

Abstract
The radical S-adenosylmethionine (SAM) protein PqqE is predicted to function in the pyrroloquinoline quinone (PQQ) biosynthetic pathway via catalysis of carbon-carbon bond formation between a glutamate and tyrosine side chain within the small peptide substrate PqqA. We report here that PqqE activity is dependent on the accessory protein PqqD, which was recently shown to bind PqqA tightly. In addition, PqqE activity in vitro requires the presence of a flavodoxin- and flavodoxin reductase-based reduction system, with other reductants leading to an uncoupled cleavage of the co-substrate SAM. These results indicate that PqqE, in conjunction with PqqD, carries out the first step in PQQ biosynthesis: a radical-mediated formation of a new carbon-carbon bond between two amino acid side chains on PqqA.
Keywords: flavoprotein, iron-sulfur protein, peptides, post-translational modification (PTM), S-adenosylmethionine (SAM), PQQ, radical SAM, SPASM domain
Introduction
Pyrroloquinoline quinone (PQQ)2 is employed by a wide variety of bacteria, where it functions as a redox cofactor in substrate oxidation via an alternate (non-glycolytic) pathway for production of cellular ATP (1). Hundreds of bacterial species are thought to produce PQQ, based on the presence in their genomes of the strongly conserved pqq operon (2) (Fig. 1A). Although the existence of this important redox cofactor has been known for decades, knowledge of the biosynthetic pathway for PQQ has remained scant (3, 4). The PQQ molecule derives from the evolutionarily conserved glutamate and tyrosine side chains within a ribosomally produced peptide substrate PqqA (Fig. 1, B and C). To produce PQQ, a large number of chemical modifications are required: (i) the formation of a new carbon-carbon bond between the γ-carbon of glutamate and the 3-position of tyrosine (labeled in green in Fig. 1C); (ii) the addition of two additional hydroxyl groups to the tyrosine ring; (iii) a condensation between the 4-OH of tyrosine and the backbone amine of glutamate to produce a heterocyclic ring; and (iv) an 8-electron oxidation catalyzed by PqqC, a cofactor-less enzyme that converts 3a-(2-amino-2-carboxyethyl)-4,5-dioxo-4,5,6,7,8,9-hexahydroquinoline-7,9-dicarboxylic acid (AHQQ) to PQQ in the final step of the pathway (5–7).
FIGURE 1.
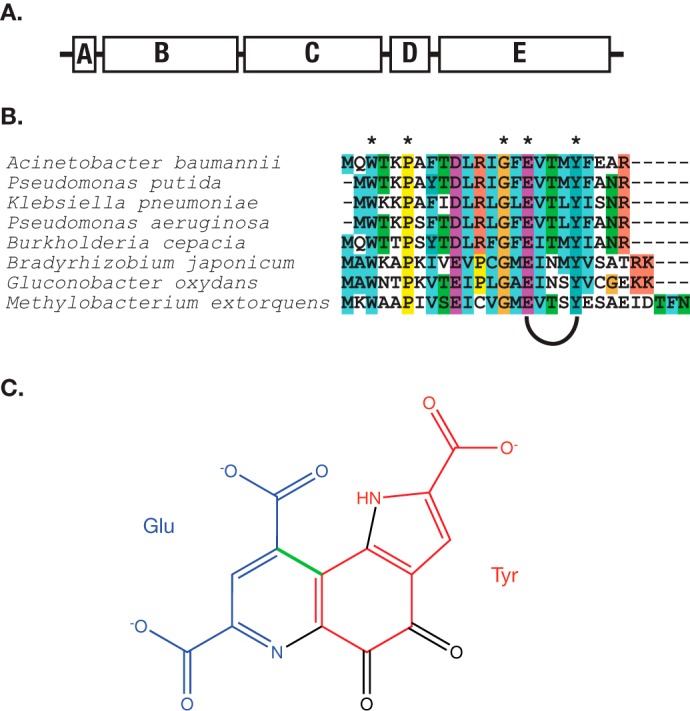
A, the pqq operon contains, at a minimum, the genes PqqA, PqqB, PqqC, PqqD, and PqqE. PqqF, a protease, is often present as well, but is not uniformly conserved. The gene order is strictly conserved. In M. extorquens AM1, there is a gene fusion between PqqC and PqqD. B, alignment of PqqA from PQQ producing organisms. Asterisks denote conserved residues. The conserved glutamate and tyrosine near the C terminus are modified to form PQQ. C, PQQ, showing the origin of the atoms that make its backbone. The bond highlighted in green is the carbon-carbon bond hypothesized to be made by PqqE.
The radical SAM enzyme PqqE has been the primary candidate as the catalyst for the formation of the new carbon-carbon bond between glutamate and tyrosine. Radical SAM proteins use the reductive cleavage of S-adenosyl methionine to initiate free radical chemistry, and can accomplish a wide variety of reactions, including carbon-carbon bond formation (8, 9). PqqE is a founding member of the SPASM domain-containing radical SAM proteins, which contain one or more auxiliary clusters in their C-terminal regions, and of which several are known to modify small peptides or proteins (10, 11). Recently, a small (∼10 kDa) protein, PqqD, has been shown to form a strong, sub-micromolar KD complex with the peptide substrate PqqA. This complex was further demonstrated to associate with PqqE (12). These findings suggested that PqqD would serve as a chaperone to deliver PqqA to PqqE, functioning as a necessary and heretofore missing component of the subsequent radical-based chemistry. This connects PqqD and the PQQ biosynthesis pathway to a growing class of proteins involved in binding and modifying small, ribosomally produced peptides, of which many are radical SAM proteins (13–15). We now show that PqqE, in association with the complex, PqqA/D, generates a functional catalyst for the cross-linking of glutamate and tyrosine within PqqA.
Experimental Procedures
Methylobacterium extorquens AM1 PqqD, a C-terminal domain of the longer PqqCD gene fusion (12), was cloned into the pGEX-6p-1 vector using the NotI and BamHI restriction sites. Purification was carried out over a glutathione-Sepharose column, followed by cleavage of the GST tag with PreScission protease overnight at 4 °C. The GST tag was removed by passing cleaved protein through a glutathione-Sepharose column. PqqA was made synthetically by CPC Scientific. A non-conserved cysteine was mutated to serine to increase the stability of the peptide. PqqE was expressed in the presence of oxygen, in TB medium supplemented with 100 μm ferric citrate. The incubation was started at 31 °C for 4 h, and then the temperature was decreased to 20 °C. An hour later, isopropyl-1-thio-β-d-galactopyranoside was added to a final concentration of 100 μm, and cysteine was added to a concentration of 50 μm. Cells were harvested 18 h later. UV-visible absorbance spectroscopy of PqqE was performed on a Cary 50 spectrophotometer (spectral bandwidth 1.5 nm), in a septum-sealed cuvette. Flavodoxin A (FldA) and flavodoxin reductase (FNR) from Escherichia coli were grown essentially as described previously (16, 17). Purification employed a DEAE-Sepharose column at pH 7.5, 50 mm Tris, with an elution gradient from 50 to 500 mm NaCl over 10 column volumes. The protein was then passed over a preparatory gel filtration column to remove high molecular weight contaminants, exchanged into oxygen-free buffer in the glove box, and frozen in small aliquots. FNR from M. extorquens AM1 was cloned from genomic DNA (ATCC 14718) using the primers CCATTCATATGCCCAAGATCACCTTCGT and CCATTCTCGAGTCAGCCCTGGCGGG, and then ligated into the plasmid pET-28a between the NdeI and XhoI restriction sites. This plasmid was used to transform BL21 (DE3) cells, which were grown overnight in LB medium supplemented with 100 μm riboflavin at 31 °C. Protein was purified over a nickel-nitrilotriacetic acid column. Azotobacter vinelandii (ATCC 478) FldA was cloned from genomic DNA using the primers GATTGGATCCATGAGCGTAACCATTGTTTATGGGTCC and ATTGCGGCCGCCTACATGAGCTGAGCAAGCCATGC and ligated into the pGEX-6p-1 plasmid between the BamHI and NotI restriction sites. This was grown as described for FNR, with the exception that 50 μm FMN was added to the medium instead of riboflavin. Protein was purified over a glutathione-Sepharose column and cleaved with PreScission protease, and the final product was separated on a size-exclusion column.
For 57Fe labeling of PqqE, the protein was expressed in M9 medium supplemented with 5 g/liter casamino acids and 20 μm iron-57 citrate. The casamino acids were passed through a Chelex column prior to addition to broth. Iron-57 was purchased as the iron oxide (Cambridge Isotopes) and reacted with 10 m HCl at 80 °C to form 57FeCl3. This was diluted 10-fold and mixed with a 2-fold excess of citric acid. Trace minerals were added, and the medium was supplemented with 10 μm pyridoxine HCl and 100 μm cysteine after the addition of isopropyl-1-thio-β-d-galactopyranoside. Purification and reconstitution were carried out as with the native form of PqqE.
Iron and Sulfide Quantification of PqqE
The method of Crack et al. (18) was used for both, with ferrozine substituting for ferene in the iron assay. The sample absorbance was compared with an iron standard, and then determined in triplicate. The ferrozine assay was found to be unperturbed by the presence of protein through the method of standard addition. For the sulfide quantification, the standard was generated from a sodium sulfide solution dissolved in 10 mm NaOH. In this case, the method of standard addition was used to correct for interference from the protein (19).
Protein Quantification Assay
PqqE was mixed with 6 m guanidinium HCl plus 0.1 m citric acid and allowed to react for 1 h to fully denature the protein. The citric acid was added to destroy the Fe-S clusters and chelate free iron. The protein was then exchanged into 6 m guanidinium HCl using a PD-10 column, and the concentration of protein was determined by absorbance at 280 nm using the method of Pace et al. (20). Protein concentration of the same sample was determined using a Bradford assay where the BSA standard curve was linearized by dividing the 590 nm absorbance by the 450 nm absorbance, according to the method of Zor and Selinger (21). The comparison of these two values provides a conversion factor for PqqE of 0.855 (i.e. the true protein concentration is the Bradford-determined value multiplied by 0.855) that was used to correct for systematic error in the Bradford assay. This conversion factor was then used to determine the concentration of subsequent PqqE preparations.
Mössbauer Spectroscopy
Zero-field, 57Fe Mössbauer spectra were recorded in a constant acceleration spectrometer (SEE Co., Edina, MN) at 4 K using a Janis Research Co. cryostat (Woburn, MA). Collected spectra were analyzed with the WMOSS software package (See Co., Edina, MN). Isomer shifts are reported relative to α-iron (27-μm foil) at room temperature. Samples of PqqE were prepared by freezing solutions in a Teflon sample holder (thickness 0.2 inch) under an inert atmosphere. The sample holder was placed snuggly in the sample rod holder and wrapped in Kapton® tape prior to introduction into the spectrometer.
Reconstitution of PqqE
The as-isolated PqqE was diluted to 100–200 μm in a buffer containing 2 mm DTT, 50 mm Tris, pH 7.9, 300 mm KCl, and 10% (v/v) glycerol in a glovebox containing <5 ppm of O2. While stirring, aliquots of Na2S and ammonium iron(II) sulfate were added to the sample in 100 μm doses every 30 min. The sample was kept at room temperature. The protein was then passed over a PD-10 column to remove excess iron and sulfide, and the protein was diluted 1:1 with water and bound to a DEAE column. Using 50 mm Tris, pH 7.9, 500 mm KCl, 10% glycerol as eluent, a black aggregate remained on the column, while soluble protein eluted as a brown fraction.
Peptide Modification Assay
Assays contained 50 μm PqqE, 50 μm PqqD, 50 μm PqqA, and 500 μm SAM, and were carried out in the glove box. The buffering system was 50 mm Tris, pH 7.9, 200 mm KCl, 10% (v/v) glycerol. For reducing reagents, the assay had either 500 μm sodium dithionite, 500 μm titanium(III) citrate, or a mixture of 1 mm NADPH, 5 μm FNR, and 20 μm FldA. FNR concentration was maintained at a concentration less than FldA concentration to ensure that FldA binding to FNR did not out-compete association of FldA with PqqE. The reaction was quenched with 5% formic acid.
Mass Spectrometric Assay for Conversion of PqqA into Its Cross-linked Product
Samples were quenched with formic acid (5%) and analyzed by an Agilent 1200 LC that was connected in-line with a Thermo LTQ-Orbitrap-XL mass spectrometer equipped with an electrospray ionization source and operated in the positive ion mode. The LC was equipped with a C4 column (Restek), and analytes were eluted using a linear water/acetonitrile gradient. Data acquisition and analysis were performed using the Xcalibur software (version 2.0.7, Thermo). The extent of cross-linking of the PqqA peptide was determined by integrating extracted ion chromatograms for the [M+2H]2+ ions of cross-linked and unmodified forms of PqqA (occurring at m/z = 1536.7 and 1537.7, respectively). Tandem mass spectrometry (MS/MS) measurements were performed using collision-induced dissociation (CID). Mass-to-charge ratios (m/z) of theoretical fragment ions of a given amino acid sequence were calculated using the MS-Product tool of the ProteinProspector software. Mass envelopes were simulated using the MassXpert software.
Results
As-isolated PqqE Contains Incomplete Fe-S Centers
As reported previously, we have expressed M. extorquens AM1 PqqE as a His6 tag fusion in E. coli, and the protein is found to be very soluble and to express in high yield (12). Quantification of iron shows that there are roughly 7–10 iron atoms per polypeptide, which, although varying among enzyme preparations, suggested multiple 4Fe-4S clusters (22). Due to a persistent lack of activity in the as-purified PqqE, we turned to Mössbauer spectroscopy to assess the status of its iron-sulfur centers. The zero-field spectrum of oxidized PqqE shows only two significant doublets: one being a [4Fe-4S]2+ signal (δ = 0.478, ΔEq = 1.214), comprising 66% of the total iron, and the other being a [2Fe-2S]2+ or [3Fe-4S]+ signal (δ = 0.324, ΔEq = 0.499), comprising 33% of the total iron (Fig. 2A). Taken together, this suggests that protein as-isolated contains a mixture of 2Fe-2S and 4Fe-4S clusters as recently reported by Saichana et al. (23).
FIGURE 2.
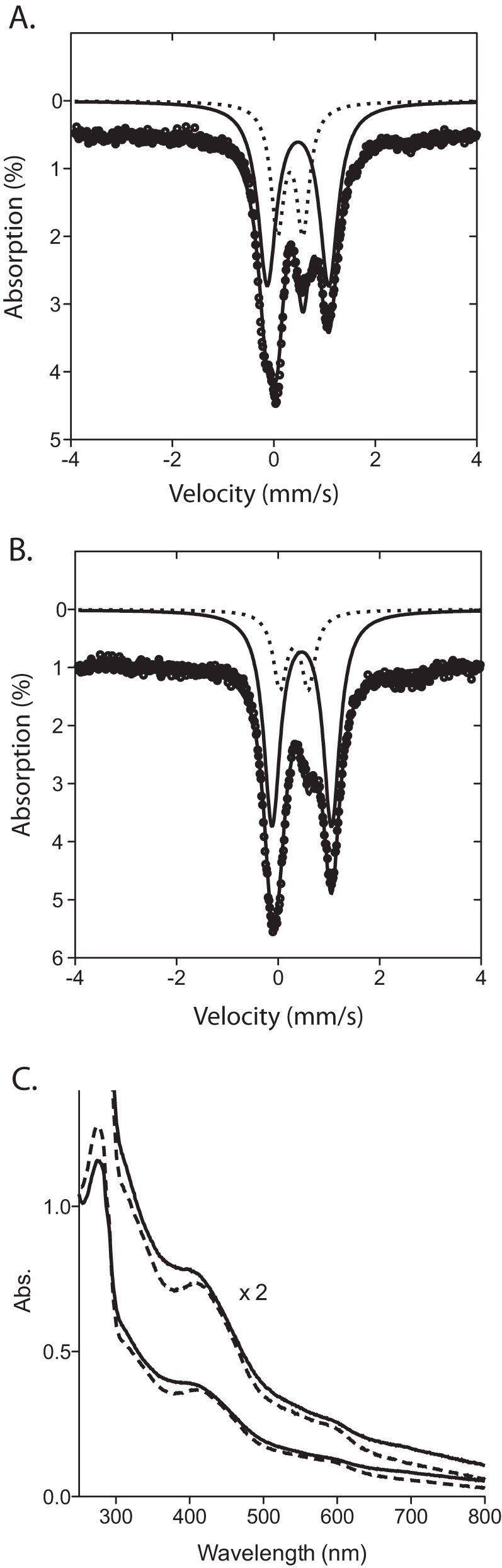
Reconstitution of M. extorquens PqqE shows a 4Fe-4S protein. A, zero-field Mössbauer spectrum of PqqE at 4 K, as recombinantly expressed in the presence of 57Fe and purified from E. coli. Black circles show the raw data, which have been offset from the axis by 0.5% for ease of interpretation. These data are decomposed into 4Fe-4S (solid line) and 2Fe-2S (dotted line) components. B, Mössbauer spectrum of as-isolated PqqE reconstituted with additional 57Fe and sulfide; the 4Fe-4S component comprises 80% of all iron in the sample. C, absorbance (Abs.) spectrum of PqqE before (dashed lines) and after (solid lines) reconstitution of the as-isolated protein.
In an effort to obtain active forms of PqqE, we turned to reconstitution of the as-purified M. extorquens PqqE, initially adding in 4 eq of iron and sulfide. Following dialysis to remove any adventitiously bound iron, an increase in both the 4Fe-4S Mössbauer signal and the total iron loading is observed, yielding 13.0 ± 0.1 total irons per polypeptide (average of four reconstituted enzyme preparations), whereas sulfide quantification shows 12.2 ± 0.5 sulfurs per polypeptide. This result, together with the fact that PqqE contains seven conserved cysteines in its C-terminal region, suggested the presence of three 4Fe-4S clusters, analogous to the SPASM domain-containing radical SAM enzyme anSME described by Drennan and co-workers (25). We then turned to the analysis of Mössbauer spectra of such reconstituted PqqE at 4 K and zero field (Fig. 2B). These data show a 4Fe-4S signal that represents ∼80% of the total iron detected. The Mössbauer spectrum of the remaining 20% irons implicates either [2Fe-2S]2+ or [3Fe-4S]+ states; the latter, which has a spectroscopic signature similar to [2Fe-2S]2+, could result from loss of a labile iron at one (or more) [4Fe-4S] clusters (24). UV-visible absorbance spectra of the reconstituted PqqE (Fig. 2C) display a spectrum more in line with known 4Fe-4S clusters: a broad, flat absorbance shoulder at ∼405 nm without the distinct absorbance peaks seen in the as-purified form (Fig. 2C). Based on the combined iron binding and spectroscopic data, we conclude that functional PqqE contains three Fe-S centers, at least two of which are present as [4Fe-4S]. Further structural and spectroscopic analyses of PqqE, and their relationship to function, are needed before the structure of the third site can be unambiguously assigned.
A Flavin-containing Reductase Is Necessary to Obtain Peptide Modification by PqqE
It has been observed in several instances that radical SAM enzymes are unable to be activated using strong reducing reagents such as sodium dithionite as an electron source. The presence of such reagents leads instead to an uncoupled reaction that produces 5′-deoxyadenosine and methionine, but no modification of the substrate. We have previously reported the uncoupled production of 5′-deoxyadenosine (dA) by Klebsiella pneumoniae PqqE using sodium dithionite as the reducing reagent (22). As an alternative to such a non-physiological reductant, researchers have employed the flavodoxin (FldA)/flavodoxin reductase (FNR) system from E. coli. In this system, NADPH functions as a source of electrons: FNR binds to NADPH and its FAD cofactor is reduced by two electrons; these electrons are then transferred to the FMN cofactor of FldA, and finally to the radical SAM enzyme itself. For the enzyme ThiH, this reduction method was found to be the sole method for productive cleavage of SAM (26), with dithionite leading to uncoupled SAM cleavage as the sole pathway. Bruender et al. (27) found that flavodoxin homologues increased the activity of 7-carboxy-7-deazaguanine (CDG) synthase 75-fold, and in this case, flavodoxin was able to accept electrons from sodium dithionite and increase the reaction rate relative to dithionite alone. Notably, the most effective reduction rate came from using the native flavodoxins, suggesting that there may be specific interactions between the radical SAM enzyme and flavodoxin, or that a highly tuned redox potential is required for productive turnover. With these results in mind, we set up reactions between PqqE, PqqD, and PqqA using different reducing reagents that utilize FNR/FldA, sodium dithionite, or titanium(III) citrate as the sole electron source. We examined both the E. coli FldA/FNR pair and a second reducing system composed of an FNR from M. extorquens FNR and an FldA from A. vinelandii. Although a flavodoxin from M. extorquens would have been preferable, we were unable to locate an open reading frame for such a gene product in the genome of M. extorquens; further, the FNR of A. vinelandii is 67% identical to that from M. extorquens. LC-MS measurements were used to analyze the products of the enzymatic reactions, to identify any mass changes that occur as a result of modification of PqqA. The most abundant peak of unmodified PqqA appears at m/z = 1537.7, with z = +2 (Fig. 3A). The elution profile of the unmodified PqqA appears as a broad peak beginning at around 13 min in the chromatogram (Fig. 3C). Upon reaction at room temperature overnight, a new, broader fraction eluted from the LC, which contained a mixture of unmodified and modified PqqA, with the latter observed in the leading edge of the fraction (Fig. 3D), and exhibiting a −2-Da molecular mass shift relative to unmodified PqqA (Fig. 3B). This mass shift is as expected for a cross-linking reaction between glutamate and tyrosine in PqqA, in which formation of the new carbon-carbon bond is followed by re-aromatization of the tyrosine ring and concomitant elimination of two hydrogen atoms (see below). Use of either titanium(III) citrate or sodium dithionite as reductant failed to lead to modified PqqA. Use of sodium dithionite resulted in complete formation of dA through an uncoupling reaction, both in the absence and in the presence of PqqA and PqqD. Surprisingly, despite the low yield of modified PqqA (estimated as ∼4% at 24 h), no uncoupled production of dA could be detected using FldA/FNR/NADPH as reductant. Reaction mixtures in which PqqD is omitted are unreactive and show no change in PqqA mass relative to starting material. This was also recently found in another radical SAM protein that contains a PqqD-like domain and modifies a small peptide (28). The slow turnover of PqqE may be related to the use of a less than optimal flavodoxin as electron carrier or PqqE inactivation in the course of reaction, and there is clearly room for further optimization. Importantly, these results indicate for the first time that an intramolecular cross-linking reaction occurs between PqqA residues, and that this process is critically dependent on PqqE, PqqD, and the flavodoxin reductase system.
FIGURE 3.

Modification of PqqA by PqqE and PqqD monitored by LC-MS. A, the initial 2+ ion mass envelope of unmodified PqqA. B, the 2+ mass envelope of a minor peak, eluting 1 min earlier, seen following a 24-h reaction under anaerobic conditions. A noticeable shift in mass by 2 Da is observed, consistent with cross-linking of residues in PqqA according to Fig. 5. In both A and B, the red spectra are calculated mass envelopes for PqqA (A) and modified PqqA (B), offset slightly for ease of comparison. Arrows indicate the most abundant ions, which were used to quantify the relative amount of modified PqqA. C, chromatograph showing the elution profile of 1537.7 (black) and 1536.7 (blue) ions in an unreacted PqqA sample. D, chromatograph showing the elution profile of 1537.7 (black) and 1536.7 (blue) ions in a 24-h reaction mixture; a small peak containing cross-linked PqqA is seen to elute earlier than the unreacted PqqA.
MS/MS Supports Cross-linking of Glu16 to a C-terminal Side Chain of PqqA
The [M+2H]2+ ions of unmodified PqqA and modified PqqA were selected for analysis by MS/MS using CID. CID of peptide ions typically results in cleavage of peptide bonds to form b-type and/or y-type fragment ions (29). The particular fragment ions that are observed in an MS/MS spectrum depend on several factors, including the primary sequence, the precursor ion charge state, the locations of charges (i.e. ionizing protons) in the peptide precursor ions, the internal energy of the precursor ions, etc. The MS/MS spectrum of unmodified PqqA exhibits a series of consecutive b-type fragment ions, b6 through b18 (Fig. 4, lower panel). For comparison, in the MS/MS spectrum of modified PqqA, fragment ions b15 through b18 are observed at markedly lower abundance (highlighted in the orange dashed box in the upper panel of Fig. 4). Formation of fragment ions b15 through b18 results from cleavage of peptide bonds in the region of PqqA encompassed by Glu15 and Tyr19, and separation of the resulting, complementary N-terminal and C-terminal fragments. In the case of a modified PqqA, formation of fragment ions b15 through b18 is hindered by the covalent, intramolecular cross-link between Glu15 and Tyr19, which prevents detection of fragments resulting from peptide bond cleavage within this region of the peptide. Moreover, in the MS/MS spectrum of unmodified PqqA, the b12 fragment ion (m/z = 1281.72) is the base peak (i.e. the peak of highest abundance) and residual precursor ion is not observed, indicating that the precursor ion of unmodified PqqA was completely fragmented under CID. In contrast, the base peak in the MS/MS spectrum of the modified PqqA is residual precursor ion (m/z = 1536.74), indicating incomplete fragmentation of the modified PqqA under CID. The collision energy settings used for CID of unmodified and modified PqqA were identical. The incomplete fragmentation of modified PqqA is consistent with a modification that stabilizes the precursor peptide ions against fragmentation. In summary, the incomplete precursor ion fragmentation and lowered abundance of fragment ions resulting from peptide backbone cleavage within the region between Glu15 and Tyr19 strongly support the presence of a covalent cross-link between Glu15 and Tyr19 in the modified PqqA.
FIGURE 4.

MS-MS analysis of modified (top) and unmodified (bottom) PqqA. The modified peptide shows cleavage protection between E and Y; few or none of the fragments detected in the parent PqqA are observed. Mass fragments are labeled according to the sequence of PqqA shown in the top panel. b ions that are not present in the cross-linked sample are outlined in the dashed box.
Discussion
Among the three major classes of enzymatic redox cofactors (flavins, nicotinamides, and quinones), there has been a singular lack of understanding regarding the biosynthetic pathway for PQQ production. The operon encoding PQQ biosynthesis (Fig. 1A) was identified more than 25 years ago (4, 30), and more recent bioinformatics studies demonstrated a conservation of gene order that includes an occasional gene fusion between PqqD with either the C terminus of PqqC or the N terminus of PqqE (2). However, the description of function for each gene product from this operon has lagged considerably, with PqqC representing the only enzyme within the pathway successfully characterized with regard to structural, spectroscopic, and kinetic properties (5, 6, 31). Although PqqE had been shown to be a radical SAM enzyme (22), it had not been possible to demonstrate a reaction between the peptide substrate PqqA and PqqE, suggesting either that PqqA must be modified before it can interact with PqqE or that key components of the PqqE system were missing (7). In a recent breakthrough, PqqD was shown to bind tightly to PqqA and to enter into a ternary complex with PqqE (12). The resulting suggestion that PqqD was a missing component for productive PqqE activity is verified in the present study. Two additional features of a functional PqqE not previously defined are the requirements for reconstitution of enzyme with additional iron equivalents, as well as the uncovering of suitable reductant systems that can support coupling between SAM cleavage and the subsequent functionalization of PqqA.
With this study, both the initial step in PQQ production, catalyzed by PqqE, and the ultimate step, catalyzed by PqqC, have been identified Further, with a function assigned to PqqD, PqqB remains the sole gene product without any clearly established catalytic role. Although hints to the function of PqqE came with its demonstration as a radical SAM enzyme, it was uncertain whether the reaction would involve simply the formation of a new carbon-carbon bond formation or, perhaps, more drastic modifications. Based on the present work and its relationship to the recently published work on the StrB protein, a radical SAM enzyme that catalyzes carbon-carbon bond formation between lysine and tryptophan residues in a small, ribosomally produced peptide (32), we conclude that the principal role of PqqE is to catalyze formation of a new carbon-carbon bond between the γ-carbon of a glutamate and the ring of a tyrosine residue (Fig. 5). A great deal of fascinating biochemistry remains to be determined that includes determining the manner in which PqqD controls the interaction between PqqA and PqqE, the catalytic function of PqqB, and the process that liberates the cross-linked Glu-Tyr product from its parent peptide. The data presented herein, demonstrating that the initial step in PQQ biosynthesis is carried out by the combined action of PqqE and PqqD, provide a critical clarification of the mechanistic underpinnings of PQQ production as well as a defined direction for future studies of this unique member of the family of ribosomally encoded, post-translationally modified peptides (13–15).
FIGURE 5.

Scheme for the formation of PQQ from glutamate and tyrosine of PqqA. PqqA's conserved glutamate undergoes a hydrogen abstraction on the γ-carbon, forming a carbon radical that reacts with the 3-position of tyrosine to form a carbon-carbon bond. At this point, re-aromatization is favored, and the modified tyrosine loses a proton and electron, potentially to either the radical SAM cluster or an auxiliary cluster.
Author Contributions
I. B. and J. P. K. designed the study and the experiments. I. B., J. A. L., A. T. I., T. C., and J. D. H. performed and analyzed the experiments. I. B. and J. P. K. wrote the manuscript, and A. T. I. and T. C. revised the manuscript and added experimental details. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Prof. Michael Waterman (Vanderbilt University) for providing the E. coli FNR/FLD plasmids and Prof. Chris Chang (University of California, Berkeley) for use of the Mössbauer spectrometer. A mass spectrometer was acquired with National Institutes of Health support (1S10RR022393-01).
This work was supported by National Institutes of Health Grant GM 039296 (to J. P. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
- PQQ
- pyrroloquinoline quinone
- SAM
- S-adenosylmethionine
- FldA
- flavodoxin A
- FNR
- flavodoxin reductase
- dA
- deoxyadenosine
- CID
- collision-induced dissociation
- MS/MS
- tandem mass spectrometry.
References
- 1. Duine J. A. (1999) The PQQ story. J. Biosci. Bioeng. 88, 231–236 [DOI] [PubMed] [Google Scholar]
- 2. Shen Y.-Q., Bonnot F., Imsand E. M., RoseFigura J. M., Sjölander K., and Klinman J. P. (2012) Distribution and properties of the genes encoding the biosynthesis of the bacterial cofactor, pyrroloquinoline quinone. Biochemistry 51, 2265–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Kleef M. A., and Duine J. A. (1988) A search for intermediates in the bacterial biosynthesis of PQQ. Biofactors 1, 297–302 [PubMed] [Google Scholar]
- 4. Goosen N., Vermaas D. A., and van de Putte P. (1987) Cloning of the genes involved in synthesis of coenzyme pyrrolo-quinoline-quinone from Acinetobacter calcoaceticus. J. Bacteriol. 169, 303–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Magnusson O. T., Toyama H., Saeki M., Rojas A., Reed J. C., Liddington R. C., Klinman J. P., and Schwarzenbacher R. (2004) Quinone biogenesis: structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc. Natl. Acad. Sci. U.S.A. 101, 7913–7918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonnot F., Iavarone A. T., and Klinman J. P. (2013) Multistep, eight-electron oxidation catalyzed by the cofactorless oxidase, PqqC: identification of chemical intermediates and their dependence on molecular oxygen. Biochemistry 52, 4667–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klinman J. P., and Bonnot F. (2014) The intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ and LTQ. Chem. Rev. 114, 4343–4365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shisler K. A., and Broderick J. B. (2012) Emerging themes in radical SAM chemistry. Curr. Opin. Struct. Biol. 22, 701–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broderick J. B., Duffus B. R., Duschene K. S., and Shepard E. M. (2014) Radical S-adenosylmethionine enzymes. Chem. Rev. 114, 4229–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haft D. H., and Basu M. K. (2011) Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 193, 2745–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grell T. A. J., Goldman P. J., and Drennan C. L. (2015) SPASM and Twitch domains in AdoMet radical enzyme structures. J. Biol. Chem. 290, 3964–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Latham J. A., Iavarone A. T., Barr I., Juthani P. V., and Klinman J. P. (2015) PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J. Biol. Chem. 290, 12908–12918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burkhart B. J., Hudson G. A., Dunbar K. L., and Mitchell D. A. (2015) A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat. Chem. Biol. 11, 564–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Link A. J. (2015) Biosynthesis: leading the way to RiPPs. Nat. Chem. Biol. 11, 551–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., et al. (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jenkins C. M., Pikuleva I., Kagawa N., and Waterman M. R. (1997) Escherichia coli flavodoxin Sepharose as an affinity resin for cytochromes P450 and use to identify a putative cytochrome P450c17/3β-hydroxysteroid dehydrogenase interaction. Arch. Biochem. Biophys. 347, 93–102 [DOI] [PubMed] [Google Scholar]
- 17. Jenkins C. M., and Waterman M. R. (1998) NADPH-flavodoxin reductase and flavodoxin from Escherichia coli: characteristics as a soluble microsomal P450 reductase. Biochemistry 37, 6106–6113 [DOI] [PubMed] [Google Scholar]
- 18. Crack J. C., Green J., Thomson A. J., and Le Brun N. E. (2014) Techniques for the production, isolation, and analysis of iron-sulfur proteins. Methods Mol Biol. 1122, 33–48 [DOI] [PubMed] [Google Scholar]
- 19. Bader M. (1980) A systematic approach to standard addition methods in instrumental analysis. J. Chem. Educ. 57, 703 [Google Scholar]
- 20. Pace C. N., Vajdos F., Fee L., Grimsley G., and Gray T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zor T., and Selinger Z. (1996) Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies. Anal. Biochem. 236, 302–308 [DOI] [PubMed] [Google Scholar]
- 22. Wecksler S. R., Stoll S., Tran H., Magnusson O. T., Wu S.-P., King D., Britt R. D., and Klinman J. P. (2009) Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-l-methionine enzyme. Biochemistry 48, 10151–10161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saichana N., Tanizawa K., Pechoušek J., Novák P., Yakushi T., Toyama H., and Frébortová J. (2016) PqqE from Methylobacterium extorquens AM1: a radical S-adenosyl-l-methionine enzyme with an unusual tolerance to oxygen. J. Biochem. 159, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pandelia M.-E., Lanz N. D., Booker S. J., and Krebs C. (2015) Mössbauer spectroscopy of Fe/S proteins. Biochim. Biophys. Acta 1853, 1395–1405 [DOI] [PubMed] [Google Scholar]
- 25. Goldman P. J., Grove T. L., Sites L. A., McLaughlin M. I., Booker S. J., and Drennan C. L. (2013) X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proc. Natl. Acad. Sci. U.S.A. 110, 8519–8524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chatterjee A., Hazra A. B., Abdelwahed S., Hilmey D. G., and Begley T. P. (2010) A “radical dance” in thiamin biosynthesis: mechanistic analysis of the bacterial hydroxymethylpyrimidine phosphate synthase. Angew. Chem. Int. Ed. Engl. 49, 8653–8656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bruender N. A., Young A. P., and Bandarian V. (2015) Chemical and biological reduction of the radical SAM enzyme CPH4 synthase. Biochemistry 54, 2903–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wieckowski B. M., Hegemann J. D., Mielcarek A., Boss L., Burghaus O., and Marahiel M. A. (2015) The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS Lett. 589, 1802–1806 [DOI] [PubMed] [Google Scholar]
- 29. Roepstorff P., and Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601. [DOI] [PubMed] [Google Scholar]
- 30. Meulenberg J. J. M., Loenen W. A. M., Sellink E., and Postma P. W. (1989) The role of PQQ in K. aerogenes and cloning of pqq genes. in PQQ and Quinoproteins: Proceedings of the First International Symposium on PQQ and Quinoproteins, Delft, The Netherlands, 1988 (Jongejan J. A., and Duine J. A. eds), pp. 187–189, Springer Netherlands, Dordrecht, Netherlands, 10.1007/978-94-009-0957-1_28 [DOI] [Google Scholar]
- 31. Schwarzenbacher R., Stenner-Liewen F., Liewen H., Reed J. C., and Liddington R. C. (2004) Crystal structure of PqqC from Klebsiella pneumoniae at 2.1 Å resolution. Proteins 56, 401–403 [DOI] [PubMed] [Google Scholar]
- 32. Schramma K. R., Bushin L. B., and Seyedsayamdost M. R. (2015) Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink. Nat. Chem. 7, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]