmic reticulum oxidoreductase α), CCAT/enhancer binding protein homologous protein (CHOP), PDI (protein disulfide isomerase), BiP, IRE1α (inositol-requiring enzyme 1 α), and calnexin were analyzed by Western blotting. Increased levels of ER chaperone immunoglobulin-binding protein (BiP) was identified in the G149R variant (Fig. 7A). BiP induction in the absence of CHOP, which is associated with induction of cell death (43), suggests that the ER stress induced with the G149R variant is most likely due to protein misfolding/accumulation of unfolded polypeptides. To confirm that the viability of cells expressing IL23Rα-protective variants was not affected, we assayed for mitochondrial dehydrogenase activity in the IL23Rα common and protective variants using the MTT assay. The IL23Rα common and protective variant-expressing cells exhibited similar levels of cell viability (Fig. 7B). Therefore, G149R variant is severely affected in receptor maturation, and its accumulation in ER increases the induction of BiP without causing cell death (44).
FIGURE 7.
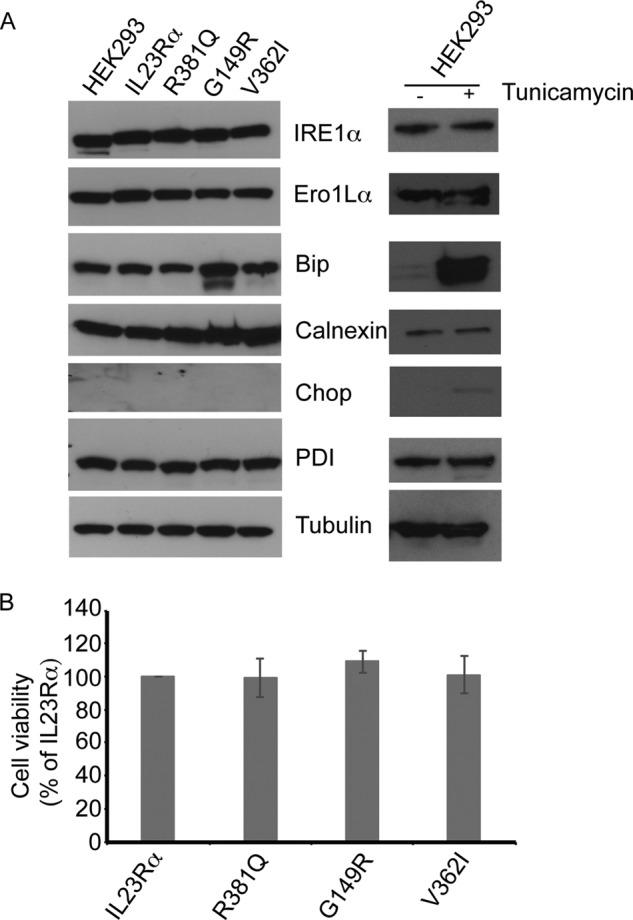
Unfolded protein response is up-regulated in cells expressing the IL23Rα G149R variant. A, HEK293 cells stably expressing IL23Rα and its variants were harvested, and cell lysates were electrophoresed on SDS-PAGE followed by Western blot. Antibodies specific to ER stress and unfolded protein response markers indicate that BiP abundance is higher in IL23Rα G149R compared with the common variant, suggesting that IL23Rα G149R is retained in the ER. BiP abundance could also be induced in HEK293 cells expressing IL23Rα when these cells are treated with tunicamycin (1 μg/ml) for 5 h. B, the viability of cells expressing IL23Rα variants is not affected. HEK293 cells stably expressing IL23Rα and variants were plated in 96-well plate (20,000 cells) in triplicate. The next day the cells were assayed for levels of mitochondrial dehydrogenase activity by using MTT (64). Data and error bars represent the S.D. from three independent experiments.
IL23Rα R381Q and V362I Show Reduction in Stability
The stability of IL23Rα and its variants was assessed by pulse-chase experiments after cycloheximide inhibition of de novo protein synthesis and detection of protein at different times by Western blotting (Fig. 8A). There is a reduction in both mature and immature IL23Rα over time, but R381Q and V362I degrade more rapidly (Fig. 8, A and B). IL23Rα had a half-life of ∼187 min, and R381Q and V362I had a half-life of 16 and 72 min, respectively (Fig. 8C). Having measured the turnover of IL23Rα common and protective variants, we turned to investigating the de novo synthesis of the receptors. Cells were treated for 4 h with cycloheximide, during which a large amount of IL23Rα was degraded, after which cycloheximide was removed by washing the cells with PBS. This was followed by monitoring the rate of mature and immature IL23Rα synthesis at various time points by Western blot using anti-IL23Rα antibodies (Fig. 9A). Interestingly, the rate of de novo mature receptor synthesis was markedly reduced for R381Q and G149R variants compared with rates for the common and V362I variants (Fig. 9, A and B). The rate of synthesis of mature versus immature IL23Rα receptor was 1:1 for the common and V362I-protective variant, whereas this ratio was severely reduced for the protective variants R381Q and G149R (Fig. 9C). These results provide further evidence that trafficking is affected in the protective variants R381Q and G149R; hence, there are fewer receptors at the cell surface. The V362I variant, however, showed a higher turnover rate than that of the common variant even if the rate of synthesis of mature versus immature receptor was similar for both. Thus, cells expressing the V362I variant appeared to have lower levels of both mature and immature forms of the receptor. To assess whether this holds true for endogenous expression of the common IL23Rα variant, as well as R381Q and V362I variants, we used lymphoblastoid cell lines derived from individuals where these haplotypes were present. Western blotting of cell lysates from these cell lines revealed that R381Q (homozygote and heterozygote) and V362I (homozygote) show a reduction in expression of the receptor at basal levels (Fig. 10). Taken together, these data demonstrate that both transfected and endogenous protein expression of IL23Rα-protective variants leads to a reduced expression relative to the common variant, suggesting a loss of function for protective variants.
FIGURE 8.

IL23Rα R381Q and V362I were not stable. A, HEK293 cells stably expressing IL23Rα and IL23Rα variants were treated with cycloheximide (500 μm) for various times. Representative Western blot analysis of the cell lysate is shown here. B, IL23Rα mature (M) and immature (IM) levels over time under cycloheximide exposure are represented in percentages, which were calculated by taking IL23Rα levels at time 0 as 100%. One-phase exponential decay fit of the data was performed using GraphPad Prism 6.0, and the R2 value for the receptors are as follows: IL23Rα M (0.7212), IL23Rα IM (0.654), R381Q M (0.7460), R381Q IM (0.8905), G149R M (0.7655), G149R IM (0.876), V362I M (0.9559), V362I IM (0.9388). Error values were S.E. where n = 3. C. Half-life of IL23Rα and variants was calculated by using GraphPad Prism 6.0. Error values were S.D. where n = 3. Statistical significance calculated by ANOVA is denoted by the asterisk (*) compared with IL23Rα where p < 0.01; in addition, R381Q was significantly different from G149R and V362I with a p < 0.01, and G149R was significantly different from V362I with a p < 0.01.
FIGURE 9.

The rates of maturation of R381Q and G149R are reduced compared with that of the common variant IL23Rα. A, HEK293 cells stably expressing IL23Rα and IL23Rα variants were treated with cycloheximide (CLX, 500 μm) for 4 h followed by washing off the cycloheximide and incubated for the indicated time points. Representative Western blot analysis of the cell lysates is presented. B, synthesis of IL23Rα mature (M) and immature (IM) levels over time after cycloheximide exposure are represented in pixels. Linear regression analysis of the data were used to calculate the rate using GraphPad Prism 6.0, and the R2 value for the receptors are as follows: IL23Rα M (0.8591), IL23Rα IM (0.7436), R381Q M (0.9256), R381Q IM (0.9054), G149R M (0.8357), G149R IM (0.9543), V362I M (0.8428), V362I IM (0.8117). The error values are S.E. where n = 3. C, ratio of the rate for IL23Rα and variants between their mature and immature receptor synthesis is represented here. Error values are S.D. where n = 3. Statistical significance calculated by ANOVA is denoted by the asterisk (*) compared with IL23Rα, where p < 0.01. In addition, R381Q was significantly different from V362I, with a p < 0.01, and G149R was significantly different from V362I, with a p < 0.01.
FIGURE 10.
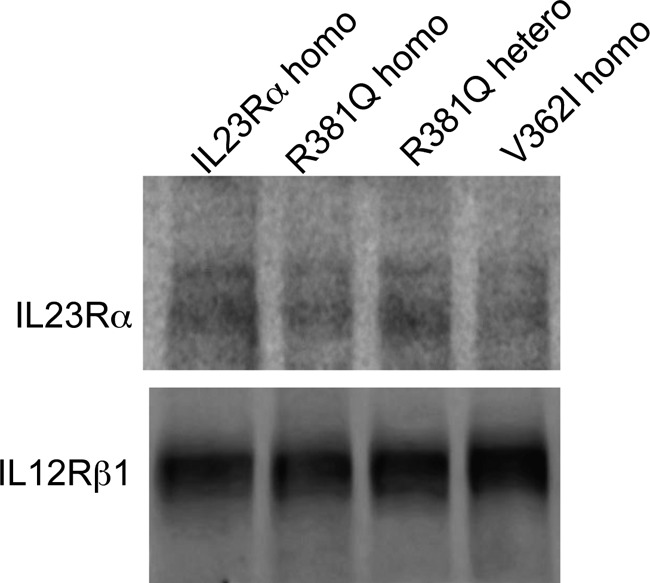
Expression levels of IL23Rα-protective variants are lower in NIDDK cell lines. Western blotting of cell lysates from NIDDK cell lines shows the expression of IL23Rα and its variants R381Q and V362I at endogenous levels using antibodies specific to IL23Rα. IL12Rβ1 antibody was used for the loading control. Representative Western blot from three independent experiments is shown, and cells that are homozygous and heterozygous to the IL23Rα allele are described as homo and hetero, respectively.
Discussion
Our results suggest that all protective coding variants of IL23Rα result in reduced cell surface expression of mature receptors due either to reduced stability and or impaired trafficking from ER. In all three cases reduced surface expression resulted in reduced IL23-mediated signaling. Previous studies showed that the IL23Rα R381Q variant displayed reduced signaling; however, mechanistic details of loss of activity were not explored (26–28). Our results are consistent with these findings. We now extend this observation to two new coding variants of IL23Rα, all described as protective variants in inflammatory bowel disease. Moreover, we provide clear mechanistic details of loss of activity for the three protective variants, all of which result in reduced cell surface expression of mature receptors due either to reduced stability and or impaired trafficking from ER. In all three cases, reduced surface expression resulted in reduced IL23-mediated signaling, such that all protective variants negatively impact IL-23 signaling.
The mutations encoding R381Q, G149R, and V362I are located in different regions of the IL23Rα receptor structure close to the transmembrane domain (C terminus of the receptor), extracellular region, and transmembrane domain, respectively. It is expected, therefore, that although the consequences of the mutations are identical, the mechanisms of reduced expression are different. We observed that the variants R381Q and G149R, unlike V362I, displayed a reduction in the mature/immature receptor ratio, and both R381Q and G149R variants were reduced in surface expression where signaling via IL23 is initiated and G149R is impaired in ER to Golgi trafficking. Similar observations have been made in other studies with variants of proteins or receptors that undergo maturation from ER to Golgi and were affected in their arrival at their destined location such as the missense mutation in very low density lipoprotein receptor, cystic fibrosis transmembrane receptor, and amyloid precursor protein (APP) (44–50). For instance, the results obtained here are reminiscent of the triple variant of APP that displayed impairment in APP maturation and trafficking from ER-Golgi. As reported for IL23Rα G149R variant, the APP triple variant also showed an increase in the level of BiP, as it was retained in the ER as misfolded polypeptide without inducing cell death. In addition, it was reduced in protein stability (44). However, unlike the APP triple variant, the IL23Rα G149R variant was not reduced in its stability. In fact, our cycloheximide studies revealed that the R381Q variant and to a lesser extent the V362I variant were unstable. The lower levels of expression of these two receptor variants in endogenously expressing cell lines further support this notion. In summary, the protective effect by the variants R381Q and V362I is due to protein instability leading to lower levels of total receptor expression coupled with a reduction in signaling. The V362I variant is unique in that the ratio of mature-to-immature protein levels is comparable with that of the common variant. However, the V362I variant exhibits a reduced half-life and presents with lower levels of endogenous receptor expression in NIDDK V362I homozygous cells.
A question that still remains is: Why would the G149R variant be unfolded in the ER but its stability not affected? A possible explanation for the observed ER retention of the G149R variant may reside in the observation that G149R creates a cleavage site, RX(L/V)X, where X is any amino acid, for PCSK-SkI-1, a convertase that is found to reside in the trans Golgi (51, 52). It is possible that the G149R variant is translocated to the Golgi, but the variant receptor may get cleaved at the extracellular domain by the convertases in the Golgi, resulting in an increase in receptor truncations and degradation.
In summary, the R381Q variant displays a higher turnover rate resulting in lower levels of receptor expression at the cell surface. This may be attributed to the fact that residue 381 of the IL23Rα chain is the fifth amino acid after the transmembrane domain. It is generally understood that if residues in this region are positively charged (positive inside rule), the proteins do not get translocated but do get inserted into the membrane (53, 54), and so mutations here may affect membrane protein insertion. The G149R variant primarily remains in the ER and does not get translocated to the plasma membrane surface, perhaps due to changes in its structure and/or due to its being a substrate for SKI-1 convertase in the Golgi. In the future, it would be interesting to obtain crystal structures of the IL23Rα receptor, which may provide some insight into how the R381Q affects protein insertion. Finally, the V362I substitution is located within the transmembrane domain of the receptor. It is possible that the added bulk of a methyl group to the side chain at this position may disrupt shape complementarity and, therefore, destabilize the interaction between IL23Rα and IL12Rβ1 receptor transmembrane domains but maintain peptide chain rigidity due to branching at the β-carbon as has been shown to be essential for other transmembrane protein-protein interactions (55). Thus the three protective variants, although found at distinct regions of the receptor, through different mechanisms may be expressed at lower levels at the plasma membrane surface, resulting in receptor-mediated signaling and thereby play a protective role in IBD.
Our results would appear to be consistent with the fact that antibodies against IL23 hormone itself present a relevant therapeutic strategy for treating IBD (56). We would argue that any strategy that directly inhibits IL23 receptor signaling could be of even greater potential therapeutic value. Targeted antagonists of IL23 receptor subunit assembly or competitive inhibitors of IL23R could prove superior therapeutics, as unlike IL23 antibodies, the number of receptors is likely much lower than that of IL23 hormone, and thus therapeutic levels of IL23R-acting agents would be lower and more effective. For instance, compounds that target protein-protein interaction for development of therapeutics has been successful in a number of cases (57, 58). As examples, competitors of IL2-IL2Rα and BcL-2/BcL-xL interactions have been identified using fragment based approach, and the compounds bind to a groove that locks the conformation of the protein and prevents its interaction with its natural partners (57). Alternatively, we have reported an allosteric peptide inhibitor of IL23R function that, in contrast to anti-p40 IL23 hormone subunit, selectively inhibits IL-23-induced inflammation in three in vivo mouse models (59). In addition, development of structure-specific synthetic antibodies could prove another alternative to developing functionally antagonistic or competitive inhibitors of IL23R function (60).
Finally our observations that the IL23R appears to be activated in a similar manner to erythropoietin receptor provides a mechanistic target for therapeutic development, and the Rluc PCA we report here or a version based on our latest infrared fluorescent protein PCA can provide an unambiguous readout for screening any class of molecules for antagonists of IL23R receptor function (29, 30, 36, 61). Inhibition of IL-23 signaling may thus have a positive impact in preventing disease onset.
Author Contributions
D. S., and S. M. designed the research. D. S. performed the research. C. B., C. Q., and J. L. contributed reagents and tools for research. D. S., S. L., S. C., J. D. R., and S. W. M. analyzed the data. D. S., and S. W. M. wrote the paper. J. L., C. B., C. Q, S. C., S. L., J. D. R., D. S., and S. W. M. participated in discussions and revised the paper.
Acknowledgments
We thank Debasmita Talapatra for assisting with experiments. We also thank the laboratory of Dr. Gerardo Ferberye for the pLpC binary vector and Monique Vasseur for help with microscopy. We extend our appreciation to Veronique Bourdeau, Emmanuelle Quierdo, Jacqueline Kowarzyk Moreno, Vincent Messier, and Jean-Francois Paradis for discussions. We also thank Dr. Nabil G. Sediah for identifying the possibility of G149R as a substrate for convertase in Golgi. We thank Katherine Harding for help with editing of the manuscript and discussion. The NIDDK Inflammatory Bowel Disease Genetics Consortium (IBDGC) study was conducted by the IBDGC Investigators and supported by the NIDDK, National Institutes of Health. The lymphoblastoid cell lines and related data from the Inflammatory Bowel Disease Genetics Consortium reported here were supplied by the NIDDK Central Repositories.
This work was supported by an award (CIHR #GPG-102170) from the Canadian Institutes of Health Research to the “CIHR Emerging Team in Integrative Biology of Inflammatory Diseases” (to J. D. R., S. C., S. L., and S. M.) with co-funding from the Crohn's and Colitis Foundation of Canada (made possible by a generous donation from the Ross McMaster Memorial Fund) in addition by an award (FRSQ#17199) from the Pfizer-FRSQ Innovation Fund (to J. D. R., S. C., S. L., and S. M.) and by an award from the Natural Science and Engineering Research Council (Discovery Grant 2015-06298; to J. L.). The authors declare that they have no conflicts of interest with the contents of this article. Other than J. D. R., this manuscript was not prepared in collaboration with Investigators of the Inflammatory Bowel Disease Genetics Consortium (IBDGC) study and does not necessarily reflect the opinions or views of the IBDGC study, the NIDDK Central Repositories, or the NIDDK.
- IBD
- inflammatory bowel disease
- IL12R
- interleukin 12 receptor
- CD
- Crohn disease
- ER
- endoplasmic reticulum
- Endo H
- endoglycosidase H
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- vYFP
- Venus YFP
- aa
- amino acid(s)
- Rluc PCA
- Renilla luciferase protein-fragment complementation assay
- BFA
- brefeldin A
- RFP
- red fluorescent protein
- EpoR
- erythropoietin receptor
- APP
- amyloid precursor protein
- ANOVA
- analysis of variance.
References
- 1. Cho J. H. (2008) The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 8, 458–466 [DOI] [PubMed] [Google Scholar]
- 2. Abraham C., and Cho J. (2009) Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 15, 1090–1100 [DOI] [PubMed] [Google Scholar]
- 3. McKenzie B. S., Kastelein R. A., and Cua D. J. (2006) Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 27, 17–23 [DOI] [PubMed] [Google Scholar]
- 4. Hue S., Ahern P., Buonocore S., Kullberg M. C., Cua D. J., McKenzie B. S., Powrie F., and Maloy K. J. (2006) Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yen D., Cheung J., Scheerens H., Poulet F., McClanahan T., McKenzie B., Kleinschek M. A., Owyang A., Mattson J., Blumenschein W., Murphy E., Sathe M., Cua D. J., Kastelein R. A., and Rennick D. (2006) IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116, 1310–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Awasthi A., Riol-Blanco L., Jäger A., Korn T., Pot C., Galileos G., Bettelli E., Kuchroo V. K., and Oukka M. (2009) Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J. Immunol. 182, 5904–5908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chognard G., Bellemare L., Pelletier A. N., Dominguez-Punaro M. C., Beauchamp C., Guyon M. J., Charron G., Morin N., Sivanesan D., Kuchroo V., Xavier R., Michnick S. W., Chemtob S., Rioux J. D., and Lesage S. (2014) the dichotomous pattern of IL-12r and IL-23R expression elucidates the role of IL-12 and IL-23 in inflammation. PLoS ONE 9, e89092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eken A., Singh A. K., Treuting P. M., and Oukka M. (2014) IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal. Immunol. 7, 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parham C., Chirica M., Timans J., Vaisberg E., Travis M., Cheung J., Pflanz S., Zhang R., Singh K. P., Vega F., To W., Wagner J., O'Farrell A. M., McClanahan T., Zurawski S., Hannum C., Gorman D., Rennick D. M., Kastelein R. A., de Waal Malefyt R., and Moore K. W. (2002) A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 168, 5699–5708 [DOI] [PubMed] [Google Scholar]
- 10. Oppmann B., Lesley R., Blom B., Timans J. C., Xu Y., Hunte B., Vega F., Yu N., Wang J., Singh K., Zonin F., Vaisberg E., Churakova T., Liu M., Gorman D., Wagner J., Zurawski S., Liu Y., Abrams J. S., Moore K. W., Rennick D., de Waal-Malefyt R., Hannum C., Bazan J. F., and Kastelein R. A. (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725 [DOI] [PubMed] [Google Scholar]
- 11. Budarf M. L., Labbé C., David G., and Rioux J. D. (2009) GWA studies: rewriting the story of IBD. Trends Genet. 25, 137–146 [DOI] [PubMed] [Google Scholar]
- 12. Ouyang W., and Valdez P. (2008) IL-22 in mucosal immunity. Mucosal. Immunol. 1, 335–338 [DOI] [PubMed] [Google Scholar]
- 13. Cua D. J., Sherlock J., Chen Y., Murphy C. A., Joyce B., Seymour B., Lucian L., To W., Kwan S., Churakova T., Zurawski S., Wiekowski M., Lira S. A., Gorman D., Kastelein R. A., and Sedgwick J. D. (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748 [DOI] [PubMed] [Google Scholar]
- 14. McGeachy M. J., Chen Y., Tato C. M., Laurence A., Joyce-Shaikh B., Blumenschein W. M., McClanahan T. K., O'Shea J. J., and Cua D. J. (2009) The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 10, 314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo W., Luo C., Wang C., Zhu Y., Wang X., Gao X., and Yao W. (2012) Protection against Th17 cells differentiation by an interleukin-23 receptor cytokine-binding homology region. PLoS ONE 7, e45625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Langrish C. L., Chen Y., Blumenschein W. M., Mattson J., Basham B., Sedgwick J. D., McClanahan T., Kastelein R. A., and Cua D. J. (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Franke A., Balschun T., Sina C., Ellinghaus D., Häsler R., Mayr G., Albrecht M., Wittig M., Buchert E., Nikolaus S., Gieger C., Wichmann H. E., Sventoraityte J., Kupcinskas L., Onnie C. M., Gazouli M., Anagnou N. P., Strachan D., McArdle W. L., Mathew C. G., Rutgeerts P., Vermeire S., Vatn M. H., IBSEN study group, Krawczak M., Rosenstiel P., Karlsen T. H., and Schreiber S. (2010) Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL). Nat. Genet. 42, 292–294 [DOI] [PubMed] [Google Scholar]
- 18. Duerr R. H., Taylor K. D., Brant S. R., Rioux J. D., Silverberg M. S., Daly M. J., Steinhart A. H., Abraham C., Regueiro M., Griffiths A., Dassopoulos T., Bitton A., Yang H., Targan S., Datta L. W., Kistner E. O., Schumm L. P., Lee A. T., Gregersen P. K., Barmada M. M., Rotter J. I., Nicolae D. L., and Cho J. H. (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barrett J. C., Hansoul S., Nicolae D. L., Cho J. H., Duerr R. H., Rioux J. D., Brant S. R., Silverberg M. S., Taylor K. D., Barmada M. M., Bitton A., Dassopoulos T., Datta L. W., Green T., Griffiths A. M., Kistner E. O., Murtha M. T., Regueiro M. D., Rotter J. I., Schumm L. P., Steinhart A. H., Targan S. R., Xavier R. J., NIDDK IBD Genetics Consortium, Libioulle C., Sandor C., Lathrop M., Belaiche J., Dewit O., Gut I., Heath S., Laukens D., Mni M., Rutgeerts P., Van Gossum A., Zelenika D., Franchimont D., Hugot J. P., de Vos M., Vermeire S., Louis E., Belgian-French IBD Consortium, Wellcome Trust Case Control Consortium, Cardon L. R., Anderson C. A., Drummond H., Nimmo E., Ahmad T., Prescott N. J., Onnie C. M., Fisher S. A., Marchini J., Ghori J., Bumpstead S., Gwilliam R., Tremelling M., Deloukas P., Mansfield J., Jewell D., Satsangi J., Mathew C. G., Parkes M., Georges M., and Daly M. J. (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 40, 955–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beaudoin M., Goyette P., Boucher G., Lo K. S., Rivas M. A., Stevens C., Alikashani A., Ladouceur M., Ellinghaus D., Törkvist L., Goel G., Lagacé C., Annese V., Bitton A., Begun J., Brant S. R., Bresso F., Cho J. H., Duerr R. H., Halfvarson J., McGovern D. P., Radford-Smith G., Schreiber S., Schumm P. L., Sharma Y., Silverberg M. S., Weersma R. K., Quebec IBD Genetics Consortium, NIDDK IBD Genetics Consortium, International IBD Genetics Consortium, D'Amato M., Vermeire S., Franke A., Lettre G., Xavier R. J., Daly M. J., and Rioux J. D. (2013) Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 9, e1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amre D. K., Mack D., Israel D., Morgan K., Lambrette P., Law L., Grimard G., Deslandres C., Krupoves A., Bucionis V., Costea I., Bissonauth V., Feguery H., D'Souza S., Levy E., and Seidman E. G. (2008) Association between genetic variants in the IL-23R gene and early-onset Crohn's disease: results from a case-control and family-based study among Canadian children. Am. J. Gastroenterol. 103, 615–620 [DOI] [PubMed] [Google Scholar]
- 22. Capon F., Di Meglio P., Szaub J., Prescott N. J., Dunster C., Baumber L., Timms K., Gutin A., Abkevic V., Burden A. D., Lanchbury J., Barker J. N., Trembath R. C., and Nestle F. O. (2007) Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum. Genet. 122, 201–206 [DOI] [PubMed] [Google Scholar]
- 23. Rioux J. D., Xavier R. J., Taylor K. D., Silverberg M. S., Goyette P., Huett A., Green T., Kuballa P., Barmada M. M., Datta L. W., Shugart Y. Y., Griffiths A. M., Targan S. R., Ippoliti A. F., Bernard E. J., Mei L., Nicolae D. L., Regueiro M., Schumm L. P., Steinhart A. H., Rotter J. I., Duerr R. H., Cho J. H., Daly M. J., and Brant S. R. (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silverberg M. S., Cho J. H., Rioux J. D., McGovern D. P., Wu J., Annese V., Achkar J. P., Goyette P., Scott R., Xu W., Barmada M. M., Klei L., Daly M. J., Abraham C., Bayless T. M., Bossa F., Griffiths A. M., Ippoliti A. F., Lahaie R. G., Latiano A., Paré P., Proctor D. D., Regueiro M. D., Steinhart A. H., Targan S. R., Schumm L. P., Kistner E. O., Lee A. T., Gregersen P. K., Rotter J. I., Brant S. R., Taylor K. D., Roeder K., and Duerr R. H. (2009) Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat. Genet. 41, 216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zwiers A., Kraal L., van de Pouw Kraan T. C., Wurdinger T., Bouma G., and Kraal G. (2012) Cutting edge: a variant of the IL-23R gene associated with inflammatory bowel disease induces loss of microRNA regulation and enhanced protein production. J. Immunol. 188, 1573–1577 [DOI] [PubMed] [Google Scholar]
- 26. Sarin R., Wu X., and Abraham C. (2011) Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc. Natl. Acad. Sci. U.S.A. 108, 9560–9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pidasheva S., Trifari S., Phillips A., Hackney J. A., Ma Y., Smith A., Sohn S. J., Spits H., Little R. D., Behrens T. W., Honigberg L., Ghilardi N., and Clark H. F. (2011) Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS ONE 6, e25038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Meglio P., Di Cesare A., Laggner U., Chu C. C., Napolitano L., Villanova F., Tosi I., Capon F., Trembath R. C., Peris K., and Nestle F. O. (2011) The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE 6, e17160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tchekanda E., Sivanesan D., and Michnick S. W. (2014) An infrared reporter to detect spatiotemporal dynamics of protein-protein interactions. Nat. Methods 11, 641–644 [DOI] [PubMed] [Google Scholar]
- 30. Stefan E., Aquin S., Berger N., Landry C. R., Nyfeler B., Bouvier M., and Michnick S. W. (2007) Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 16916–16921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Serrano M., Lin A. W., McCurrach M. E., Beach D., and Lowe S. W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 [DOI] [PubMed] [Google Scholar]
- 32. Floss D. M., Mrotzek S., Klöcker T., Schröder J., Grötzinger J., Rose-John S., and Scheller J. (2013) Identification of canonical tyrosine-dependent and non-canonical tyrosine-independent STAT3 activation sites in the intracellular domain of the interleukin 23 receptor. J. Biol. Chem. 288, 19386–19400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tong W., Sulahian R., Gross A. W., Hendon N., Lodish H. F., and Huang L. J. (2006) The membrane-proximal region of the thrombopoietin receptor confers its high surface expression by JAK2-dependent and -independent mechanisms. J. Biol. Chem. 281, 38930–38940 [DOI] [PubMed] [Google Scholar]
- 34. Roy S., Rached M., and Gallo-Payet N. (2007) Differential regulation of the human adrenocorticotropin receptor [melanocortin-2 receptor (MC2R)] by human MC2R accessory protein isoforms α and β in isogenic human embryonic kidney 293 cells. Mol. Endocrinol. 21, 1656–1669 [DOI] [PubMed] [Google Scholar]
- 35. Roy S., Perron B., and Gallo-Payet N. (2010) Role of asparagine-linked glycosylation in cell surface expression and function of the human adrenocorticotropin receptor (melanocortin 2 receptor) in 293/FRT cells. Endocrinology 151, 660–670 [DOI] [PubMed] [Google Scholar]
- 36. Remy I., Wilson I. A., and Michnick S. W. (1999) Erythropoietin receptor activation by a ligand-induced conformation change. Science 283, 990–993 [DOI] [PubMed] [Google Scholar]
- 37. Lamon S., and Russell A. P. (2013) The role and regulation of erythropoietin (EPO) and its receptor in skeletal muscle: how much do we really know? Front Physiol. 4, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Glosson-Byers N. L., Sehra S., and Kaplan M. H. (2014) STAT4 is required for IL-23 responsiveness in Th17 memory cells and NKT cells. JAKSTAT 3, e955393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wheatley M., and Hawtin S. R. (1999) Glycosylation of G-protein-coupled receptors for hormones central to normal reproductive functioning: its occurrence and role. Hum. Reprod. Update 5, 356–364 [DOI] [PubMed] [Google Scholar]
- 40. Hebert D. N., Garman S. C., and Molinari M. (2005) The glycan code of the endoplasmic reticulum: asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 15, 364–370 [DOI] [PubMed] [Google Scholar]
- 41. Nagai T., Ibata K., Park E. S., Kubota M., Mikoshiba K., and Miyawaki A. (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20, 87–90 [DOI] [PubMed] [Google Scholar]
- 42. Caporaso G. L., Gandy S. E., Buxbaum J. D., and Greengard P. (1992) Chloroquine inhibits intracellular degradation but not secretion of Alzheimer β/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 89, 2252–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 44. Spoerri L., Vella L. J., Pham C. L., Barnham K. J., and Cappai R. (2012) The amyloid precursor protein copper binding domain histidine residues 149 and 151 mediate APP stability and metabolism. J. Biol. Chem. 287, 26840–26853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., and Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 46. Bonaventure J., Gibbs L., Horne W. C., and Baron R. (2007) The localization of FGFR3 mutations causing thanatophoric dysplasia type I differentially affects phosphorylation, processing and ubiquitylation of the receptor. FEBS J. 274, 3078–3093 [DOI] [PubMed] [Google Scholar]
- 47. Eugène E., Depienne C., Baulac S., Baulac M., Fritschy J. M., Le Guern E., Miles R., and Poncer J. C. (2007) GABA(A) receptor γ2 subunit mutations linked to human epileptic syndromes differentially affect phasic and tonic inhibition. J. Neurosci. 27, 14108–14116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vogt G., Bustamante J., Chapgier A., Feinberg J., Boisson Dupuis S., Picard C., Mahlaoui N., Gineau L., Alcaïs A., Lamaze C., Puck J. M., de Saint Basile G., Khayat C. D., Mikhael R., and Casanova J. L. (2008) Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J. Exp. Med. 205, 1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ali B. R., Ben-Rebeh I., John A., Akawi N. A., Milhem R. M., Al-Shehhi N. A., Al-Ameri M. M., Al-Shamisi S. A., and Al-Gazali L. (2011) Endoplasmic reticulum quality control is involved in the mechanism of endoglin-mediated hereditary haemorrhagic telangiectasia. PLoS ONE 6, e26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kizhakkedath P., Loregger A., John A., Bleijlevens B., Al-Blooshi A. S., Al-Hosani A. H., Al-Nuaimi A. M., Al-Gazali L., Zelcer N., and Ali B. R. (2014) Impaired trafficking of the very low density lipoprotein receptor caused by missense mutations associated with dysequilibrium syndrome. Biochim. Biophys. Acta 1843, 2871–2877 [DOI] [PubMed] [Google Scholar]
- 51. Seidah N. G., Sadr M. S., Chrétien M., and Mbikay M. (2013) The multifaceted proprotein convertases: their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 288, 21473–21481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Seidah N. G. (2011) The proprotein convertases, 20 years later. Methods Mol. Biol. 768, 23–57 [DOI] [PubMed] [Google Scholar]
- 53. van Geest M., and Lolkema J. S. (2000) Membrane topology and insertion of membrane proteins: search for topogenic signals. Microbiol. Mol. Biol. Rev. 64, 13–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. von Heijne G. (1992) Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225, 487–494 [DOI] [PubMed] [Google Scholar]
- 55. Heim E. N., Marston J. L., Federman R. S., Edwards A. P., Karabadzhak A. G., Petti L. M., Engelman D. M., and DiMaio D. (2015) Biologically active LIL proteins built with minimal chemical diversity. Proc. Natl. Acad. Sci. U.S.A. 112, E4717–E4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Niederreiter L., Adolph T. E., and Kaser A. (2013) Anti-IL-12/23 in Crohn's disease: bench and bedside. Curr. Drug Targets 14, 1379–1384 [DOI] [PubMed] [Google Scholar]
- 57. Wells J. A., and McClendon C. L. (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009 [DOI] [PubMed] [Google Scholar]
- 58. Arkin M. R., and Wells J. A. (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 3, 301–317 [DOI] [PubMed] [Google Scholar]
- 59. Quiniou C., Domínguez-Punaro M., Cloutier F., Erfani A., Ennaciri J., Sivanesan D., Sanchez M., Chognard G., Hou X., Rivera J. C., Beauchamp C., Charron G., Vilquin M., Kuchroo V., Michnick S., Rioux J. D., Lesage S., and Chemtob S. (2014) Specific targeting of the IL-23 receptor, using a novel small peptide noncompetitive antagonist, decreases the inflammatory response. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R1216–R1230 [DOI] [PubMed] [Google Scholar]
- 60. Reshetnyak A. V., Nelson B., Shi X., Boggon T. J., Pavlenco A., Mandel-Bausch E. M., Tome F., Suzuki Y., Sidhu S. S., Lax I., and Schlessinger J. (2013) Structural basis for KIT receptor tyrosine kinase inhibition by antibodies targeting the D4 membrane-proximal region. Proc. Natl. Acad. Sci. U.S.A. 110, 17832–17837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stefan E., Malleshaiah M. K., Breton B., Ear P. H., Bachmann V., Beyermann M., Bouvier M., and Michnick S. W. (2011) PKA regulatory subunits mediate synergy among conserved G-protein-coupled receptor cascades. Nat. Commun. 2, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Livnah O., Stura E. A., Middleton S. A., Johnson D. L., Jolliffe L. K., and Wilson I. A. (1999) Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 283, 987–990 [DOI] [PubMed] [Google Scholar]
- 63. Loening A. M., Fenn T. D., and Gambhir S. S. (2007) Crystal structures of the luciferase and green fluorescent protein from Renilla reniformis. J. Mol. Biol. 374, 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhao X.-q., Wang T.-x., Liu W., Wang C.-d., Wang D., Shang T., Shen L.-h., and Ren L. (2011) Multifunctional Au@IPN-pNIPAAm nanogels for cancer cell imaging and combined chemo-photothermal treatment. J. Mater. Chem. 21, 7240–7247 [Google Scholar]