

Abstract
Bcr-Abl is a constitutively active kinase that causes chronic myelogenous leukemia. We have shown that a tandem fusion of two designed binding proteins, termed monobodies, directed to the interaction interface between the Src homology 2 (SH2) and kinase domains and to the phosphotyrosine-binding site of the SH2 domain, respectively, inhibits the Bcr-Abl kinase activity. Because the latter monobody inhibits processive phosphorylation by Bcr-Abl and the SH2-kinase interface is occluded in the active kinase, it remained undetermined whether targeting the SH2-kinase interface alone was sufficient for Bcr-Abl inhibition. To address this question, we generated new, higher affinity monobodies with single nanomolar KD values targeting the kinase-binding surface of SH2. Structural and mutagenesis studies revealed the molecular underpinnings of the monobody-SH2 interactions. Importantly, the new monobodies inhibited Bcr-Abl kinase activity in vitro and in cells, and they potently induced cell death in chronic myelogenous leukemia cell lines. This work provides strong evidence for the SH2-kinase interface as a pharmacologically tractable site for allosteric inhibition of Bcr-Abl.
Keywords: ABL tyrosine kinase, enzyme inhibitor, protein engineering, protein-protein interaction, Src homology 2 domain (SH2 domain), x-ray crystallography, FN3, PPI inhibitor
Introduction
Mutations conferring drug resistance are frequently observed in cancer and infectious diseases following treatment of patients with targeted drugs (1, 2). To overcome resistance, it is necessary to develop a therapeutic strategy that is based on a mechanism of action distinct from that of the first generation drug. Bcr-Abl is a fusion protein of the breakpoint cluster region (BCR)3 and Abelson tyrosine kinase 1 (ABL1) generated by the Philadelphia chromosomal translocation (3). Bcr-Abl is a constitutively active kinase that is sufficient for the initiation and maintenance of CML. Although small molecule ATP-competitive Bcr-Abl inhibitors such as imatinib (Gleevec) and its successors have dramatically improved overall survival of CML patients, the emergence of resistance mutations renders kinase inhibitors ineffective in subsets of CML patients (4, 5). Importantly, a recently developed allosteric Bcr-Abl inhibitor targeting the myristoyl-binding pocket can strongly limit the emergence of drug resistance to ATP-competitive inhibitors and has entered clinical trials (6), highlighting the interest and strong demand for new therapeutic strategies even in the case of a well established target like Bcr-Abl (5).
Bcr-Abl has multiple protein domains including the SH2 and tyrosine kinase domains originating from Abl1 (Fig. 1A) (7). Extensive mechanistic and structural studies have revealed the intramolecular interaction between the SH2 and kinase domains to be critical for full kinase activity (Fig. 1B). Consequently, a mutation that disrupts this interface, I164E, inhibits enzyme activity and Bcr-Abl-mediated oncogenesis (8, 9). These results suggest that the SH2-kinase interface is a potentially “druggable” site for allosteric inhibition of the kinase activity.
FIGURE 1.
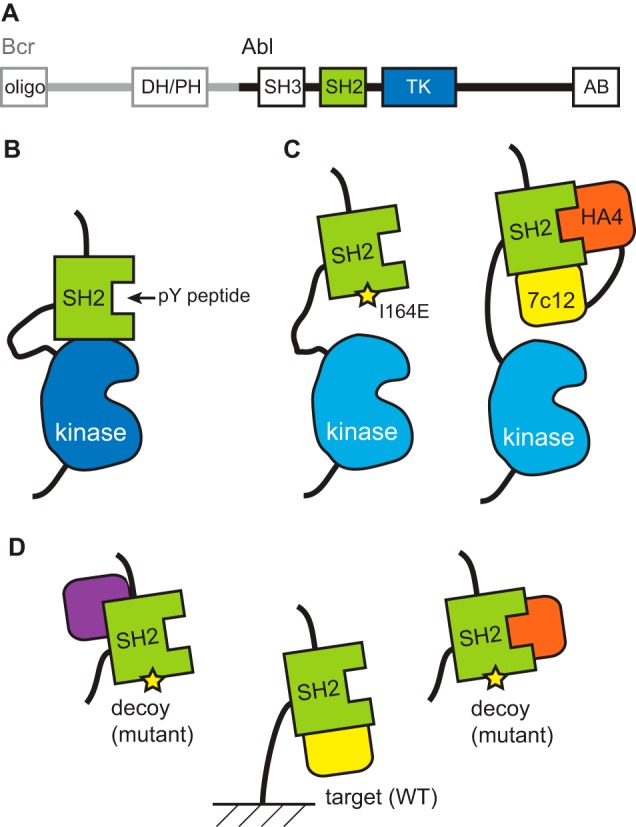
Domain architecture (A) and SH2-dependent regulatory mechanism of Bcr-Abl are shown. B and C, schematic drawings of the interface between the SH2 and kinase domains and disruptions of the interface with the I164E mutation or with the tandem monobodies. D, scheme showing a strategy for recovering monobodies binding to a predefined epitope using a decoy containing a mutation in the desired epitope. By adding the decoy in large excess over the immobilized target, monobodies binding outside the desired surface are preferentially bound to the decoys, leading to enrichment of desired monobodies (shown in yellow). PH, pleckstrin homology domain; DH, Dbl homology domain; TK, tyrosine kinase domain; AB, actin-binding domain.
In our previous work, to determine the feasibility of disrupting the SH2-kinase interface in trans, we utilized synthetic binding proteins, termed “monobodies,” to target this interface (9). Monobodies are binding proteins developed from combinatorial libraries constructed on the molecular scaffold of a fibronectin type III domain (10–12). Because monobodies are stable and, unlike antibody fragments, do not contain a disulfide bond, they are well suited as genetically encoded inhibitors of an intracellular target. We successfully generated a monobody termed “7c12” directed to the kinase-binding surface of the Abl SH2 domain (9). Because the 7c12 monobody had only moderate affinity and hence moderate biological effects in vitro and in cells, we needed to fuse it with another monobody, termed HA4, binding to a different surface of the SH2 domain, namely the binding site for phospho-Tyr-containing ligands (13). This tandem fusion approach enhanced the effective affinity so that the HA4–7c12 fusion could successfully compete with the intramolecular interaction between the SH2 and kinase domains (Fig. 1C). Expression of this tandem monobody construct suppressed Bcr-Abl-dependent oncogenic transformation of mouse bone marrow cells by inhibiting Bcr-Abl kinase activity and induced apoptosis in human cells isolated from CML patients (9). These results supported the notion that the SH2-kinase interface is a potentially druggable site.
Although the tandem monobody construct was an effective allosteric Bcr-Abl inhibitor, it remained unclear whether targeting the SH2-kinase interface alone was sufficient for Bcr-Abl inhibition because the HA4 monobody in isolation also affected downstream signaling (13). HA4 inhibits the interaction of Abl SH2 with its phospho-Tyr-containing ligands and consequently processive phosphorylation of Bcr-Abl substrates (13). Furthermore, the long linker and second monobody in the tandem monobody construct substantially increased the complexity of the molecule, diminishing the benefit of monobodies as compact protein reagents. In this work, we aimed to develop high affinity monobodies directed to the SH2-kinase interface to unambiguously determine whether Bcr-Abl could be inhibited by targeting solely this interface. We generated new monobodies and characterized their structure and recognition mechanism. These monobodies, used as single domain reagents, inhibited the in vitro and cellular activity of Bcr-Abl and strongly decreased survival of CML cells. The results presented here establish the sufficiency of pharmacologically targeting the SH2-kinase interface for the allosteric inhibition of Bcr-Abl.
Experimental Procedures
Protein Expression and Purification
The Abl SH2 domain and monobodies were produced with an N-terminal tag containing His10, FLAG, and tobacco etch virus protease recognition motifs using the pHFT2 vector (14). The I164E mutation was introduced using the mutagenesis method of Kunkel et al. (15). Proteins were expressed in Escherichia coli BL21(DE3) and purified to apparent homogeneity using nickel affinity chromatography and size exclusion chromatography (Superdex 75, GE Healthcare).
cDNAs encoding human ABL1 (Abl kinase domain (KD), residues 248–534; Abl SH2-KD, residues 138–534; splice form 1b numbering) were cloned into the NheI and XhoI restriction sites of pET-21d (Merck Millipore). Proteins were co-expressed with the YopH phosphatase in E. coli BL21(DE3). Protein purification was carried using the C-terminal hexahistidine tag by nickel affinity chromatography with further purification by anion exchange chromatography in 150 mm NaCl, 20 mm Tris-HCl, pH 7.5, 5% glycerol, and 1 mm DTT as described (16).
Monobody Generation and Characterization
General methods for phage and yeast display library sorting and gene shuffling have been described (11–13). The monobody libraries used have been reported (11). In phage display library sorting, target proteins were immobilized using a high affinity ligand for the His10 tag (17). The GG3 and GG10 monobodies were isolated after four rounds of phage display selection using target concentrations of 250, 100, 100, and 100 nm for the first through fourth rounds in the presence of a 10-fold excess of the I164E mutant SH2 domain so that monobodies binding to the wild type but not the mutant are predominantly retained. Isolation of AS25 and AS27 involved additional steps utilizing yeast surface display and has been reported (11). Combinatorial libraries for affinity maturation of AS25 were constructed in the yeast surface display format. Binding measurements in the yeast display format were performed using a Millipore Guava flow cytometer as described previously (11, 12). The dissociation constants determined from the yeast display format agreed closely with those determined using purified monobody samples with surface plasmon resonance (11).
Crystallization, Data Collection, and Structure Determination
The AS25-Abl SH2 and GG3-Abl SH2 complexes were purified with a Superdex 75 column (GE Healthcare). The complexes were concentrated to ∼7.5 (AS25-Abl SH2) and 10 mg/ml (GG3-Abl SH2); mixed 1:1 with a well solution containing 0.1 m imidazole, pH 7.8, and 3.5 m NaCl (Crystal A), 0.1 m imidazole, pH 8.5, and 3.4 m NaCl (Crystal B), or 0.1 m sodium tartrate, pH 8, and 25% (w/v) polyethylene glycol 3350 (GG3-Abl SH2); and crystallized using the hanging drop vapor diffusion method. Glycerol (20%) was used as a cryoprotectant in all cases. X-ray diffraction data were collected at the Advanced Photon Source, beamlines 24 ID-C (AS25-Abl SH2 complexes) and 24 ID-E (GG3-Abl SH2) at a wavelength of 0.97872 Å and temperature of 100 K. Data collection and structure determination statistics are given in Table 1. Diffraction data were processed and scaled with the HKL2000 package (18). The structures were solved by molecular replacement using Phaser in the CCP4 program suite (19, 20). A multicopy search was performed with the Abl SH2 domain and the fibronectin type III domain scaffold without the loop regions as the search models (Protein Data Bank codes 2ABL and 1FNA, respectively). Simulated annealing, energy minimization, B-factor refinement, and map building were carried out using CNS (21, 22). Rigid body refinement was carried out using REFMAC5 in the CCP4 program suite. Translation/libration/screw (TLS) groups were defined using the TLSMD server (23), and TLS refinement, B-factor refinement, bulk solvent parameters, final positional refinement, and the search for and refinement of water molecules were carried out using REFMAC5. Model building and evaluation were carried out using the Coot program (24), and molecular graphics were generated using PyMOL (Schrödinger, LLC). Surface area calculations were performed using the ProtorP protein-protein interaction server (25). Shape complementarity was determined using the Sc statistic (26).
TABLE 1.
Data collection and refinement statistics (molecular replacement) of Monobody-Abl1 SH2 domain complexes
N/A, not applicable; r.m.s., root mean square. Data in parentheses refer to values for the highest resolution shell.
| GG3-Abl1 SH2 | AS25-Abl1 SH2 (crystal A) | AS25-Abl1 SH2 (crystal B) | |
|---|---|---|---|
| Protein Data Bank code | 5DC0 | 5DC4 | 5DC9 |
| Data collection | |||
| Space group | C121 | P 212121 | P41212 |
| Cell dimensions | |||
| a, b, c (Å) | 131.437, 37.348, 39.705 | 61.281, 74.928, 39.229 | 66.391, 66.391, 66.391 |
| α, β, γ (°) | 90.0, 98.4, 90.0 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 2.23 | 1.48 (1.52–1.48) | 1.56 (1.62–1.56) |
| Rsym | 11.4 (37.3) | 5.6 (23.5) | 5.5 (46.3) |
| I/σI | 17.4 (5.3) | 29.7 (4.3) | 34.4 (4.3) |
| Completeness (%) | 98.7 (87.8) | 98.2 (95.1) | 98.2 (94.2) |
| Redundancy | 4.4 (3.2) | 4.9 (4.3) | 8.5 (8.3) |
| Refinement | |||
| Resolution (Å) | 2.23 | 1.48 | 1.56 |
| Total no. reflections | 41,307 | 148,388 | 308,565 |
| Unique reflections | 9,392 | 30,102 | 36,108 |
| Rwork/Rfree | 0.19/0.24 | 0.15/0.19 | 0.15/0.17 |
| No. atoms | 1,518 | 1,752 | 1,829 |
| Protein | 1,463 | 1,540 | 1,557 |
| Ligand/ion | 0 | 6 | 40 |
| Water | 55 | 206 | 232 |
| B-factors | |||
| Protein | 25.65 | 19.29 | 20.80 |
| Ligand/ion | N/A | 39.50 | 31.24 |
| Water | 27.77 | 34.52 | 42.46 |
| r.m.s. deviations | |||
| Bond lengths (Å) | 0.010 | 0.010 | 0.011 |
| Bond angles (°) | 1.386 | 1.205 | 1.376 |
| Ramachandran analysis | |||
| Favored regions (%) | 95.68 | 98.94 | 98.34 |
| Allowed regions (%) | 4.32 | 1.06 | 1.10 |
| Outliers (%) | 0 | 0 | 0.55 |
NMR Spectroscopy
Chemical shift assignments for the Abl SH2 domain were from a previous work (11). 15N heteronuclear single quantum correlation (HSQC) spectra were taken with15N-enriched Abl SH2 domain (∼90 μm) alone and in the presence of unlabeled monobodies (AS25, AS27, GG3, and GG10). All assigned peaks from the Abl SH2 domain were classified based on the degree of peak shift according to a procedure described previously (14).
In Vitro Kinase Assays
Five nanograms of recombinant Abl KD or SH2-KD protein were preincubated with the recombinant AS25 and AS27 monobodies at various concentrations or buffer controls for 10 min at room temperature in a volume of 10 μl. Kinase activity was determined by the addition of 20 μm ATP, 7 μCi of [γ-32P]ATP, and a 75 μm concentration of an optimal Abl substrate sequence carrying an N-terminal biotin (biotin-GGEAIYAAPFKK-amide) in kinase assay buffer (20 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 1 mm DTT, and 30 μm BSA) for 20 min at room temperature in a final assay volume of 20 μl. The terminated reaction (10 μl of 7.5 m guanidine hydrochloride) was spotted onto a SAM2 Biotin Capture membrane (Promega) and further treated according to the instructions of the manufacturer.
HEK293 Cell Transfection
HEK293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. Cells were co-transfected with Abl P242E/P249E (Abl PP) (27) or Bcr-Abl and 6xMyc-tagged monobody expression vectors using Polyfect transfection reagent (Qiagen) following the supplier protocol. 48 h after transfection, cells were harvested and lysed in immunoprecipitation buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 5 mm EDTA, 5 mm EGTA, 25 mm NaF, 1 mm orthovanadate, 1 mm PMSF, 10 mg/ml l-1-tosylamido-2-phenylethyl chloromethyl ketone, and protease mixture inhibitor from Roche Applied Science) and cleared by centrifugation. For immunoblotting analysis, 100 μg of cellular lysate was used.
Western Blotting and Quantification
Antibodies used for Western blotting include anti-Abl (mouse; Sigma); anti-phospho-Tyr-412 (rabbit; Cell Signaling Technology); anti-Myc (rabbit; Rockland), and anti-Tyr(P) 4G10 (mouse; in-house). Autophosphorylation levels in the activation loop (Tyr-412) were quantified using a LI-COR Odyssey system.
Retroviral Transduction and FACS Analysis of K562 Cells Stably Expressing Monobodies
K562 cells stably expressing N-terminal tandem affinity purification tag-fused monobodies (AS25, AS27, HA4, and the non-target-binding monobody HA4(Y87A)) were generated by retroviral gene transfer as described previously (13). For six consecutive days starting 24 h after the second retroviral infection of the target K562 cells and every 24 h, aliquots of 106 cells were removed from the culture and analyzed by flow cytometry (Accuri C6, BD Biosciences). Cells were analyzed for GFP expression (that is co-expressed with the tandem affinity purification-tagged monobodies) and in parallel stained with Annexin V Cy5 (BD Pharmingen, product number 559933) and 7-aminoactinomycin D (BD Pharmingen, product number 559925).
Results
Monobodies to the Abl SH2-Kinase Interaction Interface
We identify monobodies from combinatorial libraries in which positions in the fibronectin type III domain scaffold are diversified. The goal is to achieve both shape and chemical complementarity with the intended surface of the target molecule (28). We have recently developed monobody libraries using a new design that presents diversified amino acid positions in a concave topography (Fig. 2, A and B) (11). This new library design complements our original design in which diversified residues are presented on a convex surface made with three loops (BC, DE, and FG) located on one end of the scaffold (10, 13). We reasoned that monobodies from the new design are better suited for targeting the kinase-binding surface of the SH2 domain that is mostly flat and slightly convex.
FIGURE 2.

Monobodies binding to the Abl SH2 domain. A, the amino acid sequences of the monobody libraries and monobody clones. In the library designs, “X” denotes a mixture of 30% Tyr, 15% Ser, 10% Gly, 5% Phe, 5% Trp, and 2.5% each of all the other amino acids except for Cys; “B” denotes a mixture of Gly, Ser, and Tyr; “J” denotes a mixture of Ser and Tyr; “O” denotes a mixture of Asn, Asp, His, Ile, Leu, Phe, Tyr, and Val; “U” denotes a mixture of His, Leu, Phe, and Tyr; and “Z” denotes a mixture of Ala, Glu, Lys, and Thr. A hyphen indicates a deletion. B, a schematic drawing of the monobody scaffold and the locations of diversified positions in the libraries. A and B are modified from Figs. 2 and 1, respectively, of Koide et al. (11).
To enrich monobodies targeting the intended surface of the SH2 domain, we first used a strategy that utilizes a “decoy” competitor, the I164E mutant in this case, for eliminating those clones binding to surfaces outside the desired epitope (Fig. 1D). The I164E mutation is located in the kinase interaction surface of the SH2 domain and disrupts the SH2-kinase interface, thereby inhibiting kinase activity (Fig. 1B). The library used in this selection was an earlier version of the “loop-and-side” library reported previously (11) in which amino acid diversity was introduced into the FG loop and the β-strands of the scaffold (Fig. 2, A and B).
Two monobodies, GG3 and GG10, identified using this method (Fig. 2A) bound to wild-type SH2 but not to the I164E mutant as expected. To better define the targeted epitope, we conducted epitope mapping by 15N HSQC chemical shift perturbation using uniformly 15N-labeled Abl SH2 domain. This method detects changes in the 15N HSQC resonances of the 15N-labeled component, i.e. Abl SH2 in this case, that are affected by the binding of another component, i.e. a monobody (29). The effects of GG3 and GG10 on Abl SH2 backbone amide resonances were nearly identical (Fig. 3B). The residues most greatly affected were located adjacent to Ile-164 and within the kinase-interacting surface, confirming that these monobodies interact with the targeted epitope. Although we were successful in identifying monobodies interacting with the kinase-activating interface, the affinity of these monobodies as assessed by isothermal titration calorimetry was modest (KD = 2.54 and 0.90 μm for GG3 and GG10, respectively; data not shown) and weaker than that of the 7c12 monobody (KD = ∼50 nm) that failed to show strong biological effects on its own (9). This comparison suggested that the new monobodies would not be sufficiently potent although they bound to the intended surface of the SH2 domain.
FIGURE 3.

Biochemical analysis of monobody binding to Abl kinase fragments. A, binding of monobodies to Abl SH2 domain as tested in the yeast surface display format. Binding signal in terms of mean fluorescence intensity (MFI) in arbitrary (arbit.) units is plotted versus SH2 concentration. The curves are the best fit of the 1:1 binding model. The KD values are shown. The errors are the S.D. from nonlinear least square fitting. B, binding of monobodies to the SH2-kinase fragment as tested in the yeast surface display format shown in the same manner as in A. C–F, size exclusion chromatography analysis of complex formation of the AS25 monobody with wild type (C), T231R (D), and I164E (E) of the SH2-KD segment (C–E) and the wild-type KD (F) of Abl. mAu, milli-absorbance units.
Rather than engaging in extensive affinity maturation of GG3 and GG10, we next proceeded with the selection of monobodies from a next generation library that incorporated diversity in the CD loop in addition to the FG loop and β-sheet region (11) (Fig. 2, A and B). We reasoned that the additional diversified residues in the CD loop would provide increased surface area for interaction with the targeted site. We also incorporated gene shuffling and additional library sorting using yeast surface display as reported previously (11) in an effort to increase the coverage of the sequence space and improve the identification of high affinity clones. We have produced high affinity monobodies with KD values in the low nanomolar range to several targets using this strategy including two to the Abl SH2 domain, AS25 and AS27 (11, 30). Therefore, we characterized these monobodies that had been used only for the purpose of assessing the performance of the library in generating high affinity monobodies but had otherwise remained uncharacterized (Fig. 2A). AS25 and AS27 bound the Abl SH2 domain with KD values of 35 and 7.7 nm, respectively, as assessed by binding titration performed in the yeast surface display format (Fig. 3A). Note that KD values determined with the yeast surface display titration generally agree with those from biophysical measurements of purified proteins, such as surface plasmon resonance, which is the case for AS25 and AS27 (11).
15N HSQC NMR epitope mapping revealed that AS25 and AS27 also bound to an epitope predicted to overlap substantially with the kinase-interacting interface. The 15N HSQC spectra of [U-15N]SH2 domain in complex with either monobody were largely superimposable (Fig. 3B), consistent with a highly similar epitope. This similarity was expected given their closely related sequences including identical FG loops (Fig. 2A). The HSQC peak of Ile-164 of the Abl SH2 was greatly affected by the binding of either monobody. In binding assays, AS25 and AS27 interacted with the I164E mutant >100-fold more weakly than the wild-type SH2 domain (Fig. 3E and data not shown), providing independent verification of the locations of these epitopes. Together, these data demonstrate that we were able to identify high affinity monobodies that bound to the kinase interaction surface of the Abl SH2 domain.
Crystal Structures of Monobodies in Complex with Abl SH2
To better understand the mode of interaction of monobodies targeting the kinase interaction interface of Abl SH2, we determined crystal structures for the GG3-SH2 and AS25-SH2 complexes and compared them with the structures of the Abl SH2-kinase domains and the 7c12-SH2 complex (Fig. 4 and Table 1). The AS25-SH2 complex was crystallized in two distinct space groups (henceforth referred to as crystals “A” and “B”). The Abl SH2 domain maintained its structure in the GG3 and AS25 monobody complexes (root mean square deviation <0.4Å for the backbone atoms) and was also close to that in a previously determined monobody complex (Protein Data Bank code 3K2M) and multidomain Abl segments containing the SH2 domain (Protein Data Bank codes 1OPL, 1OPK, and 2FO0). The two AS25-SH2 complex structures are nearly identical (root mean square deviation of 0.567 Å for all Cα atoms) with the only substantial difference in the position of the CD loop, which is folded inward in the A structure and reflected outward in the B structure. Both GG3 and AS25 monobodies form a cradled binding surface utilizing positions in both β-sheet and loop regions (Fig. 4, C and D) as intended by the design of the library (Fig. 2B). The epitopes of the monobodies are approximately centered on Ile-164 of the Abl SH2 domain, the residue within the SH2-kinase interface that we targeted in our decoy selection strategy (Fig. 4, C and D). Ile-164 interacts with the β-sheet regions of both monobodies. These epitopes overlap substantially with the epitope of the kinase domain (Fig. 4A) as well as that of 7c12 (Fig. 4E), suggesting that these monobodies would be competitive inhibitors of the interaction between the SH2 and kinase domains. These results also rationalize the strongly decreased interaction of the I164E mutation with these monobodies.
FIGURE 4.

Structures and interactions of the Abl SH2 domain with monobodies. A, the crystal structure of the SH2-kinase fragment of Abl in the active conformation (Protein Data Bank code 1OPL; chain B). The bottom picture shows the surface of the SH2 domain with the footprint of the kinase domain in red as defined as surfaces of the atoms located within 5 Å of the kinase domain. A predicted location of a phospho-Tyr peptide based on homology to other SH2 domains is shown as the yellow object. Ile-164 is shown as a stick model. B, NMR-based epitope mapping of four monobodies. The red, yellow, and gray spheres show residues of Abl SH2 whose amide resonances in the 1H,15N HSQC spectrum were strongly affected (shift of ≥1.5 peak width), weakly affected (shift of 0.5–1.5 peak width), and minimally affected (shift of b0.5 peak width) by monobody binding, respectively. C–E, the crystal structures of the SH2 domain in complex with GG3 (C), AS25 (D), and 7c12 (E; Protein Data Bank code 3T04). The SH2 domain in the schematic models on the left is in the same orientation as that in A. The right schematic models are from an orthogonal viewing angle. The spheres show diversified residues, and yellow, gray, and pink-red denote those in the BC, CD, DE, and FG loops, respectively. The footprints of the monobodies are shown in the same manner as in A.
Whereas GG3 and AS25 use similar surfaces to interact with similar surfaces of the Abl SH2 domain, there are notable differences between the two complexes. The positions of the β-sheet core of the monobody scaffold in these structures are related by an ∼30° rotation with Ile-88, located at the N-terminal end of the G strand, as the pivot point (Fig. 5A). AS25 buries substantially larger surface areas in the interface than GG3 (Table 2). This difference comes mainly from the positioning of the CD loop and the β-sheet region of the monobody scaffold. It appears that the interactions afforded by the additionally mutated residues of AS25 brought the CD loop and the β-sheet region closer to the SH2 domain in the AS25 complex than in the GG3 complex (Fig. 5A). The shape complementarity assessed with the Sc value (26) was similarly good for both structures (Table 2), indicating that the interfaces are well packed. The GG3 complex had the highest Sc value, suggesting a better packed, concentrated binding interface. These interface characteristics are consistent with the different designs of monobody libraries from which the two monobodies are derived.
FIGURE 5.

The binding interfaces of the GG3 and AS25 complexes. A, a comparison of the binding modes of the two monobodies. The “back side” (the A, B, and E strands and loops adjacent to them) of the monobodies are omitted for clarity. The surface model shows the SH2 domain. The C and F strands and FG loop are labeled for comparison. The sphere shows Ile-88 that is the pivot point for the rotation relative to the β-sheet core of the two monobodies. B, the interaction of the FG loop of AS25 with SH2. The N-terminal half (YGYPY) of the loop is shown with carbon atoms in green. C, the interaction of the FG loop of GG3 with SH2 with the N-terminal half (LLSSS) shown with carbon atoms in green. D, Ala-scanning mutagenesis of AS25. The ΔΔG values are shown in the graph. The asterisks indicate that the ΔΔG values are the lower limit because the KD values for the mutants were too high to be determined in the assay. The “hot spot” residues with ΔΔG >3 kcal/mol are shown as red sticks. E, a portion of the AS25-SH2 interface depicting the unsatisfied charge of Arg-239 and three positions in the monobody where mutations increased the affinity. The graph shows binding titration curves measured in the yeast surface display format. arbit., arbitrary.
TABLE 2.
Interface statistics of the monobody-SH2 complexes
The buried surface areas contributed by different segments of the monobodies in Å2 and their fractions (in parentheses) with respect to the total surface area are shown.
| Region | GG3 | AS25 (crystal A) | AS25 (crystal B) |
|---|---|---|---|
| C strand | 35 (6%) | 72 (8%) | 83 (12%) |
| CD loop | 52 (9%) | 173 (21%) | 82 (12%) |
| D strand | 36 (6%) | 10 (1%) | 28 (4%) |
| DE loop and E strand | 0 (0%) | 0 (0%) | 0 (0%) |
| F strand | 59 (10%) | 120 (15%) | 102 (15%) |
| FG loop | 311 (54%) | 318 (39%) | 303 (45%) |
| G strand | 88 (15%) | 125 (15%) | 80 (12%) |
| Total, strands | 219 (38%) | 327 (40%) | 293 (43%) |
| Total, loops | 362 (62%) | 491 (60%) | 385 (57%) |
| Total | 581 | 818 | 678 |
| Sca | 0.769 | 0.738 | 0.697 |
| Hydrogen bondsb | 3 | 6 | 6 |
a The surface complementary values.
b The number of predicted hydrogen bonds in the interface.
In both structures, the FG loop contributed substantially to the buried surface areas in the interface with 311 and 303 Å2 of buried surface areas for GG3 and AS25, respectively (Table 2). However, except for a Val residue inserting its side chain into a small cavity of the SH2 domain (Fig. 5, B and C), the locations and modes of interaction of the FG loops were different between the two structures (Fig. 5A). In the AS25 complexes, the FG loop forms a broad, flat surface in which all but one residue (Tyr-84) make contact with the SH2 domain (Fig. 5B). By contrast, in the GG3 structure, only five residues located in the C-terminal half of the FG loop make contact with the SH2 domain, but each buries a large surface area (>50 Å2; Fig. 4C). Together, the crystal structures confirm that the monobodies target the intended surface of the SH2 domain and demonstrate that there are multiple, distinct solutions to the problem of recognizing the same surface of a target protein even using a single scaffold.
Interface Energetics and Affinity Maturation of AS25
To assess the energetic contributions of individual monobody residues within the AS25-Abl SH2 interface, we performed alanine-scanning mutagenesis. We constructed individual point mutants of a total of nine residues in AS25 that each buries more than 20 Å2 of surface area in both crystal structures. The mutated residues are together responsible for >55% of total monobody surface area burial. We measured their affinity to the Abl SH2 domain by surface plasmon resonance. The experiment identified four hot spot residues, Tyr-45, Tyr-79, Val-80, and Asn-83, where mutation to alanine markedly reduced binding, corresponding to a ΔΔG ≥3.0 kcal mol−1 (Fig. 5D). In addition, two residues (G76A and P87A) contributed modestly to binding (ΔΔG = ∼0.9 and 1.0 kcal mol−1, respectively). These results provide further validation of the contacts observed in the crystal structure and highlight energetic contributions from both the CD and FG loops, consistent with the cradle-like design of the library (Fig. 2B).
We next enhanced the affinity of AS25 using structure-guided design. We found that Arg-239 of the Abl SH2 domain located in the interaction interface had no negative charges in its vicinity (Fig. 5E). The T39D mutation in the C-strand of AS25 enhanced the affinity ∼8-fold to a KD value of 2.5 ± 2 nm (Fig. 5E), probably by forming electrostatic interactions with Arg-239. Similarly, from a small combinatorial library, we identified a double mutant, S89Q/N91D, that had even higher affinity with a KD value of 1.3 ± 0.2 nm (∼15-fold enhancement). Interestingly, the S89Q/N91D mutations only marginally enhanced (∼4-fold) the affinity of AS27, and T39D had no significant effect (data not shown). The inability to transfer the positive effects of these mutations on AS25 affinity to AS27 suggests that the local structure near Arg-239 is substantially different between the two monobody-Abl SH2 complexes, and thus the mutations did not form productive interactions in AS27. Although our structure-guided designs were successful in enhancing the affinity of AS25, the highest affinity achieved did not substantially exceed that of AS27. Thus, we used AS25 and AS27 in the functional assays described below.
AS25 and AS27 Bind and Inhibit Kinase Activity of Bcr-Abl
To examine the effects of the monobodies on Bcr-Abl kinase activity, we first performed binding experiments to determine whether AS25 and AS27 are able to compete with the SH2-kinase domain interaction “in trans.” We expressed and purified fragments of Bcr-Abl, one containing the SH2-KD and the other containing only the KD as described recently (16). In addition, SH2-KD proteins carrying a mutation that either disrupts or increases the SH2-KD interaction, I164E in the SH2 domain or T231R in the kinase domain, respectively, were purified (31, 32). Size exclusion chromatography experiments showed efficient formation of AS25 complexes with the wild-type and T231R-containing SH2-KD proteins, whereas no binding of AS25 to KD or SH2-KD(I164E) was observed (Fig. 3, C–F). AS25 and AS27 bound the SH2-KD protein with KD values of ∼10 nm as tested using monobodies displayed on yeast surface (Fig. 3B).
To determine whether AS25 or AS27 binding to the SH2-kinase interface also results in inhibition of kinase activity, we performed in vitro kinase assays with recombinant SH2-KD and KD. We observed substantial decreases in the in vitro kinase activity of SH2-KD with these monobodies in a dose-dependent manner, whereas KD alone was not inhibited (Fig. 6A). At the highest monobody concentrations tested, the level of SH2-KD activity was the same as that observed for KD. This degree of decrease in kinase activity is similar to that by the I164E mutation on full-length Bcr-Abl, a mutation that disrupts the SH2-kinase interface (9). In line with our previous data with the HA4–7c12 fusion monobody and other allosteric Bcr-Abl inhibitors (9, 33), higher concentrations of these inhibitors are needed to impact in vitro kinase activity than would be expected from binding affinity measurement to the allosteric pocket/domain. These data strongly suggest that the AS25 and AS27 monobodies inhibit Abl kinase by binding to the targeted surface of the SH2 domain and thereby disrupting the SH2-kinase interface as designed.
FIGURE 6.
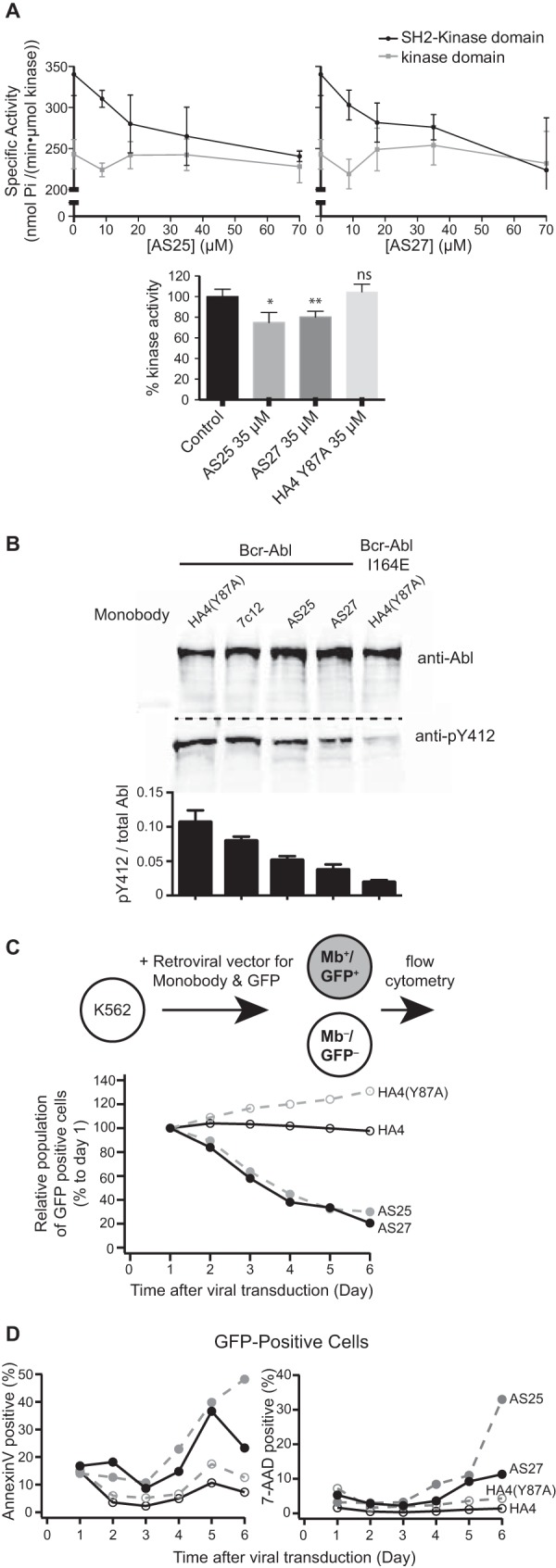
Inhibition of Bcr-Abl and Abl by monobodies. A, inhibition of Abl kinase activity in vitro. The activity of recombinant Abl kinase domain (gray) and Abl SH2-kinase domain unit (black) were assayed in the presence of the indicated concentrations of recombinant AS25 (left panel) or AS27 (right panel). The 0 μm AS25 and AS27 controls contained 70 μm HA4 harboring the Y87A mutation as a non-binding negative control (13). Specific activities for phosphorylation of an optimal Abl substrate peptide are shown. Error bars represent S.D. Significance levels in comparison with control are indicated (*, p < 0.05; **, p < 0.01; unpaired t test). The lower panel compares relative kinase activity in the presence of the indicated monobodies. Error bars represent S.D. B, inhibition of the Bcr-Abl kinase expressed in HEK293 cells by co-expressed monobodies. Besides AS25 and AS27, the first generation monobody directed to the SH2-kinase interface, 7c12 (9), and HA4(Y87A) are included. Phosphorylation of Abl Tyr-412 in the kinase activation loop was normalized to the total Bcr-Abl amount by quantitative immunoblotting. The graph shows quantification of the blots. Error bars represent S.D. C, effect of monobody expression on proliferation of the K562 cell line. The relative population of monobody (Mb)-expressing cells to non-expressing cells quantified using flow cytometry is plotted versus time after viral transduction. D, the fractions among the GFP-positive cells from C that were positive for early (Annexin V; left panel) and late (7-aminoactinomycin D; right panel) apoptosis markers are plotted versus time after viral transduction.
AS25 and AS27 Inhibit Bcr-Abl Activity in Cells and Inhibit CML Cell Survival
To determine whether AS27 is sufficiently specific to be a useful tool in the cellular context, we first examined its ability to capture endogenous Bcr-Abl from cell lysates. In this assay, a biotinylated recombinant AS27 protein was incubated with lysate from the Bcr-Abl-expressing cell line K562, and following the addition of streptavidin-coated beads and serial washing bound Abl and Bcr-Abl were detected using a fluorescently labeled monobody, HA4, that is specific to the phosphopeptide-binding interface of the Abl SH2 domain (13) or with an anti-Bcr antibody (Fig. 7A). AS27 captured Bcr-Abl kinase, whereas a control, non-binding mutant, AS27(Y45K/V80K), did not. Moreover, the capture of Bcr-Abl by AS27 was eliminated by preincubating AS27 with purified Abl SH2 domain. Likewise, co-immunoprecipitation experiments showed strong binding of AS25 and AS27 to constitutively activated full-length Abl protein (“Abl PP”) (27) in mammalian cell lysates, whereas the I164E mutation strongly decreased binding to background levels observed with a non-binding control monobody (Fig. 7B). Together, these results demonstrate that AS25 and AS27 are sufficiently specific to recognize the SH2 domain of full-length Abl and Bcr-Abl in cell lysates.
FIGURE 7.

Interactions and effects of monobodies in cellular contexts. A, a scheme showing the principle of the pulldown experiment (left) and the results (graphs) from two independent experiments. The left graph shows results obtained with an anti-Bcr antibody, and the right graph shows results obtained with a monobody binding to the Tyr(P)-binding pocket of the Abl SH2 domain. B, besides AS25 and AS27, the first generation monobody directed to the SH2-kinase interface, 7c12 (9), and HA4 harboring the Y87A mutation (13) as a non-binding negative control were co-expressed as Myc-tagged proteins with a constitutively active mutant of Abl (Abl PP) and the same mutant in which the SH2-kinase interface was disrupted in addition (Abl PP(I164E)) in HEK293 cells. Immunoprecipitation (IP) with an anti-Abl antibody revealed equal levels of Abl PP protein in the immunoprecipitates (upper panel), whereas AS25 and AS27 and to a lesser extent 7c12 were co-precipitated (lower panel). C, HA4(Y87A), AS25, and AS27 were co-expressed with Abl PP in HEK293 cells in duplicates. Equal amounts of total cell lysate were immunoblotted with the indicated antibodies. D, total Abl tyrosine phosphorylation levels (left graph) and phosphorylation of Abl Tyr-412 in the kinase activation loop (right graph) of the samples in C were normalized to the total Abl amount by quantitative immunoblotting. The graphs show quantification of the blots. Error bars represent S.D. The levels were scaled so that the level for HA4(Y87A) corresponded to 1.0. Significance levels in comparison with HA4(Y87A) are indicated (*, p < 0.05; **, p < 0.01; unpaired t test).
We next assessed the effects of the AS25 and AS27 on Bcr-Abl activity in cells by examining the activation state of Bcr-Abl by co-expressing Bcr-Abl with different monobodies in HEK293 cells. AS25 and AS27 strongly decreased the phosphorylation of Abl Tyr-412 located in the activation loop of the kinase, a marker for the activated Bcr-Abl kinase (Fig. 6B). The degree of inhibition was greater than that achieved with the previous monobody 7c12 and approached that by the interface-disrupting mutation I164E. These results strongly suggest that these new monobodies potently inhibit Bcr-Abl in cells. Similar results were obtained with constitutively active full-length Abl (Fig. 7C).
Finally, we tested whether the inhibition of Bcr-Abl kinase activity that we observed in vitro and in cells impacted survival of the CML cell line K562. Growth and survival of K562 cells are exquisitely dependent on Bcr-Abl kinase activity and signaling (34). We attempted to constitutively express AS25 or AS27 in K562 cells by retroviral gene transfer. Despite our repeated trials and in contrast to 7c12 and several other monobodies (9, 13, 30), we were unable to detect AS25 or AS27 monobody expression by immunoblotting or by flow cytometry analysis for a co-expressed GFP 6 days after retroviral infection (data not shown). In contrast, the Bcr-Abl-negative cell lines UT-7 and HEK293 could be transduced to express AS25 or AS27 constitutively, stably over time, and at comparably high expression levels as several other monobodies (data not shown). This difficulty prompted us to hypothesize that AS25 and AS27 expression might not be compatible with K562 cell proliferation or survival. To test this possibility, we monitored the relative population of GFP-positive, i.e. monobody-expressing, cells to non-expressing cells starting 24 h after retroviral infection of K562 cells. The cells expressing AS25 or AS27, but not HA4 or a non-binding mutant, were rapidly depleted from the culture (Fig. 6C), and the AS25- or AS27-expressing cell population contained much more early and late apoptotic cells as detected using Annexin V and 7-aminoactinomycin D staining, respectively (Fig. 6D). These results indicated that AS25 and AS27 indeed strongly inhibit proliferation of K562 and induce apoptosis. Taken together, these results from cellular studies strongly suggest that AS25 and AS27 potently inhibit the kinase activity of Bcr-Abl (and Abl) that is critical for downstream signaling pathways that mediate cell survival of the Bcr-Abl oncogene-addicted K562 cell line.
Discussion
This work establishes the feasibility of targeting only the SH2-kinase interface for pharmacologically inhibiting Bcr-Abl. Although one might have considered that the original tandem monobody was successful only because the binding of the HA4 monobody to a readily accessible epitope effectively anchored the 7c12 monobody to the vicinity of the SH2-kinase interface (Fig. 1B), results in this work show that such anchoring is not a prerequisite for disrupting the interface. They also show that the blockage of the SH2-Tyr(P) ligand interactions by the HA4 monobody was not required for Bcr-Abl inhibition when the SH2-kinase interface is potently inhibited. Interestingly, these monobodies bound to the SH2-kinase fragment as tightly as they did to the SH2-only fragment (Fig. 3). The negligible effect of the kinase domain is surprising as we anticipated that the kinase domain should sequester the monobody-binding site. The fact that AS25 and AS27 with low nm KD values achieved effective inhibition suggests that the SH2-kinase interface is dynamic and exhibits substantial levels of transient opening during which these monobodies can bind to the epitope. This is also in line with the very low affinity between the separated SH2 and kinase domains, i.e. in trans binding.4 These results are encouraging in terms of developing therapeutic agents targeted to the SH2-kinase interface.
Structural analyses of the two monobody-SH2 complexes revealed a small cleft of the SH2 domain that both monobodies exploited (Fig. 5, B and C). Three hot spot residues of AS25 (Tyr-79, Val-80, and Asn-83) bound to a contiguous surface including this cavity. It is tempting to speculate that one might be able to use this structural information for generating small molecule inhibitors, either by directly designing small molecule mimics of the hot spot residue or by performing site-directed screening of small molecules utilizing, for example, the tethering approach (35).
It is notable that AS25 and AS27 have been generated in an unbiased manner, i.e. without the use of a competitor, but they bound to the kinase interaction surface of the Abl SH2 domain. In contrast, our first attempt using the “loop-only” library produced monobodies predominantly targeted to the phosphopeptide-binding cleft of the Abl SH2 domain, such as the HA4 monobody (13). These two epitopes are involved in natural protein-protein interactions, but their shapes are distinct. The kinase-binding surface is flat and slightly convex, whereas the peptide-binding site forms a cleft. These observations suggest that the new library includes monobody clones that are particularly suited for binding to a flatter surface. These observations further support the importance of controlling interface topography in designing binding proteins (28). The results also illustrate the importance of utilizing complementary libraries in generating monobodies to a new target or a new site.
Although the focus of this study was to determine the sufficiency of targeting a particular surface of a target protein for inhibition, our results clearly illustrate a general approach that can identify a potentially druggable site and define cellular effects of perturbing the site. Technological advances have made it possible to rapidly generate synthetic binding proteins, such as monobodies, with high affinity and high specificity suitable for assessing cellular effects. These attractive features should make synthetic binding proteins broadly useful in drug target discovery and validation.
Author Contributions
J. W., O. H., and S. K. designed the project. J. W., G. G., A. K., and S. K. generated monobodies and performed in vitro binding experiments. J. W. and G. G. performed crystal structure determination. J. W. performed NMR characterization. L. A. and A. K. performed structure-guided affinity maturation. A. J. L. performed in vitro and cellular kinase inhibition assays. B. G. performed assays on CML cells. J. W., O. H., and S. K. wrote the paper with contributions from the other authors. All authors commented on the manuscript.
Acknowledgments
We thank S. Georgeon for the gel filtration data and technical support throughout the project. We acknowledge the use of the Northeastern Collaborative Access Team beamlines, which are funded by National Institutes of Health Grant P41 GM103403. The Pilatus 6M detector on the 24-ID-C beamline is funded by National Institutes of Health Office of Research Infrastructure Programs High-End Instrumentation Grant S10 RR029205. The Advanced Photon Source is a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
This work was supported by National Institutes of Health Grants R01-GM090324 (to S. K.), T32GM07281 (to J. W.), and P30CA014599 (to The University of Chicago Comprehensive Cancer Center), the Institut Suisse de Recherche Expérimentale sur le Cancer (ISREC) Foundation (to O. H.), Swiss National Centre of Competence in Research–Chemical Biology and Swiss Cancer League Grants KLS-3132-02-2013 and KLS-3595-02-2015 (to A. J. L., B. G., and O. H.). J. W., A. K., and S. K. are listed as inventors on a patent application filed by The University of Chicago that covers a design of monobody libraries (United States Patent application 13/813,409). The other authors declare no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5DC0, 5DC4, and 5DC9) have been deposited in the Protein Data Bank (http://wwpdb.org/).
A. J. Lamontanara and O. Hantschel, unpublished observation.
- BCR
- breakpoint cluster region
- ABL
- Abelson tyrosine kinase
- CML
- chronic myelogenous leukemia
- HSQC
- heteronuclear single quantum correlation
- KD
- kinase domain
- SH2
- Src homology 2
- TLS
- translation/libration/screw.
References
- 1. Holohan C., Van Schaeybroeck S., Longley D. B., and Johnston P. G. (2013) Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 [DOI] [PubMed] [Google Scholar]
- 2. Shah N. P., and Sawyers C. L. (2003) Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene 22, 7389–7395 [DOI] [PubMed] [Google Scholar]
- 3. Deininger M. W., Goldman J. M., and Melo J. V. (2000) The molecular biology of chronic myeloid leukemia. Blood 96, 3343–3356 [PubMed] [Google Scholar]
- 4. Shah N. P., Nicoll J. M., Nagar B., Gorre M. E., Paquette R. L., Kuriyan J., and Sawyers C. L. (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 [DOI] [PubMed] [Google Scholar]
- 5. Hantschel O., Grebien F., and Superti-Furga G. (2012) The growing arsenal of ATP-competitive and allosteric inhibitors of BCR-ABL. Cancer Res. 72, 4890–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wylie A., Schoepfer J., Berellini G., Cai H., Caravatti G., Cotesta S., Dodd S., Donovan J., Erb B., Furet P., Gangal G., Grotzfeld R., Hassan Q., Hood T., Iyer V., et al. (2014) ABL001, a potent allosteric inhibitor of BCR-ABL, prevents emergence of resistant disease when administered in combination with nilotinib in an in vivo murine model of chronic myeloid leukemia. Blood 124, 398 [Google Scholar]
- 7. Hantschel O., and Superti-Furga G. (2004) Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 5, 33–44 [DOI] [PubMed] [Google Scholar]
- 8. Filippakopoulos P., Kofler M., Hantschel O., Gish G. D., Grebien F., Salah E., Neudecker P., Kay L. E., Turk B. E., Superti-Furga G., Pawson T., and Knapp S. (2008) Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell 134, 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grebien F., Hantschel O., Wojcik J., Kaupe I., Kovacic B., Wyrzucki A. M., Gish G. D., Cerny-Reiterer S., Koide A., Beug H., Pawson T., Valent P., Koide S., and Superti-Furga G. (2011) Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell 147, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koide A., Bailey C. W., Huang X., and Koide S. (1998) The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 284, 1141–1151 [DOI] [PubMed] [Google Scholar]
- 11. Koide A., Wojcik J., Gilbreth R. N., Hoey R. J., and Koide S. (2012) Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 415, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koide S., Koide A., and Lipovšek D. (2012) Target-binding proteins based on the 10th human fibronectin type III domain ((10)Fn3). Methods Enzymol. 503, 135–156 [DOI] [PubMed] [Google Scholar]
- 13. Wojcik J., Hantschel O., Grebien F., Kaupe I., Bennett K. L., Barkinge J., Jones R. B., Koide A., Superti-Furga G., and Koide S. (2010) A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 17, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koide A., Gilbreth R. N., Esaki K., Tereshko V., and Koide S. (2007) High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. U.S.A. 104, 6632–6637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kunkel T. A., Roberts J. D., and Zakour R. A. (1987) Rapid and efficient site-directed mutagenesis without phenotypic selection. Methods Enzymol. 154, 367–382 [DOI] [PubMed] [Google Scholar]
- 16. Lamontanara A. J., Georgeon S., Tria G., Svergun D. I., and Hantschel O. (2014) The SH2 domain of Abl kinases regulates kinase autophosphorylation by controlling activation loop accessibility. Nat. Commun. 5, 5470. [DOI] [PubMed] [Google Scholar]
- 17. Koide A., Wojcik J., Gilbreth R. N., Reichel A., Piehler J., and Koide S. (2009) Accelerating phage-display library selection by reversible and site-specific biotinylation. Protein Eng. Des. Sel. 22, 685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 19. Collaborative Computational Project, Number 4 (1994) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 20. Potterton E., Briggs P., Turkenburg M., and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 21. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 22. Brünger A. T. (1992) X-PLOR: A System for X-ray Crystallography and NMR, Version 3.1, Yale University Press, New Haven, CT [Google Scholar]
- 23. Painter J., and Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 24. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Reynolds C., Damerell D., and Jones S. (2009) ProtorP: a protein-protein interaction analysis server. Bioinformatics 25, 413–414 [DOI] [PubMed] [Google Scholar]
- 26. Lawrence M. C., and Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 27. Barilá D., and Superti-Furga G. (1998) An intramolecular SH3-domain interaction regulates c-Abl activity. Nat. Genet. 18, 280–282 [DOI] [PubMed] [Google Scholar]
- 28. Gilbreth R. N., and Koide S. (2012) Structural insights for engineering binding proteins based on non-antibody scaffolds. Curr. Opin. Struct. Biol. 22, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang X., Yang X., Luft B. J., and Koide S. (1998) NMR identification of epitopes of Lyme disease antigen OspA to monoclonal antibodies. J. Mol. Biol. 281, 61–67 [DOI] [PubMed] [Google Scholar]
- 30. Sha F., Gencer E. B., Georgeon S., Koide A., Yasui N., Koide S., and Hantschel O. (2013) Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. Proc. Natl. Acad. Sci. U.S.A. 110, 14924–14929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagar B., Hantschel O., Seeliger M., Davies J. M., Weis W. I., Superti-Furga G., and Kuriyan J. (2006) Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol. Cell 21, 787–798 [DOI] [PubMed] [Google Scholar]
- 32. Sherbenou D. W., Hantschel O., Kaupe I., Willis S., Bumm T., Turaga L. P., Lange T., Dao K. H., Press R. D., Druker B. J., Superti-Furga G., and Deininger M. W. (2010) BCR-ABL SH3-SH2 domain mutations in chronic myeloid leukemia patients on imatinib. Blood 116, 3278–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adrián F. J., Ding Q., Sim T., Velentza A., Sloan C., Liu Y., Zhang G., Hur W., Ding S., Manley P., Mestan J., Fabbro D., and Gray N. S. (2006) Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol. 2, 95–102 [DOI] [PubMed] [Google Scholar]
- 34. Hantschel O., Gstoettenbauer A., Colinge J., Kaupe I., Bilban M., Burkard T. R., Valent P., and Superti-Furga G. (2008) The chemokine interleukin-8 and the surface activation protein CD69 are markers for Bcr-Abl activity in chronic myeloid leukemia. Mol. Oncol. 2, 272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Erlanson D. A., Wells J. A., and Braisted A. C. (2004) Tethering: fragment-based drug discovery. Annu. Rev. Biophys. Biomol. Struct. 33, 199–223 [DOI] [PubMed] [Google Scholar]