

Abstract
FoxO1 binds to insulin response elements located in the promoters of insulin-like growth factor-binding protein 1 (IGFBP1) and glucose-6-phosphatase (G6Pase), activating their expression. Insulin-mediated phosphorylation of FoxO1 promotes cytoplasmic translocation, inhibiting FoxO1-mediated transactivation. We have previously demonstrated that FoxO1 opens and remodels chromatin assembled from the IGFBP1 promoter via a highly conserved winged helix motif. This finding, which established FoxO1 as a “pioneer” factor, suggested a model whereby FoxO1 chromatin remodeling at regulatory targets facilitates binding and recruitment of additional regulatory factors. However, the impact of FoxO1 phosphorylation on its ability to bind chromatin and the effect of FoxO1 loss on recruitment of neighboring transcription factors at its regulatory targets in liver chromatin is unknown. In this study, we demonstrate that an amino acid substitution that mimics insulin-mediated phosphorylation of a serine in the winged helix DNA binding motif curtails FoxO1 nucleosome binding. We also demonstrate that shRNA-mediated loss of FoxO1 binding to the IGFBP1 and G6Pase promoters in HepG2 cells significantly reduces binding of RNA polymerase II and the pioneer factors FoxA1/A2. Knockdown of FoxA1 similarly reduced binding of RNA polymerase II and FoxO1. Reduction in acetylation of histone H3 Lys-27 accompanies loss of FoxO1 and FoxA1/A2 binding. Interdependent binding of FoxO1 and FoxA1/A2 possibly entails cooperative binding because FoxO1 and FoxA1/A2 facilitate one another's binding to IGFPB1 promoter DNA. These results illustrate how transcription factors can nucleate transcriptional events in chromatin in response to signaling events and suggest a model for regulation of hepatic glucose metabolism through interdependent FoxO/FoxA binding.
Keywords: chromatin, gene regulation, histone modification, insulin, nucleosome, FoxA1, FoxA2, FoxO1
Introduction
A challenge in the field of transcriptional regulation is to discover how signaling pathways exert their influence on chromatin structure through DNA binding proteins. Targeted modification of transcription factors, which provides rapid alterations in their activities in response to external and internal stimuli, is an important mechanism in the regulation of their target genes. The perturbations in chromatin structure required to poise genes for activation and modulate their expression are mediated, at least in part, by initial “pioneer” chromatin binding factors capable of binding to and remodeling chromatin in response to environmental and/or developmental cues (1–12) (reviewed in Refs. 13 and 14). Previous studies have revealed that targeted post-translational modification of these pioneer factors can substantially alter their ability to interact with cellular chromatin (15–18) and in vitro assembled chromatin templates (19, 20). However, the impact of these alterations in pioneer factor chromatin binding on epigenetic modification and regulatory factor recruitment necessary for the transcriptional response to extracellular signaling cascades is largely unstudied.
To address this, we have investigated the impact of insulin-mediated phosphorylation on chromatin binding and regulatory factor recruitment by the pioneer forkhead transcription factor FoxO1. FoxO1 (formerly FKHR) belongs to the FoxO3 subfamily of forkhead transcription factors that also includes FoxO3, FoxO4, and FoxO6 (21, 22). The FoxO factors regulate the expression of multiple genes encoding glucose metabolic enzymes, pro-apoptotic factors, and cell cycle regulators in multiple tissues (reviewed in Refs. 23–25). In particular, FoxO1 regulates the expression of the insulin-like growth factor-binding protein 1 (IGFBP1) and glucose-6-phosphatase (G6PC) genes in response to insulin signaling in liver (26–29), and ablation or inactivation of FoxO1 in mice leads to insulin-mediated repression of gluconeogenesis (30, 31).
The function of FoxO factors is dynamically regulated by post-translational modification. In response to insulin, protein kinase B (PKB)/Akt-mediated phosphorylation of FoxO1 at three conserved sites, Thr-24, Ser-253, and Ser-316, located in the N-terminal, DNA binding, and C-terminal domains of the protein, respectively, leads to the cytoplasmic retention of FoxO1 and the consequent inhibition of its transcriptional activity (32–38). In the absence of stimulation by insulin or growth factors, FoxO1 is dephosphorylated and translocates into the nucleus. In the nucleus, FoxO1 binds to an insulin response element (IRE) located in the promoters of its target genes, transactivating their expression (26–29, 39).
We have previously demonstrated that FoxO1 stably binds to its sites assembled onto nucleosome particles and opens linker histone-compacted nucleosome arrays (40). These capabilities, which establish FoxO1 as a pioneer factor capable of initiating regulatory events in chromatin, map to a winged helix “forkhead box” DNA binding motif (41, 42); sequence variants of this motif define the forkhead classes FOXA–FOXS (43). This highly conserved DNA binding domain, named for the crystal structure of the forkhead transcription factor FoxA3, is a variant of a helix-turn-helix motif in which the recognition helix is flanked by two “wings” of polypeptide chain (41). X-ray crystallographic studies have demonstrated the structural resemblance of the forkhead box and the winged helix DNA binding “globular” domain of the linker histone (41, 44), and this DNA binding motif is accordingly used by forkhead factors, including FoxO1 and FoxA, to mediate their effects on cellular processes through chromatin remodeling (5, 9, 40, 45–47).
Intriguingly, one site of Akt-mediated FoxO1 phosphorylation, serine 253, resides within a domain of the FoxO1 winged helix motif, wing 2, which stabilizes binding of the recognition helix to DNA (42, 48). Whereas cytoplasmic sequestration of phosphorylated FoxO1 in response to insulin is well established, the impact of phosphorylation on the ability of FoxO1 to bind and remodel chromatin is unknown. More important, the impact of insulin-mediated loss of FoxO1 binding at insulin-regulated FoxO1 targets on chromatin structure, epigenetic modifications, and recruitment of adjacent transcription factors has not been studied.
To answer these questions, we first determined the impact of insulin-mediated FoxO1 phosphorylation on chromatin binding by comparing binding of recombinant wild type FoxO1 and FoxO1 phosphorylation mimics with in vitro reconstituted nucleosome particles containing the IGFBP1 promoter. Remarkably, we find that an amino acid substitution that mimics insulin-mediated phosphorylation of a serine residue located in the FoxO1 winged helix motif, Ser-253, specifically curtails nucleosome binding by FoxO1 without affecting the affinity of the protein for its sites in free DNA. To more fully understand the consequence of insulin-mediated loss of FoxO1 binding at FoxO1 target genes within cellular chromatin, we next determined the impact of shRNA-mediated FoxO1 knockdown on binding of neighboring regulatory factors and epigenetic modifications in hepatic cells.
We demonstrate for the first time interdependent binding of FoxO1 and FoxA1/A2 to adjacent sites at the IGFBP1 and glucose-6-phosphatase (G6Pase) promoters, whereby shRNA or insulin-mediated loss of FoxO1 binding curtails that of FoxA1/A2 and shRNA-mediated loss of FoxA1 curtails FoxO1 binding. Failure of either FoxO1 or FoxA1/A2 binding curtails recruitment of RNA polymerase II and acetylation of H3K27. FoxO1 and FoxA1 bind cooperatively to IGFBP1 promoter DNA in footprinting assays, suggesting a mechanism for interdependent FoxO1-FoxA1/A2 binding in vivo. Taken together, these results illustrate how transcription factors can nucleate transcriptional events in chromatin in response to specific signaling events and suggest a model for regulation of hepatic glucose metabolism through interdependent FoxO/FoxA binding.
Experimental Procedures
Plasmid Construction and Mutagenesis
The bacterial expression plasmid encoding histidine-tagged full-length FoxO1 (pET-16b-His10-FoxO1) (40) was used as the substrate for mutagenesis. Mutagenesis of PKB/Akt phosphorylation sites within murine FoxO1 protein was conducted by overlap PCR. Mutagenesis primers were as follows (5′–3′): T24A, CGC TCC TGT GCC TGG CCG C and CAG CGG CCA GGC ACA GGA G; T24D, CGC TCC TGT GAC TGG CCG CTG CCC AGG CCG and CAG CGG CCA GTC ACA GGA GCG CTG CCG GGG; S253A, AGA GCT GCG GCC ATG GAC AAC AAC AGT AAA and GTT GTC CAT GGC CGC AGC TCT TCT CCG GGG; S253D, AGA GCT GCG GAC ATG GAC AAC AAC AGT AAA and GTT GTC CAT GTC CGC AGC TCT TCT CCG GGG; S316A, CGA ACC AGC GCA AAT GCT AGT ACC ATC AGT and ACT AGC ATT TGC GCT GGT TCG AGG ACG AAA; S316D, CGA ACC AGC GAC AAT GCT AGT ACC ATC AGT and ACT AGC ATT GTC GCT GGT TCG AGG ACG AAA. The bacterial expression plasmid encoding histidine-tagged full-length FoxA1 (pET28b-His6-FoxA) contains the mouse FoxA1 cDNA subcloned into the pET28b plasmid (Novagen, Gibbstown, NJ), as described previously (47).
Protein Preparation
Core histones were isolated from livers of 6-month-old Sprague-Dawley rats and purified by FPLC for footprinting experiments shown in Fig. 1. HeLa core histones were purchased for footprinting experiments shown in Fig. 7 (Vaxron, Rockaway, NJ). Purified recombinant FoxA1 and FoxO1 proteins were expressed in Escherichia coli and purified through a combination of anion exchange and nickel-agarose chromatography, as described previously (20, 40).
FIGURE 1.

Phosphorylation of Ser-253 curtails FoxO1 binding to its sites on nucleosome cores. A, substitution mutant series of FoxO1. Numbers refer to amino acid positions. SDS-PAGE analysis of 3 μg of each protein, stained with Coomassie Blue. M, molecular weight protein markers. B and C, DNase I footprinting analysis of the designated pmol amounts of wild type FoxO1 and the indicated substitution mutants bound to IGFBP1 promoter free DNA (B) and nucleosome cores (C). The position of the IRE is indicated at the left of each panel. The bracket to the right of lane 11 in C denotes loss of DNase protections induced by binding of FoxO1 S253D to the IRE. G, guanine cleavage ladder marker. D, data representing the mean ±S.E. (error bars) of six independent DNase I footprinting experiments quantitated on a phosphor imager and plotted as average percentage occupancy at each designated amount of FoxO1 protein. The 4- and 8-pmol amounts of FoxO1 S253D are bound 30% less well to the IRE assembled onto nucleosome particles compared with the wild type and S253A FoxO1 proteins (**, p < 0.02, *, p < 0.05, Student's t test).
FIGURE 7.
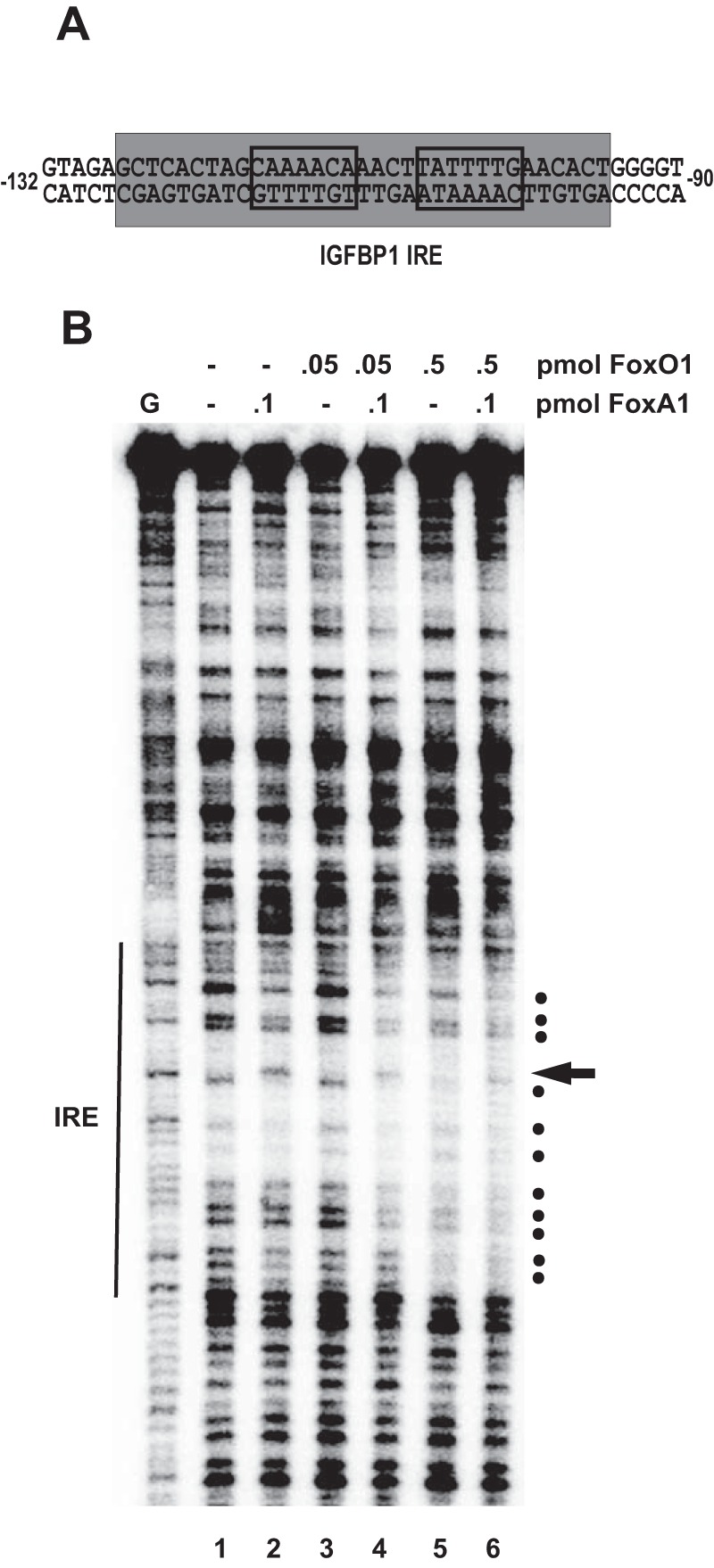
FoxO1 and FoxA1 cooperatively bind the IGFBP1 promoter. A, IGFBP1 IRE sequence. The boundaries of the footprint generated by FoxA1/A2 and FoxO1 binding are highlighted in gray, and the forkhead consensus sites are delineated by boxes. B, DNase I footprinting analysis of the designated pmol amounts of recombinant FoxO1 and FoxA1 bound to IGFBP1 promoter DNA. The position of the IRE is indicated at the left. Dots indicate footprinted sites, and the arrow indicates a hypersensitive site. This gel is representative of four independent DNase I footprinting experiments. G, guanine cleavage ladder marker.
Nucleosome Preparation
Nucleosome cores were prepared as described previously (40). The mouse IGFBP1 promoter DNA fragment corresponds to positions −204 to −25 and was created by PCR with 32P-end-labeled primers (5′–3′, TTA GCT CCT GTC CCA GTC CA and TTA TGA AGG GCT GGC TGT GC) followed by gel purification. 32P-End-labeled IGFBP1 promoter DNA was assembled onto purified core histones by salt-urea gradient dialysis. The nucleosome cores were purified by glycerol gradient sedimentation and dialyzed against 10 mm Tris-HCl, pH 8.0, 1 mm 2-mercaptoethanol.
Binding Reactions and Enzymatic Assays
Binding reactions of wild type and mutant FoxO1 proteins and FoxA1 to free DNA templates or nucleosome cores were performed in final buffer conditions of 10 mm Tris-HCl, pH 7.5, 1 mm MgCl2, 5 mm DTT, 40 mm KCl, 0.5% glycerol, 3 mg/ml BSA, and 1% Ficoll at 21–25 °C for 1 h. The binding reactions were digested with 75 ng (free DNA templates) or 300 ng (nucleosome cores) of DNase I (diluted in 20 mm MgCl2, 1 mm CaCl2) for 1 min at 21–25 °C. Digestion reactions were stopped by the addition of an equal volume of DNase stop buffer (30 mm EDTA, 0.1% SDS, 50 μg/ml tRNA, 0.35 m NaCl), and DNA fragments were extracted with phenol/chloroform (1:1) and then chloroform alone, followed by ethanol precipitation. The DNA fragments were separated on 6% polyacrylamide, 7 m urea sequencing gels in 1× Tris-borate EDTA buffer and exposed to a phosphorimaging screen. Phosphor images were subjected to densitometry using ImageGauge version 1.0.0.1 where indicated. Statistical analysis of densitometry results was performed using Student's t test.
Tissue Culture
293T cells obtained from the laboratory of Dr. Steven Duncan (Medical College of Wisconsin) were maintained in Dulbecco's modified Eagle's high glucose medium containing l-glutamine and pyruvate supplemented with 10% fetal bovine serum and 10 units of antibiotic solution (Invitrogen). HepG2 cells obtained from the American Tissue Culture Collection (Manassas, VA) were maintained in Earle's salts and l-glutamine-containing minimum essential medium lacking ribonucleosides and deoxyribonucleosides supplemented with 10% fetal bovine serum and 10 units of antibiotic solution.
Lentivirus Production and shRNA Knockdown
pLKO.1 lentiviral shRNA constructs targeting human FoxO1 and FoxA1 were purchased from GE Dharmacon (Lafayette CO). The FoxO1 targeting construct clone ID is TRCN0000020707. The FoxA1 targeting construct clone ID is TRCN0000014881. pLKO.1 expressing an unrelated scrambled shRNA (Addgene plasmid 1864) was used as a control for off-target effects (49). 293T cells were used to package infectious lentivirus particles. Lipofectamine 2000 (Invitrogen) was used to transfect 293T cells with each of the lentiviral shRNA constructs and the three-plasmid packaging system (Invitrogen). 24 h after transfection, medium was replaced with fresh HepG2 medium, and cells were grown for 48 h for virus production. Lentivirus-containing HepG2 medium was harvested and filtered through a 45-μm filter. Lentivirus infection of HepG2 cells was performed in the presence of 6 μg/ml Polybrene. A day after infection, the lentivirus-containing medium was replaced with fresh HepG2 medium. Infection was carried out for a total of 72 h. For insulin treatment of knockdown HepG2 cells, the cells were placed in serum-free medium 24 h prior to harvest. 6 h prior to harvest, the serum-free medium was replaced with fresh serum-free medium with or without 1 μg/ml insulin (Sigma).
Western Blotting
Cells were lysed using radioimmune precipitation assay buffer (1.25% Triton X-100, 1.25% sodium deoxycholate, 0.0125 m sodium phosphate, pH 7.2, 2 mm EDTA, 50 mm sodium fluoride, protease inhibitor mixture (Sigma)), and whole cell extracts were subjected to Western blotting analysis as described previously (20). Extracts were run on a 10% gel, and proteins were transferred to a PVDF membrane (Bio-Rad). Blots were incubated with primary antibodies overnight and, after washes with TBST buffer, were incubated with secondary antibodies for 1 h. Primary antibodies were anti-FoxO1 (C29H4, lot 10 (Cell Signaling, Danvers, MA)) at a 1:1,000 dilution, anti-FoxA1 (2F83, lot 0513A (Seven Hills Bioreagents, Cincinnati, OH)) at a 1:1,000 dilution, anti-FoxA2 (WRAB-1200, lot 0614A (Seven Hills Bioreagents)) at a 1:5,000 dilution, and anti-α-tubulin (sc-E-19-R, lot K2910 (Santa Cruz Biotechnology, Inc., Dallas, TX)) at a 1:1,000 dilution. Secondary antibodies were goat anti-rabbit IgG-HRP at a 1:5,000 dilution (sc-2030, lot F2613 (Santa Cruz Biotechnology)) and goat anti-mouse IgG-HRP at a 1:5,000 dilution (sc-2031, lot F0711 (Santa Cruz Biotechnology)). Detection was by enhanced chemiluminescence (GE Healthcare).
RNA Isolation and Quantitative RT-PCR
Total RNA was isolated from cells using the QIAshredder and RNeasy minikits (Qiagen, Germantown, MD). cDNA was synthesized from up to 2.5 μg of total RNA using the reverse transcriptase Maloney murine leukemia virus and random primers (Invitrogen). Quantitative RT-PCR was performed using an icycler (Bio-Rad). Relative gene expression was determined using the 2−ΔΔCt method (50) with the human hypoxanthine-guanine phosphoribosyltransferase gene serving as the internal control. Primers used were as follows (5′–3′): hypoxanthine-guanine phosphoribosyltransferase, AAT TAT GGA CAG GAC TGA ACG TC (forward) and CGT GGG GTC CTT TTC ACC AGC AAG (reverse); IGFBP1, ATC ATT CCA TCC TTT GGG ACG CCA (forward) and TGG ATG TCT CAC ACT GTC TGC TGT (reverse); G6Pase, CCA CCA AGC CTG GAA TAA CTG CAA (forward) and TCA CGG ACA CCA AGA TGA ACC AGT (reverse).
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed as described previously (51–53). HepG2 cells were cross-linked for 10 min with 1% formaldehyde and quenched with 0.125 m glycine. Cell were lysed in 85 mm KCl, 0.5% Nonidet P-40, 5 mm Pipes, and 15 mm sodium butyrate, followed by nuclear lysis in 50 mm Tris-HCl, pH 8.0, 10 mm EDTA, 1% SDS, and 15 mm sodium butyrate. Chromatin was sonicated using a Branson cell disruptor 185 sonifier at setting 4 for four pulses and 20 s/pulse with 2 min between pulses. The average DNA fragment size was ∼500 bp, and chromatin was quantified using the Bradford protein assay (Bio-Rad). Chromatin was diluted by a factor of 6 in immunoprecipitation dilution buffer (1.1% Triton X-100, 0.01% SDS, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.0, 16.7 mm NaCl, and 15 mm sodium butyrate) and precleared with protein A/G-Sepharose beads (Santa Cruz Biotechnology) blocked with BSA and salmon sperm DNA. Up to 600 μg of chromatin was used for immunoprecipitation of transcription factors, up to 136 μg was used for histone modifications, and up to 164 μg of chromatin was used as total input. Immunoprecipitation was performed overnight, followed by an additional 1.5 h of incubation with BSA and salmon sperm DNA-blocked protein A/G-Sepharose beads. For the following antibodies, 2 μg was used for immunoprecipitation: anti-FKHR (sc-11350, Santa Cruz Biotechnology), anti-HNF-3α/β (sc-6553x, Santa Cruz Biotechnology), anti-RNA polymerase II (sc-899, Santa Cruz Biotechnology), C/EBPα (sc-61, Santa Cruz Biotechnology), C/EBPβ (sc-150, Santa Cruz Biotechnology), total H3 (ab1791, Abcam, Cambridge MA), histone H3 acetylated on Lys-27 (17-683, Millipore, Billerica, MA), histone H3 acetylated on Lys-9 (17-658, Millipore), and IgG (Millipore). For the following antibodies, 1 μg was used for immunoprecipitation: histone H3 dimethylated on Lys-4 (ab7766, Abcam) and histone H3 trimethylated on Lys-4 (ab8580, Abcam). Following one wash each in low salt buffer (2 mm EDTA, 20 mm Tris, pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.1% SDS), high salt buffer (2 mm EDTA, 20 mm Tris, pH 8.0, 500 mm NaCl, 1% Triton X-100, 0.1% SDS), lithium chloride buffer (1 mm EDTA, 10 mm Tris, pH 8.0, 1% Igepal, 1% (w/v) sodium deoxycholate, 250 mm LiCl), and TE buffer (1 mm EDTA, 10 mm Tris, pH 8.0), samples were incubated at 67 °C overnight to reverse the formaldehyde cross-links. DNA was purified using the Qiaquick PCR purification kit (Qiagen, Germantown, MD). Primers used for quantitative PCR analysis on the respective promoters were as follows (5′–3′): IGFBP1, TGG ACT TTA ACT GAG GGC CTG AAC (forward) and TGC ACC AGG AGG TTA ATG ATT GGC (reverse); G6Pase, AAG GCA CAG ACT CAT AGC AGA GCA (forward) and TTC CTT GGC ACC TCA GGA AGA TGT (reverse).
Results
Phosphorylation of Ser-253 in the Winged Helix Domain Curtails Nucleosome Binding by FoxO1 on IGFBP1 Mononucleosome Particles
PKB/Akt-mediated phosphorylation of FoxO1 at three conserved sites, Thr-24, Ser-253, and Ser-316, in response to insulin signaling has been demonstrated to repress FoxO1-mediated transactivation of the IGFBP1 promoter (32–38). Phosphorylation of FoxO1 by PKB/Akt takes place in the nucleus, in the context of FoxO1 bound to insulin response elements within promoters of FoxO1-activated genes (reviewed in Refs. 54 and 55). Therefore, the ability of FoxO1 to bind to and remodel nucleosome particles suggested a model for insulin-mediated down-regulation of IGFBP1 whereby phosphorylation of the winged helix and/or other structural domains might result in the attenuation of nucleosome binding by FoxO1 at the IGFBP1 promoter.
In order to investigate this possibility, we studied the binding of FoxO1 proteins containing substitution mutations mimicking insulin-mediated serine/threonine phosphorylation to the IGFBP1 promoter assembled onto nucleosome particles. Mutant FoxO1 proteins containing substitutions of Thr-24, Ser-253, and Ser-316 with aspartic acid (Fig. 1A, lanes 2–4) and, as controls, alanine (lanes 5–7) were prepared, and the ability of each of the substitution mutants to bind to the IRE within free DNA (Fig. 1B) and on nucleosome particles (Fig. 1C) was assessed by DNase I footprinting analysis. The data shown are representative of six independent footprinting experiments.
As seen in Fig. 1B, the T24D, S253D, and S316D FoxO1 substitution mutants each protected the IRE within free DNA as well as the wild type protein (compare lanes 2–4 with lanes 8–10, lanes 14–16, and lanes 20–22, respectively). In contrast, we observed a consistent decrease in binding by the S253D mutant (Fig. 1C, lanes 14–16) at the IRE on the nucleosome particles as compared with the wild type protein (lanes 2–4) and S253A mutant (lanes 11–13). As illustrated in Fig. 1D, quantitation of the data from six separate footprinting experiments revealed that at the 4- and 8-pmol levels, the S253D mutant bound 30% less efficiently to the IRE than either the wild type or S253A FoxO1 proteins (p < 0.02 and 0.05, respectively).
To a lesser extent, occupancy of the nucleosome cores by the T24D mutant was also decreased as compared with the wild type protein or corresponding alanine substitution mutant (compare lanes 8–10 with lanes 2–4 and lanes 5–7, respectively). The S316D mutant (lanes 20–22) bound nucleosomal DNA similar to the wild type protein. We conclude from these experiments that the introduction of a negative charge at S253 curtails binding of FoxO1 to its sites on the IRE assembled onto a nucleosome.
Loss of FoxO1 Binding Attenuates Binding of Regulatory Transcription Factors at the IGFBP1 Promoter
Previous studies have shown that FoxO1 regulates IGFBP1 expression by binding to an IRE within the IGFBP1 promoter and that insulin-mediated loss of FoxO1 binding through FoxO1 phosphorylation and subsequent cytoplasmic sequestration represses IGFPB1 expression (28, 37, 38, 56, 57). The negative impact of the S253D mutation on FoxO1 nucleosome binding suggested a model for FoxO1 regulation of hepatocyte glucose metabolic functions whereby insulin-mediated FoxO1 phosphorylation and the consequent loss of FoxO1 binding will result in the partial or complete loss of chromatin remodeling and modification together with the transcriptional regulators necessary to support transcription. However, the direct impact of FoxO1 loss on binding and recruitment of adjacent regulatory factors and the general transcription machinery has not been addressed.
To address this, we used ChIP to examine the effect of shRNA-mediated FoxO1 loss on transcription factor recruitment at the IGFBP1 promoter in HepG2 human hepatoma cells. HepG2 cells were chosen for these experiments based on their use to study the impact of insulin signaling on FoxO1 transcriptional activity in previous reports (29, 58).
Transient knockdown of endogenously expressed FoxO1 was performed by infection of HepG2 cells with lentivirus expressing FoxO1 shRNA and, as a control, scrambled shRNA. FoxO1 protein expression was successfully knocked down in HepG2 cells expressing this shRNA (Fig. 2A). As expected, insulin treatment of HepG2 cells expressing either the FoxO1 or scrambled shRNA had no additional impact on FoxO1 protein expression (Fig. 2A). In agreement with previous reports of conditional FoxO1 knock-out in adult mouse liver, depletion of FoxO1 protein in HepG2 cells was accompanied by a significant drop in expression of the IGFBP1 gene, similar to that seen in insulin-treated HepG2 cells expressing the scrambled shRNA (Fig. 2B) (56).
FIGURE 2.

FoxO1 knockdown reduces IGFBP1 transcript and curtails FoxA1/A2 binding and RNA polymerase II recruitment to the IGFBP1 promoter. A, Western blot of FoxO1 protein in unstimulated/insulin stimulated scrambled or FoxO1 shRNA-infected HepG2 cells. B, quantitative RT-PCR analysis of IGFBP1 expression in unstimulated/insulin-stimulated HepG2 cells infected with lentivirus expressing scrambled or FoxO1 shRNA. Values are expressed as the -fold change over non-insulin-treated cells infected with the scrambled shRNA. Data represent the mean ± S.E. (error bars) for four independent biological experiments and two technical replicates per biological replicate. C–E, ChIP analysis was performed using antibodies directed against FoxO1 (C), RNA polymerase II (D), and FoxA1/A2 (E) on chromatin isolated from unstimulated/insulin-stimulated scrambled or FoxO1 shRNA-infected HepG2 cells. Results were normalized to total input as an internal control, and values are expressed as a ratio over non-insulin-treated cells infected with the scrambled shRNA. All data represent the mean ± S.E. for four independent biological experiments and two technical replicates per biological replicate. **, p < 0.01, Student's t test. F and G, Western blot of FoxA1 (F) and FoxA2 (G) proteins in unstimulated/insulin-stimulated scrambled or FoxO1 shRNA-infected HepG2 cells.
ChIP confirmed binding of FoxO1 (Fig. 2C), together with the liver-enriched factors FoxA1/A2 (Fig. 2D) and RNA polymerase II (Fig. 2E) to the IGFBP1 promoter in HepG2 cells infected with scrambled shRNA. Immunoprecipitation using IgG served as a negative control. In contrast, loss of FoxO1 binding to the IGFBP1 promoter in the FoxO1 knockdown cells (Fig. 2C) eliminated binding of FoxA1/A2 (Fig. 2D) and RNA polymerase II (Fig. 2E). The antibody used in the ChIP analysis detects both FoxA1 and FoxA2. Similar to FoxO1, FoxA factor binding to the IRE has been demonstrated to activate the IGFBP1 promoter (59), although insulin-mediated repression of IGFPB1 expression is not mediated through FoxA (60). The loss of FoxA1/A2 binding was totally unexpected because FoxA, like FoxO1, functions as a pioneer factor, and therefore its ability to bind cellular chromatin would not be expected to be dependent upon the presence of FoxO1.
Western analysis revealed equal levels of FoxA1 and -2 proteins in HepG2 cells infected with the scrambled and FoxO1 shRNA, showing that loss of FoxA binding is not due to loss of FoxA protein expression (Fig. 2, F and G). Insulin treatment of HepG2 cells expressing the scrambled shRNA exhibited loss of RNA polymerase II and FoxA1/A2 binding, together with the expected loss of FoxO1 binding, similar to the FoxO1 knockdown cells, whereas insulin treatment of FoxO1 knockdown cells did not lead to a further reduction in binding of RNA polymerase II and FoxA1/A2. This indicates that the impact of insulin signaling on regulatory factor binding to this promoter is primarily driven through FoxO1. We conclude that FoxO1 binding is required for recruitment of transcriptional activators, together with the general transcription machinery, to enable activation of the IGFPB1 gene.
Loss of FoxA1/A2 Binding Curtails FoxO1 and RNA Polymerase II Binding to the IGFBP1 Promoter
The IGFPB1 IRE contains two adjacent forkhead binding sites capable of binding FoxO1 and FoxA (37, 61), raising the possibility that FoxO1 and FoxA1/A2 influence each other's binding to the IRE. To investigate this, we knocked down FoxA1 in HepG2 cells by lentiviral infection with FoxA1 shRNA and determined the impact on FoxO1 binding to the IGFPB1 promoter.
FoxA1 protein expression was successfully knocked down in HepG2 cells expressing the FoxA1 shRNA, whereas FoxA2 protein levels were unchanged (Fig. 3, A and B). We were unable to knock down FoxA2 levels in HepG2 cells by expression of FoxA2 shRNA either alone or in conjunction with FoxA1 knockdown (data not shown). Despite the continued presence of FoxA2 protein, ChIP using the antibody recognizing both FoxA1 and FoxA2 demonstrated that FoxA1 knockdown eliminated binding of both FoxA1 and FoxA2 to the IGFBP1 promoter (Fig. 3D), significantly reducing IGFBP1 expression in HepG2 cells in comparison with infection with the scrambled shRNA (Fig. 3C). Loss of FoxA1/A2 binding to the IGFBP1 promoter in the FoxA1 knockdown HepG2 cells significantly reduced binding of RNA polymerase II (Fig. 3E) and completely eliminated binding of FoxO1 (Fig. 3F).
FIGURE 3.

FoxA1 knockdown curtails IGFBP1 expression and promotes loss of FoxO1 and RNA polymerase II binding to the IGFBP1 promoter. A and B, Western blot of FoxA1 (A) and FoxA2 (B) proteins in unstimulated/insulin-stimulated scrambled or FoxA1 shRNA-infected HepG2 cells. C, quantitative RT-PCR analysis of IGFBP1 expression in unstimulated/insulin stimulated scrambled or FoxA1 shRNA-infected HepG2 cells. Values are expressed as the -fold change over non-insulin-treated cells infected with the scrambled shRNA. Data represent the mean ± S.E. (error bars) for three independent biological experiments and two technical replicates per biological replicate. D–F, ChIP analysis was performed with antibodies directed against FoxA1/A2 (D), RNA polymerase II (E), or FoxO1 (F) on chromatin isolated from unstimulated/insulin-stimulated scrambled or FoxA1 shRNA-infected HepG2 cells. Results are normalized to total input, and values are expressed as a ratio over non-insulin-treated cells infected with the scrambled shRNA. Data represent the mean ± S.E. of three independent biological experiments and two technical replicates per biological replicate. ***, p < 0.001; *, p < 0.05, Student's t test. G, Western blot of FoxO1 protein in unstimulated/insulin-stimulated scrambled or FoxA1 shRNA-infected HepG2 cells.
Western blotting analysis revealed equal levels of FoxO1 in HepG2 cells infected with the scrambled and FoxA1 shRNA, showing that loss of FoxO1 binding is not due to loss of FoxO1 protein expression (Fig. 3G). Similar to the situation seen in the FoxO1 knockdown cells, insulin treatment of FoxA1 knockdown cells did not lead to a further reduction in binding of FoxO1 or RNA polymerase II to the IGFBP1 promoter. Taken together, these results suggest that FoxO1 and FoxA1/A2 interdependently bind to the IGFBP1 promoter.
FoxO1/FoxA Interdependently Bind at the G6Pase Promoter
Expression of the G6PC gene, which encodes a rate-limiting enzyme for gluconeogenesis and glycogenolysis, is also regulated by FoxO1 and FoxA1/A2 binding to an IRE present within the gene's promoter. The G6Pase IRE contains three adjacent forkhead binding sites, two of which mediate insulin regulation (62, 63). Similar to what was observed for IGFPB1, FoxO1 knockdown in HepG2 cells significantly reduced G6Pase transcription in comparison with that in HepG2 cells infected with a scrambled shRNA (Fig. 4A). ChIP analysis demonstrated that FoxO1 knockdown in HepG2 cells significantly reduced FoxO1 (Fig. 4B), RNA polymerase II (Fig. 4C), and FoxA1/A2 (Fig. 4D) binding to the G6Pase promoter.
FIGURE 4.

FoxO1/FoxA interdependently bind to the G6Pase promoter. A and E, quantitative RT-PCR analysis of G6Pase expression in unstimulated/insulin stimulated HepG2 cells infected with scrambled or FoxO1 shRNA (A) and scrambled or FoxA1 shRNA (E). Values are expressed as the -fold change over non-insulin-treated cells treated with the scrambled shRNA. Data represent the mean ± S.E. (error bars) for three independent biological experiments and two technical replicates per biological replicate for each. B–D and F–H, ChIP analysis was performed on chromatin isolated from unstimulated/insulin stimulated HepG2 cells infected with scrambled or FoxO1 shRNA (B–D) and scrambled or FoxA1 shRNA (F–H) with antibodies directed against FoxO1 (B and H), RNA polymerase II (C and G), and FoxA1/A2 (D and F). Results are normalized to total input, and values are expressed as a ratio over non-insulin-treated cells infected with the scrambled shRNA. Data represent the mean ± S.E. for three independent biological experiments and two technical replicates per biological replicate for each. **, p < 0.01; *, p < 0.05, Student's t test.
Similarly, FoxA1 knockdown in HepG2 cells significantly reduced G6Pase transcript (Fig. 4E). FoxA1/A2 (Fig. 4F), RNA polymerase II (Fig. 4G), and FoxO1 (Fig. 4H) binding to the G6Pase promoter were also significantly reduced when FoxA1 was knocked down. Insulin treatment in combination with FoxO1 or FoxA1 knockdown did not lead to a further reduction in binding of FoxO1, FoxA1/A2, or RNA polymerase II. We conclude that interdependent FoxO1-FoxA1/A2 binding to an IRE regulates expression of two insulin-regulated genes, IGFBP1 and G6PC.
FoxO1 Knockdown Does Not Impact C/EBPα or -β Binding to the IGFBP1 and G6Pase Promoters
The negative impact of FoxO1 and FoxA1/2 loss on RNA polymerase II binding to the IGFBP1 and G6Pase promoters led us to investigate whether binding of other transcriptional regulators was similarly affected. To accomplish this, we used ChIP to examine binding of C/EBPα and -β to the IGFPB1 and G6Pase promoters in HepG2 cells following FoxO1 knockdown and/or insulin treatment. The liver-enriched transcription factors C/EBPα and -β have previously been shown to bind adjacent to the IRE in the IGFBP1 and G6Pase promoters and to regulate expression of the corresponding genes (64, 65). Neither FoxO1 knockdown nor insulin treatment significantly altered binding of C/EBPα (Fig. 5A) or C/EBPβ (Fig. 5B) to the IGFBP1 promoter. Binding of C/EBPα (Fig. 5C) and C/EBPβ (Fig. 5D) to the G6Pase promoter was similarly unchanged. We conclude that loss of FoxO1 and FoxA1/A2 binding to the IGFPB1 and G6Pase promoters in response to FoxO1 knockdown and/or insulin treatment in HepG2 cells does not globally impair regulatory factor binding.
FIGURE 5.

FoxO1 knockdown does not impact C/EBPα/β binding to the IGFBP1 and G6Pase promoters. A–D, ChIP analysis was performed on chromatin isolated from unstimulated/insulin-stimulated HepG2 cells infected with scrambled or FoxO1 shRNA with antibodies directed against C/EBPα (A and C) or C/EBPβ (B and D). Results are normalized to total input, and values are expressed as a ratio over non-insulin-treated cells infected with the scrambled shRNA. Data represent the mean ± S.E. (error bars) for three independent biological experiments and two technical replicates per biological replicate.
Loss of FoxO1 and FoxA1/A2 Binding Reduces H3K27 Acetylation at the IGFBP1 Promoter
Due to their previously demonstrated function as pioneer chromatin remodeling factors, we hypothesized that loss of FoxO1 and FoxA1/A2 binding would result in alterations in chromatin structure at the IGFBP1 promoter. To investigate this, we used ChIP to assess changes in histone modification in response to FoxO1 knockdown and/or insulin treatment in HepG2 cells. FoxO1 knockdown HepG2 cells exhibited significantly reduced acetylation of H3K27, a mark of transcriptionally active chromatin, at the IGFBP1 promoter (Fig. 6A). Two other histone modifications commonly observed at promoters of transcriptionally active genes, histone H3 acetylated on Lys-9 (Fig. 6B) and histone H3 trimethylated on Lys-4 (Fig. 6C) were also negatively impacted, although the difference in the levels of these histone modifications between knockdown or control cells plus or minus insulin treatment was not significant. A small increase in histone H3 dimethylated on Lys-4, a mark of transcriptionally poised chromatin, was observed at the IGFBP1 promoter in the FoxO1 knockdown cells, although this change was also not statistically significant (Fig. 6D).
FIGURE 6.

FoxO1 knockdown reduces H3K27 acetylation at the IGFBP1 promoter. A–D, ChIP analysis of histone modifications with antibodies directed against acetylated H3K27 (H3K27Ac) (A), histone H3 acetylated at Lys-9 (H3K9Ac) (B), histone H3 trimethylated at Lys-4 (H3K4me3) (C), and histone H3 dimethylated at Lys-4 (H3K4me2) (D) on chromatin isolated from unstimulated/insulin-stimulated scrambled or FoxO1 shRNA-infected HepG2 cells. Results are first normalized to total input and then normalized to total histone H3 levels, and values are expressed as a ratio over non-insulin-treated cells infected with the scrambled shRNA. Data represent the mean ± S.E. (error bars) for five independent biological experiments and two technical replicates per biological replicate. *, p < 0.05, Student's t test.
A similar impact on histone modification (a statistically significant decrease in H3K27 acetylation, small decreases in histone H3 acetylated on Lys-9 and histone H3 trimethylated on Lys-4, and a small increase in histone H3 dimethylated on Lys-4) was observed at the IGFBP1 promoter in FoxA1 knockdown cells (data not shown). We conclude that loss of FoxO1 and FoxA1/A2 binding to the IGFPB1 promoter reduces acetylation of H3K27.
FoxO1 and FoxA1 Exhibit Cooperative Binding to IGFBP1 Promoter DNA
The interdependent binding exhibited by FoxO1 and FoxA1/A2 to adjacent sites at the IGFPB1 and G6Pase promoters in cellular chromatin raised the possibility that FoxO1 and FoxA1/A2 cooperatively bind to the adjacent forkhead sites present within the IRE. To investigate this, we performed DNase I footprinting assays in which we examined binding of increasing levels of recombinant FoxO1 protein in the presence of subsaturating amounts of recombinant FoxA1. The sequence of the IGFBP1 IRE is shown in Fig. 7A; the location of the expected FoxA/FoxO1 footprint is highlighted in gray, and the tandem forkhead consensus sites are delineated by boxes. The footprinting gel shown in Fig. 7B is representative of four independent footprinting experiments.
0.1 pmol of FoxA1 binds preferentially to the upstream forkhead consensus within the IRE of the IGFBP1 promoter, as seen by the introduction of a hypersensitive site (compare lanes 1 and 2, arrow to the right of the gel). 0.5 pmol of FoxO1 binds to the IGFBP1 IRE, as seen by the disappearance of multiple bands (compare lane 5 with lane 1; dots to the right of the gel). In contrast, 0.05 pmol of FoxO1 cannot bind to the IRE because the banding pattern looks similar to that in free DNA, where no protein is added (compare lanes 1 and 3). When 0.1 pmol of FoxA1 and 0.05 pmol of FoxO1 are added together, we see evidence of both factors binding to the IRE (compare lanes 1 and 4). The hypersensitive site (arrow) indicates FoxA1 binding, and the disappearance of the additional bands (dots) in the vicinity of the downstream forkhead consensus indicates FoxO1 binding. The ability of 0.05 pmol of FoxO1 to footprint sites when co-bound with FoxA1 is greater than that of 0.5 pmol of FoxO1 alone (compare lanes 4 and 5). Cooperative binding of FoxO1 and FoxA1 is also observed at higher concentrations of FoxO1 (compare lanes 1 and 6). From these data, we conclude that FoxO1 and FoxA1 can cooperatively bind to IGFBP1 promoter DNA in vitro. We suggest that cooperative binding of FoxO1 and FoxA1/A2 contributes to the interdependent binding of these two factors in cellular chromatin.
Discussion
In this paper, we have demonstrated that an amino acid substitution that mimics insulin-mediated phosphorylation of a serine residue located within the FoxO1 “winged helix” DNA binding motif specifically curtails nucleosome binding by this transcription factor. We have previously shown that this same protein motif, which constitutes the primary nucleosome-binding domain of the FoxA forkhead transcription factor (41, 47) and the linker histone H1 (44) directs binding of FoxO1 to its cognate sites within an IRE assembled onto IGFBP1 promoter nucleosome particles (40). Serine 253 is located within the basic region of Wing 2 at the C-terminal end of the FoxO1 winged helix motif; this basic region is essential for high affinity FoxO1 binding to target sites (42, 48, 66). Whereas Wing 2 is not present in the published crystal structure for FoxO1, the crystal structure of the closely related FoxO3 protein reveals direct contacts between the analogous serine and a phosphate group in the DNA backbone (42, 48). It has been suggested that the impact of serine 253 phosphorylation on FoxO1 binding to DNA is likely to be most important when FoxO1 interacts with its naturally occurring target sites where sequence-specific interactions with helix 3 are not optimal (48), as would be the case when these sequences are assembled into nucleosomal DNA.
We have shown that phosphorylation of Ser-253 negatively impacts FoxO1 binding to nucleosomal but not free DNA templates. Whereas phosphorylation of Ser-253 was previously reported to reduce binding of the isolated FoxO1 DNA binding domain (DBD) to free DNA templates (35), the failure of phosphorylation to negatively impact binding of full-length FoxO1 protein to free DNA is not unexpected. Boura et al. (66) have demonstrated that an N-terminal loop located just upstream of helix H1 in the winged helix motif is essential for high affinity DNA binding by another FoxO factor, FoxO4. In the presence of this loop, which was not included in the DBD fragments previously used to investigate FoxO1 binding to DNA, phosphorylation at the amino acid corresponding to Ser-253 in the FoxO4 winged helix domain had a negligible effect on DNA binding by this protein (66). Brent et al. (42) later demonstrated similar DNA binding by phosphorylated and non-phosphorylated FoxO1 DBD fragments containing this loop. Based on our data, we propose that phosphorylation of serine 253 perturbs contacts between the C-terminal region of the FoxO1 DBD and the negatively charged phosphate residues. Although these perturbations are not sufficient, in the context of the full-length FoxO1 protein, to arrest FoxO1 binding to its sites in naked DNA, they are sufficient to curtail binding to its sites on a nucleosome for which FoxO1 exhibits a much lower binding affinity (40) and for which FoxO1 would be expected to exhibit a lower tolerance to perturbations in DNA binding interactions.
Fluorescence anisotropy studies have indicated that interactions between the FoxO1 DBD and its target sites are both rapid and reversible (35), suggesting that once in the nucleus, FoxO1 interacts rapidly and reversibly with its target sites. By reducing the ability of FoxO1 to occupy its sites in chromatin, phosphorylation of Ser-253 following insulin/growth factor stimulation would tip the balance in favor of the nuclear, unbound state. This would in turn promote phosphorylation of the threonine and serine residues located in the FoxO1 N and C terminus, respectively, leading to the sequestration of FoxO1 through subsequent binding to 14-3-3 and translocation to the cytoplasm (32–36). Our model concurs with the results of previous studies that have demonstrated a “gatekeeping” function for Ser-253 phosphorylation as a requirement for phosphorylation of Thr-24 and Ser-316 (35, 67).
This is the first report to describe the direct impact of FoxO1 loss on transcription factor binding, RNA polymerase II recruitment, and histone modifications at FoxO1-activated metabolic genes and the first to demonstrate interdependent FoxO1-FoxA1/A2 binding. We show that loss of FoxO1 binding by either insulin treatment or shRNA knockdown in HepG2 cells negatively impacts binding of FoxA1/A2 and recruitment of RNA polymerase II to the IGFBP1 and G6Pase promoters. The reciprocal is also true in that knockdown of FoxA1 attenuates binding of FoxO1 and recruitment of RNA polymerase II to both promoters. A previous study has demonstrated loss of FoxO1, FoxA1/A2, and RNA pol II binding, but not that of other transcription factors, at the G6Pase promoter in hepatic cells in response to insulin treatment (68). However, this study did not examine the impact of FoxO1 and FoxA1/A2 loss on each other's binding or the recruitment of RNA polymerase II. FoxO1 (Fig. 1) and FoxA1/A2 (data not shown) are capable of binding the IRE assembled onto a nucleosome particle on their own; therefore, interdependent binding of FoxO1-FoxA1/A2 to the IRE within cellular chromatin is most likely due to cooperative binding. This cooperative binding could be facilitated by protein-protein interactions between FoxO1 and FoxA1/A2. Additionally, alterations in DNA and/or chromatin structure initiated by FoxA1/A2 binding could facilitate binding FoxO1, or vice versa. FoxA1/A2 binding bends DNA, exposing the DNA minor groove on the complementary strand to enhanced DNase digestion and creating the characteristic hypersensitive site observed within FoxA1/A2 DNase footprints (47). DNA bending indicated by the hypersensitive site generated by FoxA2 binding to the upstream forkhead consensus within the IGFBP1 IRE (Fig. 7B, arrow to the right of the gel) could facilitate binding of FoxO1 at the adjacent site. Binding of FoxO1 and FoxA1/A2 to nucleosomes has been shown to perturb underlying histone:DNA contacts (40, 47), suggesting another way in which the two factors could facilitate each other's binding. Attenuation of RNA polymerase II binding following FoxO1 and FoxA1/A2 knockdown suggests that curtailed RNA polymerase II binding following insulin treatment is due to loss of FoxO1 and/or FoxA1/A2. Failure of FoxO1 and FoxA1/2 loss to impact binding of C/EBPα and -β to the IGFBP1 and G6Pase promoters argues against FoxO1- and FoxA1/A2-mediated alterations in global chromatin accessibility as a mechanism for RNA polymerase II loss. Alternatively, FoxO1 and/or FoxA1/A2 could recruit an as yet unknown co-activator(s) necessary for recruitment and/or stabilization of RNA polymerase II binding. We are currently investigating this possibility.
FoxA2 was previously shown to respond to insulin signaling via PKB/Akt-mediated phosphorylation and to undergo cytoplasmic sequestration similar to FoxO1 (69). However, nuclear to cytoplasmic translocation of FoxA2 remains a controversial topic (70). Moreover, FoxA2 phosphorylation does not appear to impact binding to DNA or chromatin (69).4 Importantly, we observe loss of binding by both FoxA1 and FoxA2 in response to loss of FoxO1 binding following FoxO1 knockdown even in the absence of insulin treatment. When considered together with our demonstration of curtailed nucleosome binding by the S253D FoxO1 phosphorylation mimic and cooperative DNA binding of FoxO1 and FoxA1, this suggests that loss of FoxA1 and -A2 binding in response to insulin/PKB signaling is primarily due to loss of FoxO1 binding resulting from phosphorylation of FoxO1. Phosphorylation of FoxA2 might then have additional deleterious effects on this factor. This agrees with a previous report that showed that insulin repression of IGFBP1 transcription is independent of FoxA binding (60).
Loss of FoxO1 and FoxA1/A2 binding in response to either insulin treatment or FoxO1 knockdown caused a significant reduction in H3K27 acetylation, a mark of transcriptionally active chromatin, at the IGFBP1 promoter. Recruitment of p300, the acetyltransferase responsible for acetylation of histone H3 at this lysine residue, has been attributed to both FoxO1 and FoxA1/A2 (20, 39, 71, 72). We also observed slight reductions in other epigenetic marks of active transcription, including histone H3 Lys-9 acetylation and histone H3 Lys-4 trimethylation and a slight increase in histone H3 Lys-4 dimethylation, a mark of transcriptionally poised chromatin. While reproducible, these latter changes were statistically insignificant. Overall, these alterations in chromatin modification following loss of FoxO1 and FoxA1/A2 binding at the IGFBP1 promoter suggest a change from a transcriptionally active to a poised chromatin state. The transcriptionally poised chromatin would be primed for rebinding of the forkhead factors and recruitment of transcriptional machinery necessary for quick reactivation of gene expression following insulin removal to facilitate maintenance of glucose homeostasis.
In conclusion, our results illustrate how transcription factors can nucleate transcriptional events in chromatin in response to specific signaling events and suggest a model for regulation of hepatic glucose metabolism through interdependent FoxO1-FoxA1/A2 binding. Comparison of ChIP-sequencing results for FoxO1 and FoxA2 in adult mouse liver has recently revealed that FoxO1 and FoxA2 co-target multiple genes linked to metabolic pathways for glucose, carboxylic acids, lipids, and steroids in liver (73). Therefore, interdependent FoxO1-FoxA1/A2 binding revealed for the IGFPB1 and G6Pase promoters probably represents a general regulatory mechanism enabling modulation of active chromatin states in response to extracellular cues for a broad array of hepatic metabolic processes.
Author Contributions
A. Y. designed, carried out, and analyzed the FoxO1, FoxA, and RNA polymerase II ChIP experiments and contributed to the generation of the manuscript. D. S. designed, carried out, and analyzed Western blots, the histone modification ChIP, and complementary binding footprinting; wrote the manuscript; and generated the figures for the paper. M. H. designed, carried out, and analyzed the FoxO phosphomutant footprinting experiments. N. J. carried out and analyzed the C/EBPα and -β ChIP experiments. L. A. C. conceived and coordinated the study and revised the drafts of the manuscript and figures. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Karen Giles and Joshua Nord for data interpretation and thoughtful discussion.
The work was supported by National Institutes of Health Grant R01 DK093763 and American Diabetes Association Grants 1-04-IN-09 and 1-06-RA-30 (to L. A. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
D. Schill and L. A. Cirillo, unpublished observations.
- FoxO
- forkhead box O
- FoxA
- forkhead box A
- IGFBP1
- insulin-like growth factor-binding protein 1
- G6Pase
- glucose-6-phosphatase
- G6PC
- glucose-6-phosphatase gene
- IRE
- insulin response element
- DBD
- DNA binding domain
- DNase I
- deoxyribonuclease I
- H3K27
- histone H3 lysine 27.
References
- 1. Belikov S., Gelius B., and Wrange O. (2001) Hormone-induced nucleosome positioning in the MMTV promoter is reversible. EMBO J. 20, 2802–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Archer T. K., Lefebvre P., Wolford R. G., and Hager G. L. (1992) Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science 255, 1573–1576 [DOI] [PubMed] [Google Scholar]
- 3. Boyes J., Omichinski J., Clark D., Pikaart M., and Felsenfeld G. (1998) Perturbation of nucleosome structure by the erythroid transcription factor GATA-1. J. Mol. Biol. 279, 529–544 [DOI] [PubMed] [Google Scholar]
- 4. Cordingley M. G., Riegel A. T., and Hager G. L. (1987) Steroid-dependent interaction of transcription factors with the inducible promoter of mouse mammary tumor virus in vivo. Cell 48, 261–270 [DOI] [PubMed] [Google Scholar]
- 5. Cirillo L. A., Lin F. R., Cuesta I., Friedman D., Jarnik M., and Zaret K. S. (2002) Opening of compacted chromatin by early developmental transcription factors HNF3 (FOXA) and GATA-4. Mol. Cell 9, 279–289 [DOI] [PubMed] [Google Scholar]
- 6. Truss M., Bartsch J., Schelbert A., Haché R. J., and Beato M. (1995) Hormone induces binding of receptors and transcription factors to a rearranged nuclesome on the MMTV promoter in vivo. EMBO J. 14, 1737–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Svaren J., Schmitz J., and Hörz W. (1994) The transactivation domain of Pho4 is required for nucleosome disruption at the PHO5 promoter. EMBO J. 13, 4856–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berkes C. A., Bergstrom D. A., Penn B. H., Seaver K. J., Knoepfler P. S., and Tapscott S. J. (2004) Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol. Cell 14, 465–477 [DOI] [PubMed] [Google Scholar]
- 9. Cuesta I., Zaret K. S., and Santisteban P. (2007) The forkhead factor FoxE1 binds to the thyroperoxidase promoter during thyroid cell differentiation and modifies compacted chromatin structure. Mol. Cell Biol. 27, 7302–7314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., and Glass C. K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soufi A., Donahue G., and Zaret K. S. (2012) Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell 151, 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soufi A., Garcia M. F., Jaroszewicz A., Osman N., Pellegrini M., and Zaret K. S. (2015) Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zaret K. S., and Carroll J. S. (2011) Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 25, 2227–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Magnani L., Eeckhoute J., and Lupien M. (2011) Pioneer factors: directing transcriptional regulators within the chromatin environment. Trends Genet. 27, 465–474 [DOI] [PubMed] [Google Scholar]
- 15. Knights C. D., Catania J., Di Giovanni S., Muratoglu S., Perez R., Swartzbeck A., Quong A. A., Zhang X., Beerman T., Pestell R. G., and Avantaggiati M. L. (2006) Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 173, 533–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamonica J. M., Vakoc C. R., and Blobel G. A. (2006) Acetylation of GATA-1 is required for chromatin occupancy. Blood 108, 3736–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang W., Kadam S., Emerson B. M., and Bieker J. J. (2001) Site-specific acetylation by p300 or CREB binding protein regulates erythroid Kruppel-like factor transcriptional activity via its interaction with the SWI-SNF complex. Mol. Cell Biol. 21, 2413–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang R. H., Xu X., Kim H. S., Xiao Z., and Deng C. X. (2013) SIRT1 deacetylates FOXA2 and is critical for Pdx1 transcription and beta-cell formation. Int. J. Biol. Sci. 9, 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hatta M., Liu F., and Cirillo L. A. (2009) Acetylation curtails nucleosome binding, not stable nucleosome remodeling, by FoxO1. Biochem. Biophys. Res. Commun. 379, 1005–1008 [DOI] [PubMed] [Google Scholar]
- 20. Kohler S., and Cirillo L. A. (2010) Stable chromatin binding prevents FoxA acetylation, preserving FoxA chromatin remodeling. J. Biol. Chem. 285, 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Biggs W. H. 3rd, Cavenee W. K., and Arden K. C. (2001) Identification and characterization of members of the FKHR (FOX O) subclass of winged-helix transcription factors in the mouse. Mamm. Genome 12, 416–425 [DOI] [PubMed] [Google Scholar]
- 22. Jacobs F. M., van der Heide L. P., Wijchers P. J., Burbach J. P., Hoekman M. F., and Smidt M. P. (2003) FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J. Biol. Chem. 278, 35959–35967 [DOI] [PubMed] [Google Scholar]
- 23. Gross D. N., van den Heuvel A. P., and Birnbaum M. J. (2008) The role of FoxO in the regulation of metabolism. Oncogene 27, 2320–2336 [DOI] [PubMed] [Google Scholar]
- 24. Fu Z., and Tindall D. J. (2008) FOXOs, cancer and regulation of apoptosis. Oncogene 27, 2312–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ho K. K., Myatt S. S., and Lam E. W. (2008) Many forks in the path: cycling with FoxO. Oncogene 27, 2300–2311 [DOI] [PubMed] [Google Scholar]
- 26. Nakae J., Kitamura T., Silver D. L., and Accili D. (2001) The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest. 108, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ayala J. E., Streeper R. S., Desgrosellier J. S., Durham S. K., Suwanichkul A., Svitek C. A., Goldman J. K., Barr F. G., Powell D. R., and O'Brien R. M. (1999) Conservation of an insulin response unit between mouse and human glucose-6-phosphatase catalytic subunit gene promoters: transcription factor FKHR binds the insulin response sequence. Diabetes 48, 1885–1889 [DOI] [PubMed] [Google Scholar]
- 28. Yeagley D., Guo S., Unterman T., and Quinn P. G. (2001) Gene- and activation-specific mechanisms for insulin inhibition of basal and glucocorticoid-induced insulin-like growth factor binding protein-1 and phosphoenolpyruvate carboxykinase transcription. Roles of forkhead and insulin response sequences. J. Biol. Chem. 276, 33705–33710 [DOI] [PubMed] [Google Scholar]
- 29. Daitoku H., Yamagata K., Matsuzaki H., Hatta M., and Fukamizu A. (2003) Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes 52, 642–649 [DOI] [PubMed] [Google Scholar]
- 30. Zhang K., Li L., Qi Y., Zhu X., Gan B., DePinho R. A., Averitt T., and Guo S. (2012) Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 153, 631–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haeusler R. A., Kaestner K. H., and Accili D. (2010) FoxOs function synergistically to promote glucose production. J. Biol. Chem. 285, 35245–35248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Biggs W. H. 3rd, Meisenhelder J., Hunter T., Cavenee W. K., and Arden K. C. (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. U.S.A. 96, 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakae J., Park B. C., and Accili D. (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a wortmannin-sensitive pathway. J. Biol. Chem. 274, 15982–15985 [DOI] [PubMed] [Google Scholar]
- 34. Tang E. D., Nuñez G., Barr F. G., and Guan K.-L. (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16741–16746 [DOI] [PubMed] [Google Scholar]
- 35. Zhang X., Gan L., Pan H., Guo S., He X., Olson S. T., Mesecar A., Adam S., and Unterman T. G. (2002) Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 277, 45276–45284 [DOI] [PubMed] [Google Scholar]
- 36. Rena G., Guo S., Cichy S. C., Unterman T. G., and Cohen P. (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 274, 17179–17183 [DOI] [PubMed] [Google Scholar]
- 37. Guo S., Rena G., Cichy S., He X., Cohen P., and Unterman T. (1999) Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274, 17184–17192 [DOI] [PubMed] [Google Scholar]
- 38. Tomizawa M., Kumar A., Perrot V., Nakae J., Accili D., Rechler M. M., and Kumaro A. (2000) Insulin inhibits the activation of transcription by a C-terminal fragment of the forkhead transcription factor FKHR: a mechanism for insulin inhibition of insulin-like growth factor-binding protein-1 transcription. J. Biol. Chem. 275, 7289–7295 [DOI] [PubMed] [Google Scholar]
- 39. Nasrin N., Ogg S., Cahill C. M., Biggs W., Nui S., Dore J., Calvo D., Shi Y., Ruvkun G., and Alexander-Bridges M. C. (2000) DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor binding protein 1 promoter in HepG2 cells. Proc. Natl. Acad. Sci. U.S.A. 97, 10412–10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hatta M., and Cirillo L. A. (2007) Chromatin opening and stable perturbation of core histone:DNA contacts by FoxO1. J. Biol. Chem. 282, 35583–35593 [DOI] [PubMed] [Google Scholar]
- 41. Clark K. L., Halay E. D., Lai E., and Burley S. K. (1993) Co-crystal structure of the HNF3/fork head DNA recognition motif resembles histone H5. Nature 364, 412–420 [DOI] [PubMed] [Google Scholar]
- 42. Brent M. M., Anand R., and Marmorstein R. (2008) Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 16, 1407–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hannenhalli S., and Kaestner K. H. (2009) The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 10, 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ramakrishnan V., Finch J. T., Graziano V., Lee P. L., and Sweet R. M. (1993) Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 362, 219–223 [DOI] [PubMed] [Google Scholar]
- 45. Cirillo L. A., and Zaret K. S. (1999) An early developmental transcription factor complex that is more stable on nucleosome core particles than on free DNA. Mol. Cell 4, 961–969 [DOI] [PubMed] [Google Scholar]
- 46. Koranda M., Schleiffer A., Endler L., and Ammerer G. (2000) Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature 406, 94–98 [DOI] [PubMed] [Google Scholar]
- 47. Cirillo L. A., McPherson C. E., Bossard P., Stevens K., Cherian S., Shim E. Y., Clark K. L., Burley S. K., and Zaret K. S. (1998) Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 17, 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsai K. L., Sun Y. J., Huang C. Y., Yang J. Y., Hung M. C., and Hsiao C. D. (2007) Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 35, 6984–6994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 50. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 51. Oduro A. K., Fritsch M. K., and Murdoch F. E. (2008) Chromatin context dominates estrogen regulation of pS2 gene expression. Exp. Cell Res. 314, 2796–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weinmann A. S., and Farnham P. J. (2002) Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods 26, 37–47 [DOI] [PubMed] [Google Scholar]
- 53. Métivier R., Penot G., Hübner M. R., Reid G., Brand H., Kos M., and Gannon F. (2003) Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763 [DOI] [PubMed] [Google Scholar]
- 54. Tzivion G., Dobson M., and Ramakrishnan G. (2011) FoxO transcription factors: regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945 [DOI] [PubMed] [Google Scholar]
- 55. Martelli A. M., Tabellini G., Bressanin D., Ognibene A., Goto K., Cocco L., and Evangelisti C. (2012) The emerging multiple roles of nuclear Akt. Biochim. Biophys. Acta 1823, 2168–2178 [DOI] [PubMed] [Google Scholar]
- 56. Matsumoto M., Pocai A., Rossetti L., Depinho R. A., and Accili D. (2007) Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 6, 208–216 [DOI] [PubMed] [Google Scholar]
- 57. Dong X. C., Copps K. D., Guo S., Li Y., Kollipara R., DePinho R. A., and White M. F. (2008) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matsuzaki H., Daitoku H., Hatta M., Tanaka K., and Fukamizu A. (2003) Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 100, 11285–11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Unterman T. G., Fareeduddin A., Harris M. A., Goswami R. G., Porcella A., Costa R. H., and Lacson R. G. (1994) Hepatocyte nuclear factor-3 (HNF-3) binds to the insulin response sequence in the IGF binding protein-1 (IGFBP-1) promoter and enhances promoter function. Biochem. Biophys. Res. Commun. 203, 1835–1841 [DOI] [PubMed] [Google Scholar]
- 60. Hall R. K., Yamasaki T., Kucera T., Waltner-Law M., O'Brien R., and Granner D. K. (2000) Regulation of phosphoenolpyruvate carboxykinase and insulin-like growth factor-binding protein-1 gene expression by insulin: the role of winged helix/forkhead proteins. J. Biol. Chem. 275, 30169–30175 [DOI] [PubMed] [Google Scholar]
- 61. Allander S. V., Durham S. K., Scheimann A. O., Wasserman R. M., Suwanichkul A., and Powell D. R. (1997) Hepatic nuclear factor 3 and high mobility group I/Y proteins bind the insulin response element of the insulin-like growth factor-binding protein-1 promoter. Endocrinology 138, 4291–4300 [DOI] [PubMed] [Google Scholar]
- 62. Vander Kooi B. T., Streeper R. S., Svitek C. A., Oeser J. K., Powell D. R., and O'Brien R. M. (2003) The three insulin response sequences in the glucose-6-phosphatase catalytic subunit gene promoter are functionally distinct. J. Biol. Chem. 278, 11782–11793 [DOI] [PubMed] [Google Scholar]
- 63. Streeper R. S., Svitek C. A., Chapman S., Greenbaum L. E., Taub R., and O'Brien R. M. (1997) A multicomponent insulin response sequence mediates a strong repression of mouse glucose-6-phosphatase gene transcription by insulin. J. Biol. Chem. 272, 11698–11701 [DOI] [PubMed] [Google Scholar]
- 64. Ghosh A. K., Lacson R., Liu P., Cichy S. B., Danilkovich A., Guo S., and Unterman T. G. (2001) A nucleoprotein complex containing CCAAT/enhancer-binding protein beta interacts with an insulin response sequence in the insulin-like growth factor-binding protein-1 gene and contributes to insulin-regulated gene expression. J. Biol. Chem. 276, 8507–8515 [DOI] [PubMed] [Google Scholar]
- 65. Gautier-Stein A., Mithieux G., and Rajas F. (2005) A distal region involving hepatocyte nuclear factor 4α and CAAT/enhancer binding protein markedly potentiates the protein kinase A stimulation of the glucose-6-phosphatase promoter. Mol. Endocrinol. 19, 163–174 [DOI] [PubMed] [Google Scholar]
- 66. Boura E., Silhan J., Herman P., Vecer J., Sulc M., Teisinger J., Obsilova V., and Obsil T. (2007) Both N-terminal loop and wing W2 of forkhead domain of transcription factor FoxO4 are important for DNA binding. J. Biol. Chem. 282, 8265–8275 [DOI] [PubMed] [Google Scholar]
- 67. Nakae J., Barr V., and Accili D. (2000) Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 19, 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hall R. K., Wang X. L., George L., Koch S. R., and Granner D. K. (2007) Insulin represses phosphoenolpyruvate carboxykinase gene transcription by causing the rapid disruption of an active transcription complex: a potential epigenetic effect. Mol. Endocrinol. 21, 550–563 [DOI] [PubMed] [Google Scholar]
- 69. Wolfrum C., Besser D., Luca E., and Stoffel M. (2003) Insulin regulates the activity of forkhead transcription factor Hnf-3β/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc. Natl. Acad. Sci. U.S.A. 100, 11624–11629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Friedman J. R., and Kaestner K. H. (2006) The Foxa family of transcription factors in development and metabolism. Cell Mol. Life Sci. 63, 2317–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mahmud D. L., G-Amlak M., Deb D. K., Platanias L. C., Uddin S., and Wickrema A. (2002) Phosphorylation of forkhead transcription factors by erythropoietin and stem cell factor prevents acetylation and their interaction with coactivator p300 in erythroid progenitor cells. Oncogene 21, 1556–1562 [DOI] [PubMed] [Google Scholar]
- 72. Liu Y. N., Lee W. W., Wang C. Y., Chao T. H., Chen Y., and Chen J. H. (2005) Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene 24, 8277–8290 [DOI] [PubMed] [Google Scholar]
- 73. Shin D. J., Joshi P., Hong S. H., Mosure K., Shin D. G., and Osborne T. F. (2012) Genome-wide analysis of FoxO1 binding in hepatic chromatin: potential involvement of FoxO1 in linking retinoid signaling to hepatic gluconeogenesis. Nucleic Acids Res. 40, 11499–11509 [DOI] [PMC free article] [PubMed] [Google Scholar]