

Abstract
The signaling pathways involved in the generation and maintenance of exocrine gland acinar cells have not yet been established. Primary human salivary gland epithelial cells, derived from salivary gland biopsies, acquired an acinar-like phenotype when the [Ca2+] in the serum-free medium (keratinocyte growth medium, KGM) was increased from 0.05 mm (KGM-L) to 1.2 mm (KGM-H). Here we examined the mechanism underlying this Ca2+-dependent generation of the acinar cell phenotype. Compared with cells in KGM-L, those in KGM-H display enhancement of Orai1, STIM1, STIM2, and nuclear factor of activated T cells 1 (NFAT1) expression together with an increase in store-operated Ca2+ entry (SOCE), SOCE-dependent nuclear translocation of pGFP-NFAT1, and NFAT-dependent but not NFκB-dependent gene expression. Importantly, AQP5, an acinar-specific protein critical for function, is up-regulated in KGM-H via SOCE/NFAT-dependent gene expression. We identified critical NFAT binding motifs in the AQP5 promoter that are involved in Ca2+-dependent up-regulation of AQP5. These important findings reveal that the Ca2+-induced switch of salivary epithelial cells to an acinar-like phenotype involves remodeling of SOCE and NFAT signaling, which together control the expression of proteins critically relevant for acinar cell function. Our data provide a novel strategy for generating and maintaining acinar cells in culture.
Keywords: calcium release-activated calcium channel protein 1 (ORAI1), gene expression, NFAT transcription factor, stromal interaction molecule 1 (STIM1), water channel
Introduction
Fluid secretion from salivary glands is initiated by a neurotransmitter-stimulated elevation of cytosolic [Ca2+] ([Ca2+]i) in acinar cells, the primary site of fluid secretion in the gland (1–4). This increase in [Ca2+]i is dependent on a Ca2+ entry mechanism called store-operated Ca2+ entry (SOCE)3 and provides crucial Ca2+ signals that are required for activation of critical ion channels such as Ca2+-activated K+ channels (5) and Cl− channels (TMEM16A) as well as increasing Na+/K+/2Cl− co-transporter activity. The net result of this is the generation of an osmotic gradient that drives fluid secretion of the cell via an apically localized water channel, AQP5, which is a marker protein for salivary acinar cells (3, 6). Physiologically, SOCE is activated in response to the release of Ca2+ from the endoplasmic reticulum by inositol 1,4,5-trisphosphate generated by neurotransmitter stimulation of acinar cells. In salivary gland cells, SOCE involves activation of the plasma membrane Ca2+ channels Orai1 and TRPC1 by the endoplasmic reticulum-Ca2+ sensor protein STIM1 (7–9).
It is now well established that changes in [Ca2+]i acutely regulate physiological functions in acinar cells. However, long-term effects of Ca2+ signals on cellular processes such as regulation of gene expression have not yet been described in this cell type. Ca2+-dependent regulation of gene expression has an important role in cell proliferation and differentiation in a number of other cell types (10). The nuclear factor of activated T cells (NFAT) family is a well characterized and critical group of Ca2+-dependent transcription factors (11, 12). NFAT1 plays a critical role in the activation and differentiation of not only T cells but also in other immune cells, such as dendritic cells, B cells, and megakaryocytes (13–16). There is strong evidence that Ca2+ entry via SOCE is a key trigger for activation of NFAT1, resulting in the binding of Ca2+ to calmodulin, which subsequently leads to the activation of calcineurin and dephosphorylation of inactive NFAT in the cytosol. Dephosphorylated NFAT1 translocates from the cytosol into the nucleus, where it binds to specific promoter regions in genes and regulates their expression. Other factors that mediate Ca2+-dependent gene expression include cAMP response element-binding protein, serum response factor, and NFκB (17, 18).
Studies to delineate intracellular signaling mechanisms involved in salivary gland development, disease, and dysfunction have been hampered by the lack of functional cultures of salivary gland acinar cells. Unlike dispersed pancreatic acini, those from salivary glands dedifferentiate in a matter of hours. Although a few studies have described successful cultures of primary epithelial cells from salivary gland explants (19–22), there has been little success in maintaining the acinar phenotype or preserving the functionality of the cells in culture (23). In our previous study (24), we described optimal conditions for maintaining primary human salivary gland (phSG) cell cultures derived from biopsies of human salivary glands and for promoting an acinar-like phenotype with enhanced expression of acinar-specific proteins (AQP5, NKCC1, and CST3) but not ductal cell markers (KLK1 and KRT19). Importantly, we found that the [Ca2+] in the culture medium modulates the phenotypic change of the cells with a relatively high, more physiological [Ca2+] (0.8–1.2 mm), triggering the acinus-like phenotype, which included an increase in acinar cell markers as well as vectorial secretion of amylase upon stimulation. Furthermore, the expression levels of Orai1, STIM1, and STIM2, key proteins involved in salivary gland physiology and Ca2+ signaling, were increased in cells that were maintained in medium with a higher [Ca2+] (24).
On the basis of these previous findings, we hypothesized that the switch in the cellular phenotype triggered by extracellular [Ca2+] could be mediated via changes in intracellular Ca2+ signaling events likely associated with SOCE. In this study, we examined SOCE and SOCE-dependent gene expression in phSG cells maintained in medium containing 0.05 or 1.2 mm Ca2+, with particular focus on the regulation of acinar marker protein AQP5 expression. We report here that up-regulation of the SOCE proteins Orai1, STIM1, and STIM2 in cells cultured with the higher [Ca2+] condition contributes to an enhancement of SOCE in these cells. Importantly, the increase in SOCE accounts for the enhancement in NFAT activation and NFAT-dependent gene expression. Finally, we present data to show that regulation of AQP5 expression is dependent on Orai1+STIM-SOCE and is mediated by NFAT1-dependent regulation of the AQP5 promoter. Together, the findings suggest that SOCE, via the Orai1/STIM1/STIM2 proteins, governs AQP5 expression and thus regulates a critical process in the generation of functional acinar cells. There has been relatively little success in establishing primary cultures of salivary gland acinar cells that recapitulate the characteristics of the cell in vivo. We propose that modulation of SOCE and SOCE-dependent Ca2+ signaling might provide a unique approach for the generation of functionally relevant acinar cells. This could have potentially important therapeutic significance for restoring the functionality of acinar cells in salivary glands patients with xerostomia.
Experimental Procedures
Cell Culture, Western Blotting, and Immunofluorescence
Primary human salivary gland epithelial cells were isolated and grown on collagen-coated plates (BioCoat, BD Biosciences) as described previously (24). Briefly, phSG cells were maintained in complete KGM (Lonza) supplemented with bovine pituitary extracts, recombinant human EGF, insulin, hydrocortisone, gentamicin, epinephrine, and transferrin, and the calcium concentration was adjusted to 0.05 mm with CaCl2 solution. For the calcium-dependent gene expression study, the indicated final calcium concentration in completed KGM was adjusted with calcium chloride, and the phSG cultures were maintained for 3 days before isolation of total RNAs or cellular protein extracts. For Western blotting, total cellular extracts were prepared as described previously and equal amounts of protein lysates were resolved in 4–12% NuPAGE gel (Invitrogen), transferred to PVDF membranes, and probed with the corresponding primary antibodies against specific proteins overnight at 4 °C. The primary antibodies used included anti-α-amylase (D55H10, Cell Signaling Technology), anti-AQP5 antibody (H-200, Santa Cruz Biotechnology), anti-cystatin C antibody (Abcam), anti-STIM1 antibody (Cell Signaling Technology), and anti-β-actin (Sigma). After washing three times with TBST (TBS with 0.05% Tween 20), the membranes were incubated with HRP-conjugated secondary antibody for 1 h at room temperature and then washed three times with PBST. The complexes were detected with SuperSignal West Pico chemiluminescent substrate (Pierce) and exposed to x-ray films (MR, Kodak). The immunofluorescence staining was performed as described previously (24). The antibodies used were AQP5, cystatin C, and an Alexa Fluor 594-conjugated goat anti-rabbit antibody (Molecular Probes). Nuclei were counterstained with DAPI in mounting solution (Vector).
Plasmid Construction and Site-directed Mutagenesis
A 1.8-kb DNA fragment from the human AQP5 upstream promoter region was amplified by PCR using human genomic DNA as a template and primers with SacI and XhoI linkers attached (forward primer, 5′-GAACTGGAGGCGAGCTCAGCAGCAAAGGAGC (SacI is underlined); reverse primer, 5′-CTTCACTCCGACCTCGAGGCGTCTAGCTCCGC (XhoI is underlined)). The PCR products were digested with SacI/XhoI, gel-cleaned, and cloned into the SacI/XhoI sites of the pGL3-Basic luciferase reporter plasmid (Promega) to generate pAQP5 (F1) plasmid. This plasmid was used as a template for subsequent PCR amplification and obtained various lengths of shorter AQP5 promoter fragments for plasmids of pAQP5 (F2), pAQP5 (F3), and pAQP5 (F4), as shown in the figure legends. To generate the mutation plasmid carrying two putative NFAT motifs (at −75 and −287 upstream of the transcription start site of the AQP5 gene) in the context of the pAQP5 (F4) construct, a site-directed mutagenesis kit (QuikChange, Stratagene) was used together with oligonucleotides of 5′-CGCAGGAGtAcAAGGAGGAGCTGG (the NFAT core binding motif (−75) is underlined, and the mutation is lowercase) and 5′-TGCCCACTTgTaCCAAAACGCCAGC (the NFAT core binding motif (−287) is underlined, and the mutation is lowercase) to make the pAQP5(F4/N1) and pAQP5(F4/N2) plasmids, respectively. All PCR fragments and the presence of the mutated sequences were confirmed by DNA sequencing.
RNA Interference (siRNA), Luciferase Assays, and Quantitative Real-time PCR
RNA interference (siRNA) was applied to knock down the transcripts of interest. The siRNAs of NFATC2, Orail1, STIM1, and STIM2 were obtained from Qiagen. The indicated siRNAs (50 nm) were transfected into phSG cells using HiPerFect transfection reagent (Qiagen) following the protocol of the manufacturer. All Star negative control siRNA (Qiagen) was included as a control. 48 h post-transfection, total RNAs were isolated as described above, and expression of AQP5 was monitored by real-time PCR. All luciferase plasmids described below were co-transfected with the pRL-TK plasmid (Promega) using Lipofectamine 2000 (Life Technologies) in KGM-L (without antibiotics). Luciferase constructs containing NFAT response motifs (pGL-4.30, Promega) or NFκB response motifs (pGL-4.32, Promega) were transfected into phSG cells for 16 h and then switched to either KGM-L or KGM-H for another 6 or 8 h before cells were lysed for the luciferase assay. For luciferase constructs that contained various truncations of the AQP5 promoter region, the duration of transfection was 8 h before the medium was changed to either KGH-L or KGM-H. Cells were maintained in their respective media until 48 h post-transfection, when the cells were lysed for the luciferase assay. Briefly, total cellular lysates were prepared by treating the cells with lysis buffer (Promega) and harvested according to the protocol of the manufacturer. Firefly and Renilla luciferase activity was determined using the Dual-Luciferase reagent assay system (Promega) and a Fluostar Omega microplate reader (BMG Labtech).
For the quantitative real-time PCR study, cells were transfected with the indicated siRNA (50 nm) using HiPerFect reagent for 48 h, and total RNAs were isolated using the miRCURY RNA isolation kit (Exiqon). Total RNAs were reverse-transcribed into cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the instructions of the manufacturer. Quantitative real-time PCR was performed using the indicated TaqMan probes with a cDNA template (100 ng of each) and run on StepOnePlus (Applied Biosystems). Triplicate reactions were carried out for each sample at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. GAPDH was used as an internal control for normalization, and the difference of the cycle threshold (Ct) of the gene of interest was calculated with the ΔCt method (ΔCt = (Ctsample − CtGAPDH)) and used to determine the relative quantitation values (2−−ΔΔCt), which represent the relative level of -fold change over control. For each study, three independent experiments with duplicate samples were performed.
Monitoring NFAT Translocation into the Nucleus
Primary phSG cells were transfected with either the AdCMV-GFP or AdCMV-GFP-NFAT1 construct for 24 h. Nucleus translocation of NFAT was imaged following stimulation with thapsigargin (1 μm) in standard extracellular solution containing 1 mm CaCl2 (25). The expressed GFP proteins were excited using a 488-nm laser, and the emission was detected using the 525-band pass emission filter. The GFP signal was observed using an Olympus IX51 microscope (Olympus) with a ×20 fluorescence objective lens. The signal was collected by a CoolSNAP HQ2 camera (Photometrics) using MetaFluor imaging software (Molecular Devices, Sunnyvale, CA). Quantification of the GFP signal in the cell nucleus and cytosol was done using MetaMorph imaging software. Briefly, regions of interest were drawn within the nucleus and cytosol. Fluorescence intensity was measured in arbitrary units and plotted using Origin graphing software (version 2015).
Measurement of Ca2+ Responses
The phSG cells were incubated with Fura-2/AM (1 μm) in its growth medium for 30 min in a 37 °C CO2 incubator. These cultures were then washed twice with standard extracellular solution, and measurements were conducted in standard extracellular solution with or without calcium. Thapsigargin (Tg, 1 μm) was added as indicated in the figures, either alone or followed by subsequent addition of CaCl2 (1 mm) to the medium. The fluorescence signal of Fura-2 was recorded using an Olympus IX51 microscope operated using Metafluor imaging software (Molecular Devices), with a Till Photonics-Polychrome V spectrofluorimeter as the light source for ratiometric excitation at 340 and 380 nm and a CoolSNAP HQ2 camera (Photometrics) to capture the emission at 510 nm (using a Fura-2 filter cube, Chroma Technology).
Electrophysiology
Cells were cultured on coverslips, transferred to the recording chamber, and perfused with a standard external solution (145 mm NaCl, 5 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 10 mm Hepes, and 10 mm glucose (pH 7.4) (NaOH)). The patch pipette had resistances between 3 and 5 mΩ after filling with the standard intracellular solution that contained the following: 145 mm cesium methane sulfonate, 8 mm NaCl, 10 mm MgCl2, 10 mm Hepes, and 10 mm EGTA (pH 7.2) (cesium hydroxide). Whole cell patch clamp experiments were performed in standard whole cell configuration at room temperature (22–25 °C) using an Axopatch 200B amplifier (Molecular Devices). Generation of the current was assessed by the amplitude at −80 mV, taken from the currents recorded during voltage ramps ranging from −90–90 mV over a period of 1 s imposed every 2 s (the holding potential was 0 mV) and digitized at a rate of 1 kHz. Liquid junction potentials were <8 mV and were not corrected. Capacitive currents and series resistance were determined and minimized. For analysis, the current recorded during the first ramp was used for leak subtraction of the subsequent current records.
ChIP Assays
Cultures of phSG cells grown in 150-mm culture dishes with 90% confluence were treated with phorbol 12-myristate 13-acetate (PMA, 10 ng/ml) and ionomycin (0.1 μm) with or without cyclosporin A (CsA, 10 nm) for 45 min. The medium was replaced with fresh growth medium containing 1% formaldehyde and incubated for 10 min to cross-link DNA and proteins. ChIP assays were conducted using the ChIP-IT Express kit (Active Motif) following the protocol of the manufacturer. In short, the nuclear extracts were isolated and sonicated on ice to achieve chromatin fragments ∼400 bp in length. After centrifugation for 10 min at 18,000 × g at 4 °C, the supernatant was collected and used for immunoprecipitation. One-tenth of each chromatin solution was used as input control. Ten micrograms of sheared chromatin were incubated with 4 μg of NFAT1 antibody (sc-7296X, Santa Cruz Biotechnology) and protein G magnetic beads at 4 °C overnight. Normal mouse IgG (Santa Cruz Biotechnology) was used as a control.
The immunocomplexes were washed, eluted, reverse-cross-linked, and treated with proteinase K (0.5 μg/μl), and then DNA was collected for the PCR reaction. Primers used to amplified the −75 and −287 region of the NFAT putative binding motifs were 5′-ACCGCCCTGCAGGACCCAGC (forward) and 5′-TGCGCGGCCTTCCCCTTTTCT (reverse) and 5′-TACCGGGCGTCGAGGATTGC (forward) and 5′-GCGCCGCTCACCTCCCTCA (reverse), respectively. PCR was conducted at one cycle of 94 °C for 2 min, 35 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s; and one cycle of 72 °C for 5 min, and the products were run on 1.5% agarose gels.
Statistical Analysis
Statistical significance was evaluated using Student's t test.
Results
Switching from Low to Relatively High [Ca2+]-containing Medium Increases the Expression of AQP5 and Critical Ca2+ Signaling Proteins
Primary human salivary gland cells obtained from biopsies of minor salivary glands were maintained in modified KGM medium as described earlier (24). The cells can be maintained in KGM containing low [Ca2+] (0.05 mm, KGM-L) for up to 8–10 passages. However, increasing the [Ca2+] in the external medium to 1.2 mm, a more physiologically relevant condition (KGM-H), induces an acinar cell-like phenotype of the cells with up-regulation of AQP5 expression (24). To determine the underlying mechanism involved in this Ca2+-dependent up-regulation of acinar cell-specific proteins, we examined the transcript levels of various proteins that could contribute to the Ca2+-dependent switch in cell phenotype and gene expression. Consistent with our previous findings, increasing the [Ca2+] in the cellular growth medium from 0.5 to 1.2 mm induced a significant increase (about 3-fold) in the level of AQP5 transcript (Fig. 1A) but not that of cystatin 3 (CST3), another marker for acinar cells. Importantly, there were significant increases in the transcript levels of Orai1, STIM1, and STIM2, which are critical proteins involved in the regulation of SOCE. Because NFAT1 is an important mediator of Ca2+-dependent gene regulation, we also examined the effect of KGM-H on its expression. Surprisingly, NFAT transcript was also increased about 2-fold. Thus, there appears to be a remodeling of the basic Ca2+ signaling toolkit that is involved in Ca2+-dependent gene expression. These findings were validated by Western blotting (Fig. 1B), and, consistent with the increase in the levels of transcripts, expression of STIM1, STIM2, and Orai1 proteins was also increased in KGM-H (1.2 mm Ca2+) relative to that in KGM-L (0.05 mm, -fold changes in proteins are given in the figure legends). Finally, phSG cells in KGM-H showed a relatively higher expression of AQP5 than cells in KGM-L (Fig. 1, C and D). However, consistent with the transcript levels of CST3, there was no difference between the level of this protein in cells grown in KGM-L and KGM-H (-fold changes are given in the figure legends).
FIGURE 1.

Ca2+-dependent up-regulation of key Ca2+ signaling proteins and AQP5 in phSG cells. phSG cells cultured from minor salivary gland biopsies as described under “Experimental Procedures” were maintained in KGM-L. Cells were shifted to 1.2 mm [Ca2+]-containing medium for 3 days before use. A, quantitative real-time PCR analysis (see “Experimental Procedures” for details) of gene expression in KGM-L and KGM-H. Expression of NFAT, AQP5, Orai1, STIM1, STIM2, and CST3 is shown relative to their level in KGM-L medium. The data are mean ± S.E. from triplicate samples from three different experiments. B, representative Western blotting analysis showing levels of Orai1, STIM1, and STIM2 in cells maintained in KGM-H and KGM-L media. Average -fold changes (mean ± S.E.) of proteins in KGM-H versus KGM-L are STIM2 = 2.1 + 0.2; STIM1 = 1.8 + 0.2; and Orail = 3.2 + 0.3. p < 0.05 (data obtained from three experiments). C, immunofluorescence detection of AQP5 (a and b) and CST3 (c and d) in phSG cultures grown in KGM-L and KGM-H media. Scale bar = 50 μm. D, Western blotting analysis showing levels of AQP5 and CST3 in phSG cells in both media. Average -fold changes (mean ± S.E.) in KGM-H versus KGM-L were AQP5 = 3.5 + 0.4 and CST3 = 0.8 + 0.3. p < 0.05 (data obtained from three experiments).
NFAT Activation Is Enhanced in phSG Cells in High [Ca2+] Medium
SOCE mediated by the Orai1-STIM1 complex generates intracellular Ca2+ signals that trigger Ca2+-calmodulin-dependent activation of the phosphatase calcineurin, leading to dephosphorylation and activation of NFAT (26, 27). On the basis of the increased expression of NFAT as well as SOCE proteins, we examined SOCE-dependent activation of NFAT. GFP-NFAT was expressed in phSG cells grown in KGM-L and KGM-H, and nuclear translocation was triggered by treating cells with Tg (1 μm) to stimulate SOCE. As shown in Fig. 2, A–C, Tg induced a robust translocation of NFAT from the cytosol into the nucleus. This response was greater and occurred at a faster rate in cells maintained in KGM-H compared with those in KGM-L. The difference in efficiency of NFAT translocation can be explained by the difference in the levels of Tg-stimulated [Ca2+]i increases in the two sets of cells (Fig. 2, D and E). Although Tg stimulated a sustained [Ca2+]i increase in both sets of cells, the magnitude was significantly lower in cells grown in KGM-L than in KGM-H. This might be due to lower levels of expression of the key SOCE components (Orai1, STIM1, and STIM2) in phSG cells in KGM-L compared with those in KGM-H. Note that although cells were grown in different media, the assays described above in both cases were performed in regular Hanks' balanced salt solution medium containing 1.2 mm Ca2+.
FIGURE 2.

NFAT1 activation and NFAT-dependent regulation of gene expression are increased by switching phSG cells to KGM-H medium. A and B, AdCMVGFP-NFAT1 (7.5 × 106 MOI) was used to mediate expression of NFAT1 in phSG cells. Cells were infected with the vector and maintained in either high (A) or low (B) calcium medium for 48 h. Cells were treated with Tg (1 μm), and GFP-NFAT fluorescence was measured in the cytosol (insets) and nucleus for the indicated time period. Values are shown relative to the initial fluorescence in these two cellular areas. C, the average intensity of the GFP signal in the nuclei in cells in KGM-H and KGM-L recorded at 30 min was calculated as (F30 − F0)/F0. The number of cells examined was as indicated. D, Tg-stimulated [Ca2+]i increases in cells maintained in KGM-L or KGM-H. E, bar graph showing the amplitude of sustained [Ca2+]i at t = 400 s in both sets of cells. Amplitude was calculated as (F400 − F0) in arbitrary fluorescence units. F, NFAT-RE-Luc (pGL-4.30, white) or NFκB-RE-Luc (pGL-4.32, black) was co-transfected with TK-Ren into phSG cells for 16 h before the medium was replaced with either KGM-L or KGM-H. Cells were then further incubated for another 6 or 8 h prior to luciferase measurements (see “Experimental Procedures” for details of the assay). *, p < 0.05. G, effect of knocking down endogenous NFAT on NFAT-Luc activity. Cells were co-transfected with the NFAT-Luc plasmid and either the scrambled siRNA (siN.C., white) or siNFAT1 siRNA (black) in KGM-L or KGM-H medium for 48 h. Data are shown relative to activity in control (cells transfected with siN.C. in KGM-L). Data are mean ± S.E. from triplicate samples of three separate experiments. *, p < 0.05. H, Tg stimulation of NFAT-dependent gene expression in phSG cells in KGM-H. Cells expressing NFAT-Luc were treated with (black) or without (white) Tg (1 μm) for 30 min, washed, and maintained in KGM-H for 16 h before being used in the luciferase assay. *, p < 0.05.
NFAT-dependent gene expression was further examined by transfecting cells with a plasmid encoding the luciferase gene driven by a promoter containing NFAT response elements (pGL-4.30). Because both NFAT and NFκB signaling pathways can be activated by changes in SOCE-dependent increases in [Ca2+]i (28, 29), the contribution of NFκB-dependent gene expression was determined by using a plasmid encoding the luciferase gene driven by a promoter containing NF-κB response elements (pGL-4.32). phSG cells in KGM-H or KGM-L were transfected with both plasmids. Fig. 2F shows that although the luciferase activity driven by the NFAT-binding promoter was greater (70% and 120% at 6 and 8 h, respectively) in KGM-H medium (cf. activity in KGM-L), the NFκB-driven promoter activity was not significantly affected by the ambient [Ca2+] nor the experimental time period. Note that cells were transfected with the luciferase constructs while in low [Ca2+] medium and maintained in the same medium for 16 h, after which they either remained in KGM-L or shifted to KGM-H. Thus the 6-h time point in KGM-L, the steady-state value in this medium, is the level of NFAT and NFκB promoter activities at the time of switch to KGM-H. Fig. 2G demonstrates that the activity of NFAT-driven luciferase is dependent on endogenous NFAT because knockdown of endogenous NFAT significantly decreased NFAT-dependent gene expression in both sets of cells. Additionally, consistent with the robust nuclear translocation of NFAT in response to Tg stimulation of cells in KGM-H, NFAT-dependent luciferase activity was also enhanced about 2-fold by Tg treatment (Fig. 2H).
Store-operated Ca2+ Entry Is Increased When phSG Cells Are Switched from Low to Physiological [Ca2+]-containing Medium
SOCE was measured in phSG cells grown in KGM-H and KGM-L media (as noted above, the assay medium was similar in both cases). Tg stimulation resulted in intracellular Ca2+ release and Ca2+ entry in both sets of cells (Fig. 3, A and C). Furthermore, although intracellular release was minimally affected by knockdown of Orai1 and STIM1, Ca2+ entry was substantially and significantly reduced by knockdown of both proteins (Fig. 3, A–D). As suggested by the data in Fig. 2, the Ca2+ entry component was significantly smaller in cells maintained in KGM-L medium compared with those in KGM-H (0.173 ± 0.003 versus 0.339 ± 0.007, p < 0.05). Because STIM2 was also increased by switching phSG cells to KGM-H medium and has been associated with SOCE, its contribution SOCE was examined in cells in KGM-H. Knockdown of STIM2 also induced a decrease in SOCE (Fig. 3, C and D). Together, these data demonstrate that Orai1, STIM1, and STIM2 contribute to the enhanced SOCE seen in phSG cells maintained in KGM-H. It has been well established that NFAT activation is dependent on SOCE, specifically on Ca2+ entry via active Orai1 channels. Thus, we suggest that the higher level of NFAT nuclear translocation as well as NFAT-dependent gene expression seen in cells grown in KGM-H is due to enhanced SOCE. The exact contributions of STIM1 and STIM2 in the regulation of SOCE have not yet been elucidated.
FIGURE 3.
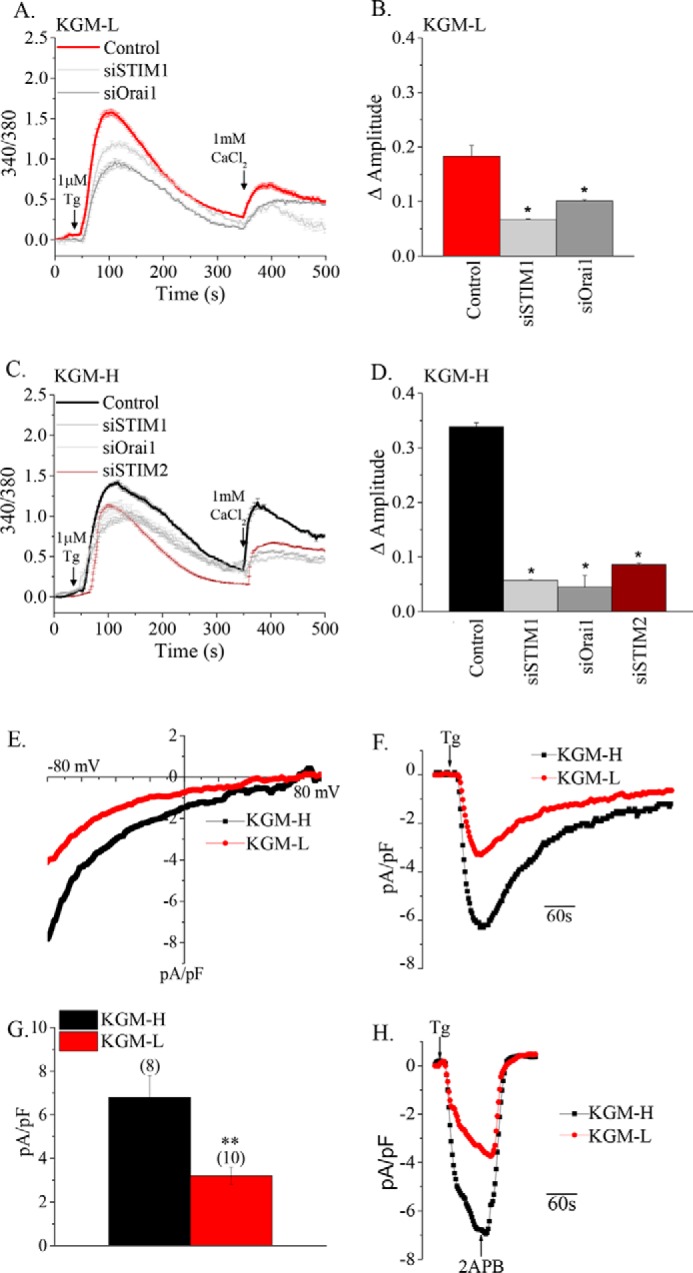
Store-operated Ca2+ entry is enhanced in phSG cells in KGM-H and is dependent on the Orai1 and STIM proteins. A and C, Tg-stimulated SOCE in phSG cells maintained in KGM-L or KGM-H medium, respectively. Cells were transfected with siRNAs as indicated. Additions of Tg and Ca2+ to cells are also indicated. B and D, the amplitude of Ca2+ influx in Tg-treated cells. Data are mean ± S.E. from n = 44–317 cells. *, p < 0.05. E–H, measurement of store-operated current in phSG cells. E and F, current-voltage relationship and representative traces of Tg-induced currents in cells grown in KGM-L and KGM-H media. pF, picofarad. G, average of current densities obtained from (number of cells) in each case. H, the inhibition of Tg-activated currents with 2APB in both sets of cells. Note that Tg and 2APB were perfused to the cells at about 5 ml/min. The data were obtained from at least three different experiments.
To further examine the increase in SOCE, the whole cell patch clamp technique was used to measure currents activated by Tg stimulation of cells. The data in Fig. 3, E–G, show that the inwardly rectifying current, ICRAC (calcium release-activated channel current), was measured in both sets of cells. Furthermore, cells in KGM-H displayed a 2-fold higher amplitude of the current than cells in KGM-L. The current, in either case, was inhibited by the SOCE blocker 2-aminoethoxydiphenyl borate (2APB) (Fig. 3H). Together, our data strongly indicate an association between up-regulation of AQP5 and enhancement of SOCE and NFAT activation in phSG cells switched to KGM-H. Similar differences in current amplitudes were measured when cells were patched with pipette solution containing 10 mm EGTA in the absence of Tg in the external solution (data not shown). The activation of current in both cases was somewhat slower than with Tg stimulation.
SOCE-dependent NFAT Activation Regulates AQP5 Expression
Enhancement of SOCE and NFAT activation was associated with increased AQP5 expression during the phenotypic switch of phSG cells. Thus, we examined a possible role for NFAT in regulating AQP5 expression. Knockdown of endogenous NFAT by increasing amounts of NFAT-specific siRNA (siNFAT) induced a dose-dependent reduction in AQP5 transcript levels between 5–25 nm, with maximal inhibition (about 80%) between 50–100 nm (Fig. 4A). Hence, cells were treated with 100 nm siNFAT for 48 h in all subsequent experiments (note that 100 nm siNFAT reduced NFAT transcript levels by >90%, Fig. 4C). To confirm the role of endogenous NFAT in AQP5 expression, cells were treated with the calcineurin inhibitor CsA, which prevents dephosphorylation of NFAT. Although CsA induced a reduction of AQP5 expression in cells cultured in either KGM-L or KGM-H, the decrease was greater in KGM-H-cultured cells, with a 50% and 98% reduction in AQP5 transcript levels at 1 and 10 μm CsA, respectively (cf. KGM-L, Fig. 4B). Moreover, consistent with the role of SOCE in NFAT activation, knockdown of Orai1, STIM1, STIM2, or STIM1+STIM2 reduced AQP5 expression to similar levels as NFAT knockdown (Fig. 4C). Note that knockdown of NFAT did not alter the expression of Orai1, STIM1, and STIM2 (data not shown). Together, these data provide strong evidence for the involvement of SOCE and NFAT in the regulation of AQP5 expression in phSG cells and in the enhancement of AQP5 expression when cells are switched from KGM-L to KGM-H.
FIGURE 4.
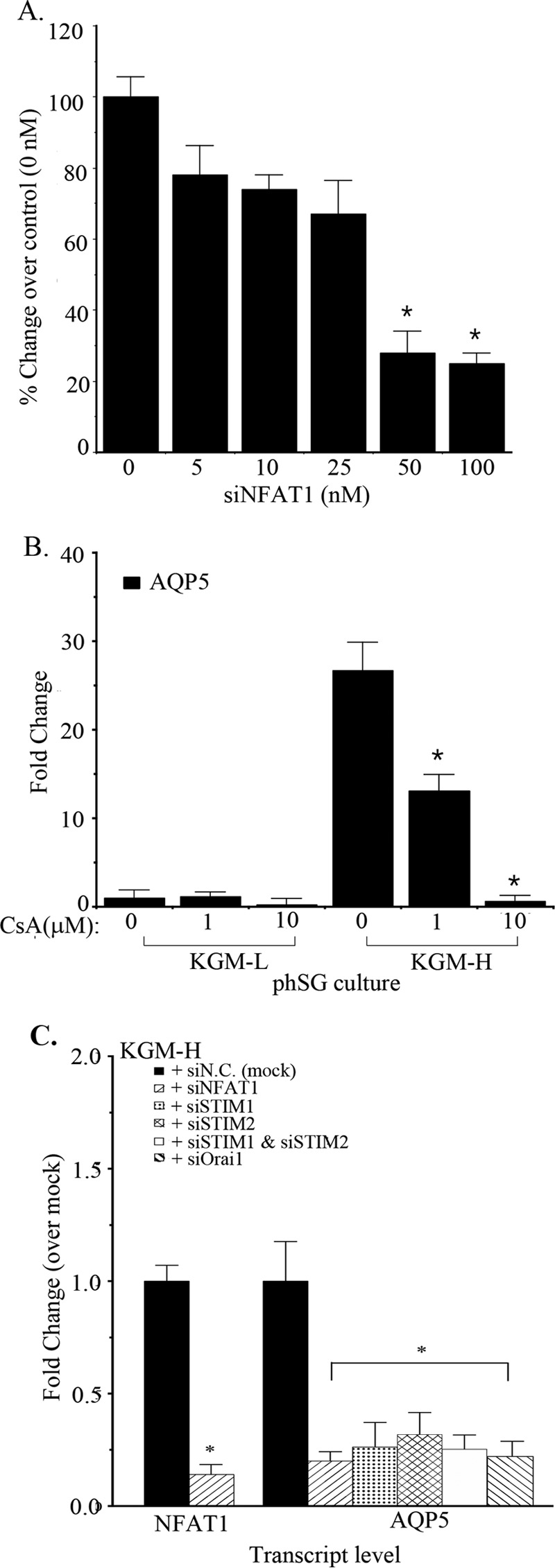
Role of NFAT in Ca2+-dependent up-regulation of AQP5 expression in phSG cells. A and B, phSG cells were transfected with indicated concentration of (A) siNFAT1 or (B) cyclosporin A and maintained in KGM-H for 48 h. The expression level of AQP5 was monitored by TaqMan probes and real-time PCR. The data are mean ± S.E. of triplicate samples from three independent experiments and expressed relative to the value in control cells (0 nm siNFAT in A and 0 μm cyclosporine in B). *, p < 0.05. C, knockdown of key Ca2+ signaling proteins on Ca2+-dependent up-regulation of AQP5 expression. phSG cells were transfected with the indicated siRNA (50 nm) and maintained in KGM-H for 48 h. TaqMan probes and real-time PCR were used to monitor the expression of NFAT1 and AQP5. The data are mean ± S.E. of triplicate samples from three separate experiments and presented relative to the value obtained with control siN.C.-transfected samples in each set. *, p < 0.05 (compared with a control sample with siN.C.).
The Presence of Functional NFAT Motifs in the AQP5 Proximal Promoter Mediates Up-regulation of AQP5 Expression in Response to Calcium Signaling
Following its translocation into the nucleus, NFAT functions as a transcription factor to induce the expression of various genes. Thus, we investigated whether the AQP5 promoter contains any regulatory regions that are dependent on NFAT. Six potential NFAT-binding sites with a core GGAAA sequence were identified within the first 2 kb of the AQP5 promoter, upstream from transcription initiation site (Fig. 5A), including one on the minus strand (−287). To evaluate the role of these putative NFAT-binding sites in regulating AQP5 promoter activity, a luciferase reporter was cloned downstream from the promoter region (F1). In addition, various truncations were made, as shown in Fig. 5, where the sites were successively deleted to generate four mutant plasmids (F2–F4). These various constructs were then transfected into cells in KGM-L and KGM-H media, and the effects of these deletions on AQP5 promoter-driven luciferase expression were evaluated. In all cases (F1–F4), substantial luciferase activities were measured with significantly higher activities (about 50%) in cells cultured in KGM-H medium than those in KGM-L (basic vector without any AQP promoter sequences were used to measure background luciferase activity) (Fig. 5B). Interestingly, the F3 (0.9 kb) construct exhibited the least activity, whereas F1 (1.8 kb) and F2 (1.1 kb) expressed relatively higher promoter activities. Thus, deleting the upstream 200 bp resulted in a 50% reduction of promoter activity, indicating the presence of positive regulatory elements within the −1100 and −900 region, in which two consensus NFAT binding motifs were found at −975 and −920, suggesting possible involvement of these elements in the regulation of AQP5 promoter activity. Intriguingly, deletion of another 450 bp in the F4 (0.45 kb) construct resulted in an ∼60% increase in luciferase activity compared with F3, which suggests the presence of negative regulatory elements within the −900 and −450 promoter region (Fig. 5B).
FIGURE 5.

Functional analysis of NFAT1-binding motifs in the AQP5 promoter and activation in KGM-H. A, schematic of consensus NFAT-binding motifs in the AQP5 promoter region 5 kb upstream of the transcription initiation site (open square, 5′-GGAAA; striped square, 5′-TTTCC). B, cells were transfected with luciferase reporter constructs containing various length of the AQP5 promoter regions and maintained either in KGM-L (white) or in KGM-H (black) medium for 48 h. Firefly luciferase activity was normalized with Renilla luciferase activity and presented as -fold change (mean ± S.E.) over that of the pGL3 Basic construct in KGM-L culture. *, p < 0.05 versus KGM-L culture. C, cells were co-transfected with siN.C. (white and black) or siNFAT (dots and bricks) together with AQP5-reporter constructs, either wild-type or containing mutations as shown in either NFAT binding motif. Cells transfected with these constructs were maintained in KGM-L (white and dots) or KGM-H (black and bricks) for 48 h following transfection with control siRNA (white and black) or siNFAT1 siRNA (dots and bricks). D, HSG cells were transfected with the indicated pAQP5 luciferase constructs and either treated with PMA (10 ng/ml) and ionomycin (0.1 μm) (black) or untreated (control, blank) as described under “Experimental Procedures.” All data shown represent mean ± S.E. from triplicate samples of three independent experiments.
The shortest promoter fragment (in F4) displayed a similar degree of up-regulation in promoter activity as the full-length fragment (in F1) in response to the higher [Ca2+] in the culture medium. We hypothesized that the proximal end of the AQP5 promoter may contain (an) NFAT-response element(s). Because this proximal end contains two putative NFAT-binding motifs (−287 and −75), we investigated whether the activation of F4 was dependent upon binding of NFAT to one or both of these two motifs. The data in Fig. 5C show that mutation of the NFAT-binding motif at the −75 position (F4/N1) resulted in a 40% and 60% reduction of luciferase expression in phSG cells maintained in KGM-L and KGM-H, respectively. The increase in promoter activity (KGM-H versus KGM-L) was 2.25-fold in F4-expressing cells and 1.6-fold in F4/N1-expressing cells (cf. control cells expressing the basic vector). In contrast, mutating the −287 motif (F4/N2) reduced luciferase activity by 35% in cells grown in KGM-H but had no effect on those in KGM-L (similar to the level in F4-expressing cells). In this case, the increase in luciferase activity was about 1.5-fold over that in KGM-L (Fig. 5C). Thus, both putative NFAT-binding sites appear to be important for regulating AQP5 expression under the high [Ca2+] condition. In addition, we knocked down endogenous NFAT to conclusively demonstrate that AQP5 promoter-driven luciferase activity in phSG cells was regulated by NFAT. Treatment with siNFAT induced a significant reduction in AQP5 promoter-driven luciferase activity in cells expressing either F4 or its mutants, F4/N1 and F4/N2 (Fig. 5C). However, significant reduction was observed with these constructs only in cells cultured in KGM-H and not those in KGM-L. Notably, siNFAT did not alter luciferase activities in cells in KGM-L expressing F4, F4/N1, and F4/N2. Furthermore, luciferase activities with these constructs in cells in KGM-H were similar to those in KGM-L. Thus, knockdown of NFAT eliminated the enhancement of AQP5 promoter activity seen in KGM-H medium, confirming that it is regulated by NFAT. Together, these results suggest that the Ca2+-dependent increase in AQP5 promoter activity is due to NFAT-dependent regulation of the proximal end of this region (as shown in the F4 construct) and that both the N1 and N2 motifs allow binding of NFAT to the proximal end.
Ca2+-dependent regulation of the AQP5 promoter was also investigated in human submandibular gland (HSG) cells (an immortalized human salivary gland cell line). HSG cells expressing the pAQP5 promoter constructs F1 and F4 as well as F4 mutants were treated with ionomycin and PMA to significantly elevate the [Ca2+]i that is required for activation of NFAT-dependent luciferase activity. All constructs showed relatively high promoter activities in untreated HSG cells compared with control cells expressing the basic vector (Fig. 5D). Nonetheless, a significant increase in luciferase activity was observed in HSG cells expressing either the F1 or F4 construct following PMA/ionomycin treatment. However, no further increase was observed in cells expressing the F4/N1 and F4/N2 constructs following treatment. These findings further confirm the involvement of the putative NFAT-binding motifs in the N1 and N2 sites within the AQP5 promoter in Ca2+-dependent regulation of AQP5 expression.
NFAT1 Binds to AQP5 Proximal Promoter in Response to Calcium Signaling in phSG Cells
To unequivocally demonstrate that NFAT indeed binds to the N1 and N2 sites within the AQP5 promoter, we performed a ChIP assay with anti-NFAT1 antibody using nuclear preparations from phSG cells. Cells were maintained in either KGM-L or KGM-H and treated with PMA/ionomycin in the presence or absence of CsA. DNA-protein complexes were pulled down using anti-NFAT1 antibody, and the DNA was used as a template to amplify a 200-bp fragment encompassing the putative NFAT-binding motifs, N1 and N2, by PCR. No significant NFAT binding was detected at the N1 site (−75 position) when phSG cells were maintained under the KGM-L condition (Fig. 6A, lane 1). However, the binding was higher when cells were maintained in KGM-H medium (Fig. 6A, lane 4), i.e. greater NFAT interaction with the AQP5 promoter under steady-state conditions in KGM-H medium. Under these conditions, treatment with PMA/ionomycin further increased the binding of NFAT1 to the N1 site in cells cultured in both KGM-L and KGM-H (Fig. 6A, lanes 2 and 5 (cf. lanes 1 and 4 respectively)), indicating that activation of SOCE promotes NFAT1 binding to the N1 motif within the endogenous AQP5 promoter. Notably, the increase in binding was greatly diminished when phSG cells were treated with ionomycin + PMA in the presence of CsA (Fig. 6A, lanes 3 and 6). On the other hand, the N2 motif did not exhibit any detectable NFAT binding under any conditions, as described above. These results directly demonstrate that NFAT1 binds to the motif at −75 (N1) but not at −287 (N2), both of which lie in the proximal end of the AQP5 promoter in phSG cells. The binding of NFAT occurred in a calcineurin-dependent manner because CsA prevented any interaction between NFAT and the N1 motif, consistent with the data shown in Fig. 4B. Furthermore, we show that this binding was increased in cells maintained in a high [Ca2+] medium, which was consistent with the marked effects of KGM-H and siNFAT on F4/N1 compared with F4/N2 activities. This finding is somewhat different from that in Fig. 5, which shows that both sites contribute to NFAT-dependent AQP5 promoter activity. Further studies will be required to elucidate factors that determine NFAT binding to the N1 and N2 sites of the AQP5 promoter and regulation of reporter expression.
FIGURE 6.

A, high [Ca2+] medium promotes binding of NFAT1 to the AQP5 promoter in response to activation of Ca2+ signaling. phSG cells were treated for 45 min either with PMA + ionomycin (10 ng/ml and 0.1 μm, respectively) (B), or together with cyclosporin A (10 nm) (C), or without treatment, control (A), in KGM-L or in KGM-H medium. Cells were collected for ChIP analysis using NFAT1 antibody or normal mouse IgG as control for pull-down and assessed using PCR (described under “Experimental Procedures”). Mr, DNA molecular weight ladder; N1 and N2, the positions of NFAT consensus motifs 75 and 287 bp upstream of the transcription initiation site, respectively. B, expression of AQP5 in phSG cells treated with PMA, ionomycin, and Tg. phSG cells were cultured in KGM-L or KGM-H for 2 days and then treated with PMA, ionomycin, or PMA/ionomycin for 4 h or with PMA/Tg or Tg for 15 min. Following this incubation, the reagent-containing media were replaced with the respective growth media (minus reagent), and cells were further maintained for 48 h. Total RNA was isolated, and the expression level of AQP5 transcript was monitored by TaqMan probe or quantitative real-time PCR assay. Data are presented as -fold change (mean ± S.E.) of triplicate samples from three separate experiments relative to that of control conditions in KGM-L.
To further define the individual contributions of PKC and SOCE to the regulation of AQP5 expression, we examined the steady-state levels of AQP5 transcript by quantitative real-time PCR in cells maintained in either KGM-L or KGM-H and treated with either PMA alone, ionomycin alone, or ionomycin + PMA. Fig. 6B shows that PMA and SOCE independently enhanced AQP5 expression in KGM-H medium. Together, their effects appear to be additive. Similar results were obtained with Tg alone or Tg + PMA. These data suggest that PMA can contribute to the enhancement of AQP5 expression. As shown in Figs. 4B and 6A, steady-state levels of AQP5 in cells in KGM-H are primarily determined by NFAT. However, the mechanisms involved in PMA-dependent activation of NFAT need to be further evaluated.
Discussion
Salivary glands consist of two morphologically and functionally distinct populations of terminally differentiated cells, acinar and ductal. It has been proposed that epithelial morphogenesis involves a series of cell fate decisions that trigger progenitor cells to differentiate. Several studies utilize explants of embryonic glands to study the morphogenesis of salivary glands. Although these studies have provided some clues regarding the mechanisms involved in early events associated with branching morphogenesis and salivary gland end bud formation, these cultures do not reach the stage where cells in the end bud region differentiate into acinar cells. Furthermore, the intracellular signaling mechanisms involved in ductal or acinar cell generation have not yet been fully established.
Here we report that SOCE and Ca2+-dependent gene expression are involved in promoting the switch of primary human salivary gland epithelial cells (phSG, established from explant cultures of minor salivary gland biopsies) to an acinus-like phenotype. Our previous findings demonstrated that phenotypic modulation of these primary salivary epithelial cells is determined by the ambient [Ca2+] of the extracellular growth medium. Although phSG cells could be maintained in a proliferative, non-differentiated state, in culture medium containing low (0.05 mm) [Ca2+], increasing the [Ca2+] in the medium to a relatively higher but more physiological level (>0.8–1.2 mm) induced an acinar cell-like phenotype with up-regulation of acinus-specific proteins like AQP5 (24). In this study, we show that this increase in extracellular [Ca2+] induces remodeling of the Ca2+ signaling toolkit, which results in increased SOCE as well as NFAT activation, both of which are critical for the up-regulation of AQP5. Importantly, our findings show that increases in Orai1, STIM1, and STIM2 contribute to the increase in SOCE. Furthermore, the Orai1-STIM generated CRAC channel appears to be the primary mediator of Ca2+ entry. The increase in SOCE drives activation of NFAT-dependent gene expression. Our data also demonstrate that up-regulation of AQP5 is mediated by the binding of activated NFAT to consensus sequences in the proximal end of the human AQP5 promoter region. We provide evidence for one essential NFAT-binding site located 75 bp upstream of the transcription initiation site. In addition, we also identified another site (at position −287) that appears to be involved in NFAT-dependent regulation of the AQP5 promoter but does not display direct binding to NFAT. One possibility is that the −287 motif may stabilize the NFAT binding on the −75 motif when forming transcription complexes. A significant reduction in NFAT-driven expression was found when the −975 and −920 motifs were deleted. This indicates that other functional NFAT binding motifs might be present on the AQP5 promoter. Further studies will be required to resolve the exact role of these domains in NFAT regulation of AQP5 expression. It is interesting to note that several NFAT-binding sites have been identified in the proximal promoter region of AQP2 that regulate gene expression in response to Ca2+ signals and osmotic stress (30).
A key event triggered by switching the cells to the relatively high [Ca2+] medium is the increase in levels of STIM1, STIM2, and Orai1, which can account for the enhancements in SOCE and SOCE-dependent activation of NFAT. This increase in Ca2+ signaling determines the enhancement of AQP5 expression in cells in KGM-H. However, the [Ca2+]i increase because of SOCE is insufficient to induce calcineurin activation and trigger NFAT translocation in cells maintained in KGM-L. Our findings suggest that this is most likely because of the lower levels of Orai1/STIM1/and STIM2 and, consequently, less functional CRAC channels in these cells. It will be important in future studies to determine the mechanisms that are involved in the regulation of Orai1, STIM1 and STIM2 expression when phSG cells are switched to a KGM-H medium.
Calcium signaling plays a critical role in the acute regulation of physiological functions in a broad range of cell types. In addition, there is strong evidence that SOCE also exerts specific long-term effects on gene expression by regulating the activation of key Ca2+-dependent transcription factors such as NFAT or cFos. Importantly, SOCE and NFAT signaling have been reported to have a role in the self-renewal and differentiation of several cell types, including lymphocytes, keratinocytes, and neural and bone cells (31–34). We and others have shown that SOCE is critical for acute regulation of ion channels that are required for neurotransmitter stimulation of fluid secretion (35–37). The data presented here provide evidence that SOCE also exerts critical long-term effects on gene expression that not only determine the switch of phSG cells to a more acinus-like phenotype but also confer functionality by up-regulating the expression of key proteins required for fluid secretion.
In conclusion, these findings demonstrate a critical link between SOCE, NFAT activation, and up-regulation of the acinus-specific protein, AQP5, during the Ca2+-dependent switch of primary human salivary gland cells to an acinus-like phenotype. We show that SOCE, NFAT, and AQP5 are increased in phSG cells maintained in 1.2 mm Ca2+ compared with cells maintained in low [Ca2+] medium. Thus, the switch of phSG cells to a more acinus-like phenotype involves the coordinated up-regulation of critical elements that together contribute to fluid secretion, a function specifically associated with acinar cells. In aggregate, these findings suggest that modulation of Ca2+ signaling could be a useful approach to establish functional secretory acinar cells under conditions of salivary gland dysfunction.
Author Contributions
S. I. J. and H. L. O. planned and carried out experiments, interpreted data, and wrote the paper. X. L. planned and carried out the electrophysiology experiments, interpreted data, and prepared figures and text for the manuscript. I. A. discussed data, provided reagents, and contributed to manuscript preparation. I. S. A. planned experiments, interpreted data, provided reagents, and wrote the paper.
Acknowledgments
We thank Dr. Changyu Zheng (NIDCR/National Institutes of Health) for AdCMVGFP-NFAT1.
The authors declare that they have no conflicts of interest with the contents of this article.
- SOCE
- store-operated Ca2+ entry
- NFAT
- nuclear factor of activated T cells
- phSG
- primary human salivary gland
- Tg
- thapsigargin
- PMA
- phorbol 12-myristate 13-acetate
- CsA
- cyclosporin A
- HSG
- human submandibular gland.
References
- 1. Ambudkar I. S. (2000) Regulation of calcium in salivary gland secretion. Crit. Rev. Oral Biol. Med. 11, 4–25 [DOI] [PubMed] [Google Scholar]
- 2. Kiselyov K., Wang X., Shin D. M., Zang W., and Muallem S. (2006) Calcium signaling complexes in microdomains of polarized secretory cells. Cell Calcium 40, 451–459 [DOI] [PubMed] [Google Scholar]
- 3. Melvin J. E., Yule D., Shuttleworth T., and Begenisich T. (2005) Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu. Rev. Physiol. 67, 445–469 [DOI] [PubMed] [Google Scholar]
- 4. Yule D. I. (2001) Subtype-specific regulation of inositol 1,4,5-trisphosphate receptors: controlling calcium signals in time and space. J. Gen. Physiol. 117, 431–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berkefeld H., Fakler B., and Schulte U. (2010) Ca2+-activated K+ channels: from protein complexes to function. Physiol. Rev. 90, 1437–1459 [DOI] [PubMed] [Google Scholar]
- 6. Ma T., Song Y., Gillespie A., Carlson E. J., Epstein C. J., and Verkman A. S. (1999) Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J. Biol. Chem. 274, 20071–20074 [DOI] [PubMed] [Google Scholar]
- 7. Ong H. L., Cheng K. T., Liu X., Bandyopadhyay B. C., Paria B. C., Soboloff J., Pani B., Gwack Y., Srikanth S., Singh B. B., Gill D. L., Gill D., and Ambudkar I. S. (2007) Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx: evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 282, 9105–9116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parekh A. B. (2010) Store-operated CRAC channels: function in health and disease. Nat. Rev. Drug Discov. 9, 399–410 [DOI] [PubMed] [Google Scholar]
- 9. Zhang S. L., Kozak J. A., Jiang W., Yeromin A. V., Chen J., Yu Y., Penna A., Shen W., Chi V., and Cahalan M. D. (2008) Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J. Biol. Chem. 283, 17662–17671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuspa S. H., Kilkenny A. E., Steinert P. M., and Roop D. R. (1989) Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J. Cell Biol. 109, 1207–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shaw J. P., Utz P. J., Durand D. B., Toole J. J., Emmel E. A., and Crabtree G. R. (1988) Identification of a putative regulator of early T cell activation genes. Science 241, 202–205 [DOI] [PubMed] [Google Scholar]
- 12. Müller M. R., and Rao A. (2010) NFAT, immunity and cancer: a transcription factor comes of age. Nat. Rev. Immunol. 10, 645–656 [DOI] [PubMed] [Google Scholar]
- 13. Crist S. A., Sprague D. L., and Ratliff T. L. (2008) Nuclear factor of activated T cells (NFAT) mediates CD154 expression in megakaryocytes. Blood 111, 3553–3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shukla U., Hatani T., Nakashima K., Ogi K., and Sada K. (2009) Tyrosine phosphorylation of 3BP2 regulates B cell receptor-mediated activation of NFAT. J. Biol. Chem. 284, 33719–33728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zanoni I., Ostuni R., Capuano G., Collini M., Caccia M., Ronchi A. E., Rocchetti M., Mingozzi F., Foti M., Chirico G., Costa B., Zaza A., Ricciardi-Castagnoli P., and Granucci F. (2009) CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature 460, 264–268 [DOI] [PubMed] [Google Scholar]
- 16. Kar P., and Parekh A. B. (2015) Distinct spatial Ca2+ signatures selectively activate different NFAT transcription factor isoforms. Mol. Cell 58, 232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kudryavtseva O., Aalkjaer C., and Matchkov V. V. (2013) Vascular smooth muscle cell phenotype is defined by Ca2+-dependent transcription factors. FEBS J. 280, 5488–5499 [DOI] [PubMed] [Google Scholar]
- 18. Lewis R. S. (2003) Calcium oscillations in T-cells: mechanisms and consequences for gene expression. Biochem. Soc. Trans. 31, 925–929 [DOI] [PubMed] [Google Scholar]
- 19. Sens D. A., Hintz D. S., Rudisill M. T., Sens M. A., and Spicer S. S. (1985) Explant culture of human submandibular gland epithelial cells: evidence of epithelial origin. Lab. Invest. 52, 559–567 [PubMed] [Google Scholar]
- 20. Sabatini L. M., Allen-Hoffmann B. L., Warner T. F., and Azen E. A. (1991) Serial cultivation of epithelial cells from human and macaque salivary glands. In Vitro Cell Dev. Biol. 27, 939–948 [DOI] [PubMed] [Google Scholar]
- 21. Okura M., Shirasuna K., Hiranuma T., Yoshioka H., Nakahara H., Aikawa T., and Matsuya T. (1993) Characterization of growth and differentiation of normal human submandibular gland epithelial cells in a serum-free medium. Differentiation 54, 143–153 [DOI] [PubMed] [Google Scholar]
- 22. Dimitriou I. D., Kapsogeorgou E. K., Abu-Helu R. F., Moutsopoulos H. M., and Manoussakis M. N. (2002) Establishment of a convenient system for the long-term culture and study of non-neoplastic human salivary gland epithelial cells. Eur. J. Oral Sci. 110, 21–30 [DOI] [PubMed] [Google Scholar]
- 23. Redman R. S., and Quissell D. O. (1993) Isolation and Maintenance of Submandibular Gland Cells. in Biology of Salivary Glands (Dobrosielski-Vergona K., ed), pp. 285–306, CRC Press, Boca Raton, FL [Google Scholar]
- 24. Jang S. I., Ong H. L., Gallo A., Liu X., Illei G., and Alevizos I. (2015) Establishment of functional acinar-like cultures from human salivary glands. J. Dent. Res. 94, 304–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu X., Bandyopadhyay B. C., Bandyopadhyay B., Nakamoto T., Singh B., Liedtke W., Melvin J. E., and Ambudkar I. (2006) A role for AQP5 in activation of TRPV4 by hypotonicity: concerted involvement of AQP5 and TRPV4 in regulation of cell volume recovery. J. Biol. Chem. 281, 15485–15495 [DOI] [PubMed] [Google Scholar]
- 26. Gallo E. M., Canté-Barrett K., and Crabtree G. R. (2006) Lymphocyte calcium signaling from membrane to nucleus. Nat. Immunol. 7, 25–32 [DOI] [PubMed] [Google Scholar]
- 27. Hogan P. G., Chen L., Nardone J., and Rao A. (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 [DOI] [PubMed] [Google Scholar]
- 28. Coudronniere N., Villalba M., Englund N., and Altman A. (2000) NF-κ B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-θ. Proc. Natl. Acad. Sci. U.S.A. 97, 3394–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rao A., Luo C., and Hogan P. G. (1997) Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15, 707–747 [DOI] [PubMed] [Google Scholar]
- 30. Li S. Z., McDill B. W., Kovach P. A., Ding L., Go W. Y., Ho S. N., and Chen F. (2007) Calcineurin-NFATc signaling pathway regulates AQP2 expression in response to calcium signals and osmotic stress. Am. J. Physiol. Cell Physiol. 292, C1606–1616 [DOI] [PubMed] [Google Scholar]
- 31. Gu X., and Spitzer N. C. (1995) Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature 375, 784–787 [DOI] [PubMed] [Google Scholar]
- 32. Maric D., Maric I., Chang Y. H., and Barker J. L. (2003) Prospective cell sorting of embryonic rat neural stem cells and neuronal and glial progenitors reveals selective effects of basic fibroblast growth factor and epidermal growth factor on self-renewal and differentiation. J. Neurosci. 23, 240–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mishra S. K., Braun N., Shukla V., Füllgrabe M., Schomerus C., Korf H. W., Gachet C., Ikehara Y., Sévigny J., Robson S. C., and Zimmermann H. (2006) Extracellular nucleotide signaling in adult neural stem cells: synergism with growth factor-mediated cellular proliferation. Development 133, 675–684 [DOI] [PubMed] [Google Scholar]
- 34. Platel J. C., Dave K. A., and Bordey A. (2008) Control of neuroblast production and migration by converging GABA and glutamate signals in the postnatal forebrain. J. Physiol. 586, 3739–3743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ambudkar I. S. (2014) Ca2+ signaling and regulation of fluid secretion in salivary gland acinar cells. Cell Calcium 55, 297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stauffer P. L., Zhao H., Luby-Phelps K., Moss R. L., Star R. A., and Muallem S. (1993) Gap junction communication modulates [Ca2+]i oscillations and enzyme secretion in pancreatic acini. J. Biol. Chem. 268, 19769–19775 [PubMed] [Google Scholar]
- 37. Lee M. G., Ohana E., Park H. W., Yang D., and Muallem S. (2012) Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol. Rev. 92, 39–74 [DOI] [PMC free article] [PubMed] [Google Scholar]