

Abstract
Human CD8+ cytotoxic T lymphocytes can mediate tumor regression in melanoma through the specific recognition of HLA-restricted peptides. Because of the relatively weak affinity of most anti-cancer T-cell receptors (TCRs), there is growing emphasis on immunizing melanoma patients with altered peptide ligands in order to induce strong anti-tumor immunity capable of breaking tolerance toward these self-antigens. However, previous studies have shown that these immunogenic designer peptides are not always effective. The melanocyte differentiation protein, glycoprotein 100 (gp100), encodes a naturally processed epitope that is an attractive target for melanoma immunotherapies, in particular peptide-based vaccines. Previous studies have shown that substitutions at peptide residue Glu3 have a broad negative impact on polyclonal T-cell responses. Here, we describe the first atomic structure of a natural cognate TCR in complex with this gp100 epitope and highlight the relatively high affinity of the interaction. Alanine scan mutagenesis performed across the gp100280–288 peptide showed that Glu3 was critically important for TCR binding. Unexpectedly, structural analysis demonstrated that the Glu3 → Ala substitution resulted in a molecular switch that was transmitted to adjacent residues, abrogating TCR binding and T-cell recognition. These findings help to clarify the mechanism of T-cell recognition of gp100 during melanoma responses and could direct the development of altered peptides for vaccination.
Keywords: cancer, melanoma, surface plasmon resonance (SPR), T-cell receptor (TCR), X-ray crystallography, CD8+ T-cells, gp100, heteroclitic peptides, peptide human leukocyte antigen (pHLA)
Introduction
Cytotoxic T-cells can mediate a specific response against autologous melanoma cells by recognizing tumor-derived peptides presented at the cell surface by human leukocyte antigen (pHLA).6 In particular, epitopes encoded by differentiation melanocyte proteins may represent shared melanoma-associated antigens targeted by T-cell receptors (TCRs) on patients' lymphocytes (1). Glycoprotein 100 (gp100) has been a widely studied target for melanoma immunotherapy. This 661-amino acid long melanoma differentiation antigen is a melanosome matrix protein involved in melanosome maturation and melanin synthesis (2). In vivo, the protein has significantly differential expression between tumor cells, being often overexpressed in all stages of melanoma progression, compared with normal melanocytes (3).
Previous studies showed that gp100 encoded epitopes are recognized by tumor-infiltrating lymphocytes and circulating T-cells, associated with tumor regression in metastatic melanoma patients after adoptive therapy (4–7). Among these, the nonamer epitope gp100280–288 (YLEPGPVTA) was originally shown to be recognized by HLA-A*0201+ tumor-infiltrating lymphocytes from melanoma patients (8) and subsequently eluted from HLA-A*0201 molecules on melanoma cells (9). Immunization with gp100280–288 peptide has been shown to stimulate an in vitro polyclonal T-cell response in the context of HLA-A*0201, present in 49% of Caucasian individuals (10). These findings generated renewed interest in developing gp100-based anti-melanoma vaccines. However, we and others have previously shown, through direct biophysical measurements, that anti-cancer TCRs bind to their cognate pHLA with affinities that are approximately 1 order of magnitude weaker than those of pathogen-specific TCRs (11, 12). Thus, altered peptide ligands, with improved primary HLA anchor residues (heteroclitic peptides), have been designed for a few melanoma-associated antigens in order to increase immunogenicity (6, 10, 13). Among these, the heteroclitic version of gp100280–288 (in which a valine replaces alanine at anchor position 9 to improve pHLA stability (14)) enhanced the induction of melanoma-reactive cytotoxic T lymphocytes in vitro and has been successfully used in clinical trials (15). Another heteroclitic form of gp100280–288, in which peptide residue Glu3 was substituted to Ala, abrogated recognition by a polyclonal population of gp100280–288-specific T-cells (16, 17). Thus, a more complete understanding of the molecular mechanisms underlying gp100280–288 targeting by specific TCRs is needed to direct the design of improved altered peptide ligands.
Previous studies using another HLA-A*0201-restricted melanoma-derived epitope have demonstrated that even minor changes in peptide anchor residues can substantially alter T-cell recognition in unpredictable ways (13, 18). In order to aid in the future design of enhanced peptide vaccines based on gp100280–288, we solved the ternary atomic structure of a human TCR in complex with the heteroclitic gp100280–288 peptide. We then used a peptide scanning approach to demonstrate the impact of peptide substitutions on TCRs from two different T-cell clones by performing in depth biophysical and functional experiments. These data demonstrate that modification of peptide residues outside of the TCR binding motif can have unpredictable knock-on effects (a modification to a residue that affects an adjacent residue indirectly) on adjacent peptide residues that abrogate TCR binding and T-cell recognition. Indeed, even conservative peptide substitutions can have unexpected consequences for T-cell recognition due to knock-on structural changes in the HLA-bound peptide. Our findings provide a molecular explanation for the sensitivity to substitutions at gp100280–288 peptide residue Glu3 (16, 17) and represent the first example of the structural mechanisms underlying T-cell recognition of this important therapeutic target for melanoma.
Experimental Procedures
Generation of Expression Plasmids
The PMEL17 TCR (TRAV21 TRBV7-3) (12) and gp100 TCR (TRAV17 TRBV19) (11) are both specific for the human HLA-A*0201 restricted YLE epitope (gp100280–288, sequence YLEPGPVTA). For each TCR, a disulfide-linked construct was used to produce the soluble α- and β-chain domains (variable and constant) (19, 20). The HLA*0201 α-chain and β2m sequences were generated by PCR cloning. All sequences were confirmed by automated DNA sequencing (Cardiff Biotechnology Service). For surface plasmon resonance (SPR) experiments, the soluble HLA-A*0201 α-chain was tagged with a biotinylation sequence, as described previously (21). All four constructs (TCRα, TCRβ, HLA-A*0201 α-chain, and β2m) were inserted into separate pGMT7 expression plasmids under the control of the T7 promoter (20).
Protein Expression, Refolding, and Purification
Competent Rosetta DE3 Escherichia coli cells were used to produce the PMEL17 TCR, gp100 TCR α and β chains, and HLA-A*0201 α and β2m chains in the form of inclusion bodies, using 0.5 m isopropyl 1-thio-β-d-galactopyranoside to induce expression. Soluble PMEL17 TCR, gp100 TCR, and pHLA proteins were refolded as described previously (19, 20), purified by anion exchange (Poros 50HQ, Life Technologies, Cheshire, UK) and size exclusion chromatography (S200 GR, GE Healthcare, Buckinghamshire, UK). For the pHLAs, HLA-A*0201 was refolded with YLEPGPVTA (A2-YLE), YLEPGPVTV (A2-YLE-9V), ALEPGPVTA (A2-YLE-1A), YLAPGPVTA (A2-YLE-3A), YLEAGPVTA (A2-YLE-4A), YLEPAPVTA (A2-YLE-5A), YLEPGAVTA (A2-YLE-6A), YLEPGPATA (A2-YLE-7A), or YLEPGPVAA (A2-YLE-8A).
SPR Analysis
The binding analysis was performed using a BIAcore 3000 or Biacore T100 equipped with a CM5 sensor chip as reported previously (21). Briefly, 500–600 response units of biotinylated pHLA-I complexes were immobilized to streptavidin, which was chemically linked by amine coupling to the chip surface. Biotinylated pHLA-I complexes were prepared as described previously (21) and injected at a slow flow rate (10 μl/min) to ensure a uniform distribution on the chip surface. Results were analyzed using BIAevaluationTM version 3.1, Microsoft ExcelTM, and OriginTM version 6.0. For equilibrium analysis, 9–10 serial dilutions of concentrated TCR were injected over the relevant sensor chip. The equilibrium-binding constant (KD(E)) values were calculated using a nonlinear curve fit (y = (P1x)/(P2 + x)). For the thermodynamics experiments, KD values determined by SPR at different temperatures were used with the standard thermodynamic equationΔG0 = −RTlnKD. The thermodynamic parameters were calculated according to the Gibbs-Helmholtz equation (ΔG0 = ΔH - TΔS0). The binding free energies, ΔG0 (ΔG0 = −RTlnKD), were plotted against temperature (K) using nonlinear regression to fit the three-parameter equation, (y = ΔH + ΔCp*(x − 298) − x*ΔS − x*ΔCp*ln(x/298)). For kinetics analysis, the Kon and Koff values were calculated assuming 1:1 Langmuir binding, and the data were analyzed using a global fit algorithm (BIAevaluationTM version 3.1). All SPR experiments were conducted in triplicate.
Crystallization, Diffraction Data Collection, and Model Refinement
All protein crystals were grown at 18 °C by vapor diffusion via the sitting drop technique. 200 nl of 1:1 molar ratio TCR and pHLA-I (10 mg/ml) in crystallization buffer (10 mm Tris, pH 8.1, and 10 mm NaCl) was added to 200 nl of reservoir solution. PMEL17 TCR·A2-YLE-9V crystals were grown in 0.2 m sodium sulfate, 0.1 m Bistris propane, pH 6.5, 20% (w/v) PEG 3350. Crystals of pHLA complexes were grown at 18 °C by seeding into hanging drops of 0.5 μl of seeding solution + 1 μl of complex + 1 μl of 0.1 m Hepes, pH 7.5, 0.2 m ammonium sulfate, 25% PEG 4000 (22). Data were collected at 100 K at the Diamond Light Source (Oxfordshire, UK). All data sets were collected at a wavelength of 0.976 Å using an ADSC Q315 CCD detector. Reflection intensities were estimated with the XIA2 package (23), and the data were scaled, reduced, and analyzed with SCALA and the CCP4 package (24). Structures were solved by molecular replacement using PHASER (25). Sequences were adjusted with COOT (26) and the models refined with REFMAC5 (27). Graphical representations were prepared with PyMOL (28). Data reduction and refinement statistics are shown in Table 1. The reflection data and final model coordinates were deposited in the Protein Data Bank (entries 5EU6 (PMEL17 TCR·A2-YLE-9V), 5EU3 (A2-YLE), 5EU4 (A2-YLE-3A), and 5EU5 (A2-YLE-5A)).
TABLE 1.
| TCR | CDR1α | CDR2α | CDR3α | CDR1β | CDR1β | CDR1β |
|---|---|---|---|---|---|---|
| PMEL17 | DSAIYN | IQSSQRE | CAVLSSGGSNYKLTFG | SGHTA | FQGTGA | CASSFIGGTDTQYFG |
| gp100 | TSINN | IRSNERE | CATDGDTPLVFG | LNHDA | SQIVND | CASSIGGPYEQYFG |
| MPD | KALYS | LLKGGEQ | CGTETNTGNQFYFG | SGHDY | FNNNVP | CASSLGRYNEQFFG |
| 296 | DSASNY | IRSNVGE | CAASTSGGTSYGKLTFG | MNHEY | SMNVEV | CASSLGSSYEQYFG |
Isothermal Titration Calorimetry (ITC)
ITC experiments were performed using a Microcal VP-ITC (GE Healthcare) as described previously (29), with 30 μm pHLA-I in the calorimeter cell and 210 μm soluble PMEL17 TCR in the syringe. Buffer conditions were 20 mm Hepes (pH 7.4) containing 150 mm NaCl, and 20 injections of 2 μl each were performed. Results were processed and integrated with the OriginTM version 6.0 software distributed with the instrument. ITC experiments were performed in duplicate.
Lentivirus Generation and Transduction of CD8+ T-cells
Lentivirus particles were generated by combining packaging plasmids pRSV, pMDL, and pVSG-V with a lentivirus plasmid bearing the gp100 TCR construct (provided by Immunocore Ltd., Oxford, UK) and used to CaCl2-transfect HEK293T/17 (ATCC) cells. Supernatant was collected after 24- and 48-h incubations, and lentiviral stocks were concentrated by ultracentrifugation. Primary CD8+ T-cells were obtained by standard density gradient centrifugation from donor blood bags, followed by positive selection using CD8 microbeads (Miltenyi Biotec). Cells were activated overnight with anti-CD3/CD28 Dynabeads (Invitrogen) (1:1) and transduced with concentrated lentivirus particles. Transduction efficiency was determined after 72 h by flow cytometry after staining with the relevant anti-TCRVβ mAb (BD Biosciences). Untransduced cells or MEL5 TCR (specific for the Melan-A/MART-1 epitope ELAGIGILTV)-transduced cells were used as controls (data not shown). Transductions were performed using primary CD8+ T-cells from three different anonymous donors.
Measurement of MIP-1β and TNFα by ELISA
To quantify the production of MIP-1β and TNFα, 6 × 104 T2 target cells were pulsed with peptide as indicated for 1 h and added to 3 × 104 overnight rested T-cells. Following overnight incubation, cells were pelleted, and the culture supernatant was harvested for measurement of MIP-1β and TNFα by ELISA according to the manufacturer's protocol (R&D Systems). Each data point represents the average of duplicate measurements.
Cytotoxicity Assay
Cytotoxic assays in this study were performed in a standard 4-h 51Cr release assay. Briefly, 2 × 103 T2 cells were labeled with 51Cr (PerkinElmer Life Sciences) and then pulsed with peptide at the indicated concentration and used as target cells. Effector and target cells, at an effector/target ratio of 5, were incubated for 4 h at 37 °C, and the supernatant was harvested. Target cells were also incubated alone or with 5% Triton X-100 detergent to give the spontaneous and total 51Cr released from the target cells, respectively. 51Cr release was determined by γ-counting (1450 Microbeta counter, PerkinElmer Life Sciences). The percentage of specific cell lysis was calculated according to the following formula: (experimental release (with effector and target cells) − spontaneous release from target cells)/(total release from target cells − spontaneous release from target cells) × 100. Each data point represents the average of duplicate measurements.
Results
Two Distinct Anti-gp100 TCRs Share Similar Binding Hot Spots
The CD8+ T-cell responses directed against gp100280–288have been shown to be polyclonal in nature (16, 17). Along with the two TCRs under investigation here, the sequences of a further two TCRs have been published, demonstrating diverse gene usage and different CDR3 loop sequences (Table 1). Despite these differences, previous studies of T-cell responses to gp100280–288 have demonstrated that modifications to peptide residue Glu3 can broadly effect activation of gp100280–288-specific T-cells (16, 17). Thus, in order to study the individual contribution of the peptide residues involved in TCR recognition of gp100280–288, particularly in relation to peptide residues Glu3, an alanine scan mutagenesis was performed across the peptide backbone, and TCR binding affinity was evaluated by SPR experiments. Residues P2 and P9, which are known to be important for HLA-A*0201 binding (30) were not assessed; in addition, the P9 residue was an Ala in the native sequence. The heteroclitic YLE-9V peptide, which has been shown to be a better agonist than the wild type sequence (10), was included in the experiment. SPR experiments were conducted with two distinct gp100280–288-specific TCRs: PMEL17 TCR (TRAV21 TRBV7-3) and gp100 TCR (TRAV17 TRBV19). PMEL17 TCR bound both A2-YLE and A2-YLE-9V with similar affinities (KD = 7.6 and 6.3 μm, respectively), consistent with the fact that the YLE-9V variant was originally designed in such a way as to increase peptide-HLA binding affinity without significantly altering TCR recognition of the pHLA complex (10) (Table 2). The gp100 TCR demonstrated a similar pattern, although at weaker affinities, of KD = 26.5 and 21.9 μm, for A2-YLE and A2-YLE-9V, respectively. With the exception of A2-YLE-3A and A2-YLE-5A, both the PMEL17 and gp100 TCRs tolerated substitutions in the native gp100280–288 peptide, albeit with reduced binding affinity, although substitutions at the peptide C terminus generally reduced binding affinity to a greater extent than at the N terminus. Substitution of peptide residue 5 to alanine reduced the affinity for both TCRs to KD >1 mm. Interestingly, replacement of Glu by Ala in position 3 completely abrogated binding by PMEL17 and gp100 TCRs, suggesting that the Glu at p3 was a dominant contact for both TCRs. Our results are supported by a recent study of gp100280–288altered peptide ligands, which described YLE-3A as a null agonist for a different TCR (17). Our data indicated that both PMEL17 and gp100 TCRs used a similar overall binding mode, focused around peptide residues 3 and 5 with supporting interactions toward the N terminus of the peptide. In combination with other data in this system (17), alanine substitution data suggest that disparate TCRs adopt a similar binding mode on A2-YLE, where position 3 dominates recognition. In order to confirm this hypothesis, we crystallized the PMEL17 TCR in complex with A2-YLE-9V.
TABLE 2.
Affinity analysis (KD) of PMEL17 TCR and gp100 TCR to gp100280–288 peptide variants
| Peptide sequence | Peptide | PMEL17 TCR (TRAV21 TRBV7-3), affinity KD | gp100 TCR (TRAV17 TRBV19), affinity KD |
|---|---|---|---|
| YLEPGPVTA | YLE | 7.6 ± 2 μm | 26.5 ± 2.3 μm |
| YLEPGPVTV | YLE-9V | 6.3 ± 1.2 μm | 21.9 ± 2.4 μm |
| ALEPGPVTA | YLE-1A | 15.9 ± 4.1 μm | 60.6 ± 5.4 μm |
| YLAPGPVTA | YLE-3A | No binding | No binding |
| YLEAGPVTA | YLE-4A | 19.7 ± 1.3 μm | 144.1 ± 7.8 μm |
| YLEPAPVTA | YLE-5A | >1 mm | >1 mm |
| YLEPGAVTA | YLE-6A | 11.4 ± 2.7 μm | 954.9 ± 97.8 μm |
| YLEPGPATA | YLE-7A | 31.1 ± 4 μm | 102.0 ± 9.2 μm |
| YLEPGPVAA | YLE-8A | 38.1 ± 7.4 μm | 121.0 ± 7.5 μm |
The PMEL17 TCR Utilizes a Peptide-centric Binding Mode to Engage A2-YLE-9V
To understand why TCR recognition of gp100280–288 was highly sensitive to some of the substitutions in the native peptide sequence, we determined the crystal structure of the PMEL17 TCR in complex with A2-YLE-9V at 2.02 Å resolution with crystallographic Rwork/Rfree ratios within accepted limits (Table 3) as shown in the theoretically expected distribution (31). Electron density around the TCR·pHLA contact interface was unambiguous (Fig. 1). The PMEL17 TCR was centrally positioned over the exposed residues of the peptide (Fig. 2, A and B) and used a conventional diagonal orientation (crossing angle = 46.15°, calculated as in Ref. 32), with the α-chain positioned over the α2 helix of the HLA-I binding groove and the β-chain over the α1 helix. All but the CDR2α loop were involved in contacting A2-YLE-9V, with the CDR3α and CDR3β “sitting” on the central axis of the antigen-binding cleft, making contacts with both the peptide and α-helices of the HLA (Fig. 1B). The PMEL17 TCR made approximately the same number of peptide-mediated contacts and HLA-A*0201 interactions, forming 53 of 125 (42.4%) van der Waals contacts and 3 of 8 (37.5%) hydrogen bonds between the TCR and the peptide (Table 4). The HLA helices were contacted by residues within the CDR3α, CDR3β, and CDR2β loops (with additional support of CDR1α residue Tyr32), which focused on Arg65, Ala69, Gln72, and Gln155 (Fig. 1C). HLA residues at positions 65, 69, and 155 are conserved TCR-mediated contact points in several TCR·pHLA-I structures determined so far and are referred to as the “restriction triad” (33).
TABLE 3.
Data reduction and refinement statistics (molecular replacement)
| Parameters | PMEL17 TCR·A2-YLE-9V | A2-YLE | A2-YLE-3A | A2-YLE-5A |
|---|---|---|---|---|
| Protein Data Bank code | 5EU6 | 5EU3 | 5EU4 | 5EU5 |
| Data set statistics | ||||
| Space group | P1 | P1 21 1 | P1 | P1 21 1 |
| Unit cell parameters (Å) | a = 45.52, b = 54.41, c = 112.12, α = 85.0°, β = 81.6°, γ = 72.6° | a = 52.81, b = 80.37, c = 56.06, β = 112.8° | a = 56.08, b = 57.63, c = 79.93, α = 90.0°, β = 89.8°, γ = 63.8° | a = 56.33, b = 79.64, c = 57.74, β = 116.2° |
| Radiation source | DIAMOND I03 | DIAMOND I03 | DIAMOND I02 | DIAMOND I02 |
| Wavelength (Å) | 0.9763 | 0.9999 | 0.9763 | 0.9763 |
| Measured resolution range (Å) | 51.87–2.02 | 45.25–1.97 | 43.39–2.12 | 43.42–1.54 |
| Outer Resolution Shell (Å) | 2.07–2.02 | 2.02–1.97 | 2.18–2.12 | 1.58–154 |
| Reflections observed | 128,191 (8,955) | 99,442 (7,056) | 99,386 (7,463) | 244,577 (17,745) |
| Unique reflections | 64,983 (4,785) | 30,103 (2,249) | 49,667 (3,636) | 67,308 (4,962) |
| Completeness (%) | 97.7 (96.7) | 98.5 (99.3) | 97.4 (96.7) | 99.6 (99.9) |
| Multiplicity | 2.0 (1.9) | 3.3 (3.1) | 2.0 (2.1) | 3.6 (3.6) |
| I/σ(I) | 5.5 (1.9) | 7.2 (1.9) | 6.7 (2.3) | 13 (2.3) |
| Rpim (%) | 5.7 (39.8) | 8.8 (44.7) | 8.7 (41.6) | 4.5 (35.4) |
| Rmerge (%) | 7.8 (39.6) | 9.8 (50.2) | 8.7 (41.6) | 5.0 (53.2) |
| Refinement statistics | ||||
| Resolution (Å) | 2.02 | 1.97 | 2.12 | 1.54 |
| No. of reflections used | 61,688 | 28,557 | 47,153 | 63,875 |
| No. of reflections in Rfree set | 3294 | 1526 | 2514 | 3406 |
| Rcryst (no cut-off) (%) | 18.1 | 19.7 | 17.2 | 17.0 |
| Rfree | 22.2 | 25.5 | 21.1 | 20.1 |
| Root mean square deviation from ideal geometry | ||||
| Bond lengths (Å) | 0.018 (0.019)a | 0.019 (0.019) | 0.021 (0.019) | 0.018 (0.019) |
| Bond angles (degrees) | 1.964 (1.939) | 1.961 (1.926) | 2.067 (1.927) | 1.914 (1.936) |
| Overall coordinate error (Å) | 0.122 | 0.153 | 0.147 | .055 |
| Ramachandran statistics | ||||
| Most favored | 791 (96%) | 371 (98%) | 749 (99%) | 384 (98%) |
| Allowed | 32 (4%) | 6 (2%) | 10 (1%) | 5 (1%) |
| Outliers | 2 (0%) | 3 (1%) | 1 (0%) | 2 (0%) |
a Values in parentheses are for the highest resolution shell.
FIGURE 1.
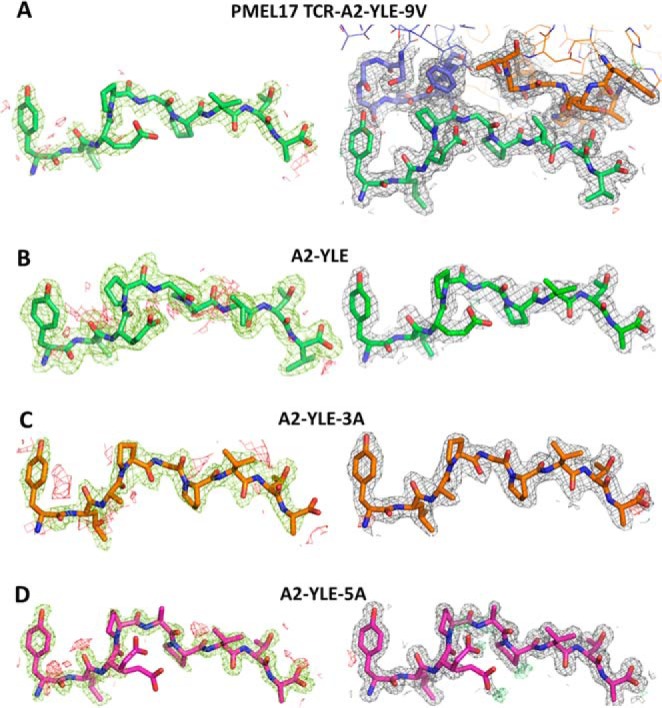
Density plot analysis. The left column shows omit maps in which the model was refined in the absence of the peptide. Difference density is contoured at 3.0 σ; positive contours are shown in green, and negative contours are red. The right-hand column shows the observed map at 1.0 σ (shown as gray mesh around stick representations of the protein chains) after subsequent refinement using automatic non-crystallographic symmetry restraints applied by REFMAC5. A, model for PMEL17 TCR·A2-YLE-9V with the TCR CDR3 loops colored blue (α chain) and orange (β chain) and the peptide in green; B, model for A2-YLE with the peptide colored dark green; C, model for A2-YLE-3A with the peptide colored orange (for A2-YLE-3A, there were two molecules in the asymmetric unit, but these were virtually identical in terms of omit and density maps, so only copy 1 is shown here); D, model for A2-YLE-5A with the peptide colored pink.
FIGURE 2.
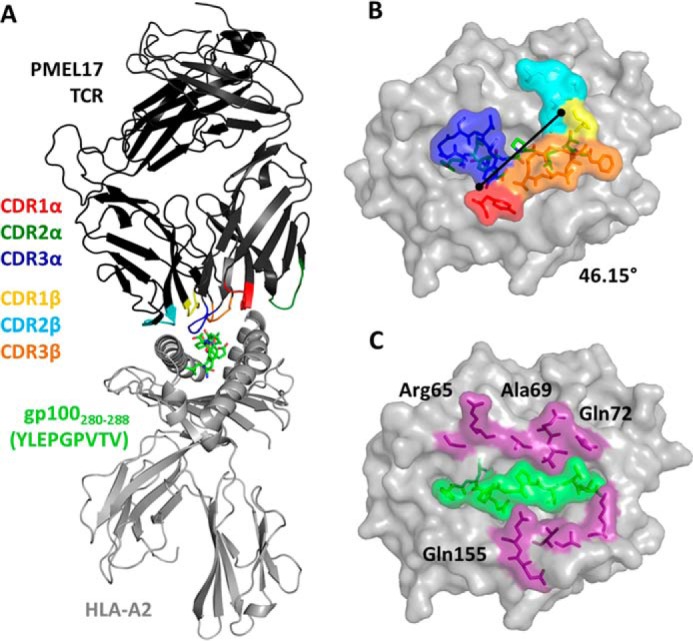
Overview of the PMEL17 TCR in complex with A2-YLE-9V. A, schematic representation of the PMEL17 TCR·A2-YLE-9V complex. The TCR α-chain and TCR β-chain are dark gray and black, respectively; TCR CDR loops are shown (red, CDR1α; dark green, CDR2α; blue, CDR3α; yellow, CDR1β; aqua, CDR2β; orange, CDR3β); the HLA-A*0201 is depicted in gray. The YLE-9V peptide is represented in green sticks. B, surface and stick representation of residues of the PMEL17 TCR CDR loops (color-coded as in A) that contact the A2-YLE surface (A2, gray; YLE-9V, green sticks). Black diagonal line, crossing angle of the TCR with respect to the long axis of the YLEPGPVTV peptide (46.15°). C, contact footprint of the PMEL17 TCR on the A2-YLE-9V surface (A2, gray); purple and green (surface and sticks) indicate HLA-A*0201 and YLE residues, respectively, contacted by the gp100 TCR (cut-off of 3.4 Å for hydrogen bonds and 4 Å for van der Waals contacts).
TABLE 4.
PMEL17 TCR·A2-YLE-9V contact table
| HLA/peptide residue | TCR residue | No. of van der Waals contacts (≤4 Å) | No. of hydrogen bonds (≤3.4 Å) |
|---|---|---|---|
| Gly62 | αGly98 | 3 | |
| αSer99 | 1 | ||
| Arg65 | αSer99 | 2 | |
| Arg65 O | αAsn100 Nδ2 | 2 | 1 |
| Arg65 NH1 | βAsp58 Oδ2 | 1 | |
| βSer59 | 8 | ||
| Lys66 | αGly98 | 1 | |
| αSer99 | 4 | ||
| αAsn100 | 4 | ||
| Ala69 | αAsn100 | 2 | |
| βAla56 | 2 | ||
| Gln72 Nϵ2 | βGln51 O | 3 | 1 |
| βGly54 | 7 | ||
| βAla55 | 1 | ||
| Thr73 | βGln51 | 1 | |
| Val76 | βGln51 | 3 | |
| βGly52 | 2 | ||
| Lys146 | βPhe97 | 3 | |
| βIle98 | 3 | ||
| Ala150 | βIle98 | 1 | |
| βAsp102 | 3 | ||
| Val152 | βIle98 | 1 | |
| Glu154 | αTyr32 | 1 | |
| Gln155 N | αTyr32 OH | 4 | 1 |
| Gln155 Oϵ1 | βThr101 N | 10 | 1 |
| Tyr1 OH | αGly97 O | 1 | 1 |
| αGly98 | 1 | ||
| αSer96 | 1 | ||
| Glu3 | αTyr101 | 1 | |
| Pro4 | αSer96 | 1 | |
| αSer99 | 1 | ||
| αAsn100 | 4 | ||
| Pro4 O | αTyr101 N | 14 | 1 |
| Gly5 | αTyr101 | 3 | |
| βGly100 | 2 | ||
| Val7 | βIle98 | 7 | |
| βGly99 | 2 | ||
| βGly100 | 2 | ||
| Thr8 | βThr31 | 5 | |
| βGln51 | 1 | ||
| βPhe97 | 1 | ||
| Thr8 N | βIle98 O | 6 | 1 |
To complement information gained from the crystal structure, we studied the affinity and thermodynamic parameters of the PMEL17 TCR·A2-YLE complex. The binding strength of the complex was measured at 5, 12, 18, 25, and 37 °C by SPR (Fig. 3A). The PMEL17 TCR·A2-YLE interaction at 25 °C (the standard temperature for TCR·pHLA parameter measurements) was characterized by a binding ΔG of −7.5 kcal/mol, which is within the normal range of TCR·pHLA interactions (34). PMEL17 TCR·A2-YLE binding was characterized by a very small, favorable enthalpy change (ΔH = −0.6 kcal/mol) and a larger, positive entropy change (TΔS = 6.9 cal/mol) (Fig. 3B). Therefore, order to disorder drove the interaction, probably through the expulsion of ordered water molecules at the interface (i.e. solvation effects). ITC was also performed because it provides a direct measure of enthalpy and is therefore considered the most reliable determination of thermodynamic parameters (29). ITC measurements (ΔH = −0.3 kcal/mol and TΔS = 5.6 cal/mol) confirmed observations made with SPR thermodynamics (Fig. 3C). The favorable enthalpy of this TCR·pHLA system shows that the overall number of formed bonds is equal to the number of disrupted ones upon PMEL17 TCR binding.
FIGURE 3.

Thermodynamic analysis of the PMEL17 TCR·A2-YLE interaction. A, PMEL17 TCR equilibrium-binding responses to A2-YLE at 5, 12, 18, 25, and 37 °C across 9–10 TCR serial dilutions. SPR raw and fitted data (assuming 1:1 Langmuir binding) are shown in the inset of each curve and were used to calculate Kon and Koff values using a global fit algorithm (BIAevaluation version 3.1). The table shows equilibrium-binding (KD(E)) and kinetic-binding constant (KD(K) = Koff/Kon) at each temperature. The equilibrium binding constant (KD, μm) values were calculated using a nonlinear fit (y = (P1x)/(P2 + x)). B, the thermodynamic parameters were calculated according to the Gibbs-Helmholtz equation (ΔG0 = ΔH − TΔS0). The binding free energies, ΔG0 (ΔG0 = −RTlnKD), were plotted against temperature (K) using nonlinear regression to fit the three-parameter equation (y = dH + dCp*(x − 298) − x*dS − x*dCp*ln(x/298)). Enthalpy (ΔH0) and entropy (TΔS0) at 298 K (25 °C) are shown in kcal/mol and were calculated by a non-linear regression of temperature (K) plotted against the free energy (ΔG0). C, ITC measurements for PMEL17 TCR·A2-YLE interaction. Enthalpy (ΔH0) and entropy (TΔS0) at 298 K (25 °C) are shown in kcal/mol.
The PMEL17 CDR Loops Focus on Peptide Residues Pro4, Val7, and Thr8
The central positioning of the PMEL17 TCR enabled contacts with 6 of 9 amino acids in the peptide (Fig. 4A). Peptide residues Pro4, Val7, and Thr8 represented the focal points of the PMEL17 TCR. Pro4 made a sizeable network of interactions (1 hydrogen bond and 14 van der Waal contacts) (Fig. 4B). Interestingly, Pro6 was the only central residue that did not interact with the PMEL17 TCR because of its reduced surface exposure. Therefore, the relative insensitivity of the PMEL17 TCR to alanine substitution at position 6 is consistent with its lack of involvement in TCR binding. In contrast, the gp100 TCR seemed to be more sensitive to this same mutation, causing a ∼40-fold drop in binding affinity compared with the unaltered peptide (Table 2). This might be explained by the different TRAV and TRBV gene usage of the two gp100-specific TCRs and the very different residues of the CDR3 loops possibly contacting the central part of the gp100280–288 peptide. However, the PMEL17 TCR complex structure did not provide any clear mechanisms to explain the reduction in binding observed when peptide residues 3 and 5 were mutated to alanine.
FIGURE 4.
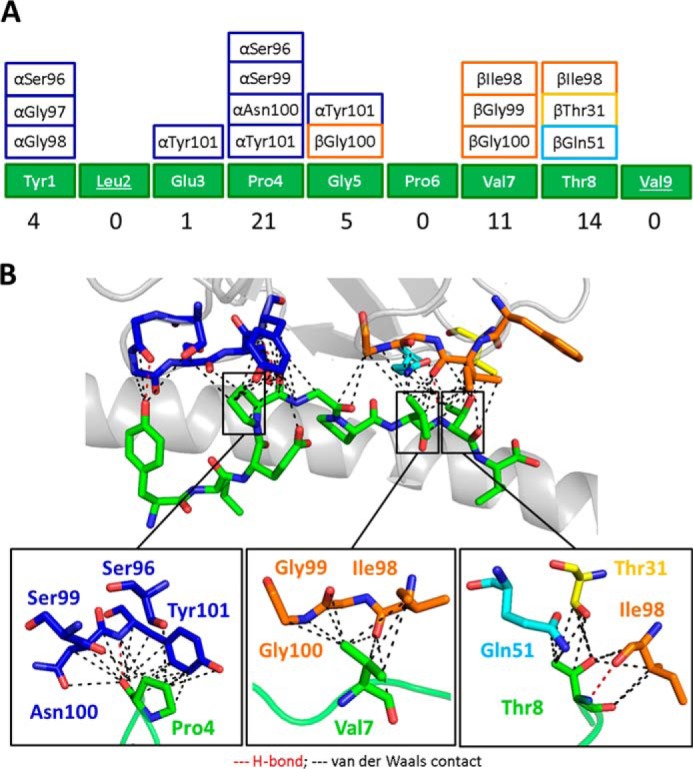
The PMEL17 CDR loops focus on peptide residues Pro4, Val7, and Thr8. A, schematic representation of contacts between YLE-9V peptide and PMEL17 CDR loop residues (color-coded as in Fig. 2A). The numbers at the bottom show total contacts between the TCR and peptide. B, contacts between the PMEL17 TCR and the YLE-9V peptide (green sticks), showing the van der Waals contacts (black dashed lines) and hydrogen bonds (red dashed lines) made by the TCR CDR3α (blue), CDR1β (yellow), CDR2β (aqua), and CDR3β (orange) loops. Bottom panel, close view of contacts between YLE Pro4, Val7, and Thr8, respectively, and TCR CDR loop residues (sticks color-coded as in Fig. 1A) (cut-off of 3.4 Å for hydrogen bonds and a cut-off of 4 Å for van der Waals contacts).
Peptide Substitutions Can Induce Perturbation at Adjacent Peptide Residues Abrogating T-cell Recognition
In order to understand the structural basis underlying the large changes in affinity of PMEL17 TCR·A2-YLE binding resulting from Glu3 → Ala and Gly5 → Ala substitutions, we also solved the unligated structures of A2-YLE, A2-YLE-3A, and A2-YLE-5A. The structures were solved between 1.54 and 2.12 Å resolution with crystallographic Rwork/Rfree ratios within accepted limits (Table 3) (31). Electron density around the peptide was unambiguous (Fig. 1). Comparison of the crystallographic structure of A2-YLE and A2-YLE-3A or A2-YLE-5A complexes did not reveal major changes in the peptide backbone conformation (Fig. 5, A and B). However, in the A2-YLE structure, Glu3 bridges across to the main chain at position 4–5; the mutation of Glu3 into a shorter alanine side chain, which is no longer able to bridge across the void, caused a knock-on effect on the central Pro4 residue (Fig. 5A). This difference could not be explained by the difference in resolution and coordinate errors in the A2-YLE-3A structure (A2-YLE-3A was solved at 2.12 Å, compared with 1.97 Å for A2-YLE and 1.54 Å for A2-YLE-5A), demonstrated by omit map analysis shown in Fig. 1. Pro4 in the A2-YLE-3A structure lost restraint, causing the oxygen atom to flip in the opposite direction. Because of this unanticipated displacement of the Pro4 oxygen, the outward facing side of the Pro4 residue was no longer in an optimal position to be contacted by the TCR, therefore potentially losing the dominant network of interactions (Fig. 4B). These findings explain the complete absence of measurable binding of the YLE-3A mutant in the alanine scan. Gly5 was the only gp100280–288 peptide residue contacted by both CDR3 loops through αTyr101 and βGly100 in the PMEL17 TCR·A2-YLE-9V structure (Fig. 5B). In the A2-YLE-5A structure, steric hindrance in the center of the peptide may explain the substantial reduction in binding affinity observed in the alanine scan. As with YLE-3A, the YLE-5A substitution did not distort the overall conformation of the YLE nonamer.
FIGURE 5.
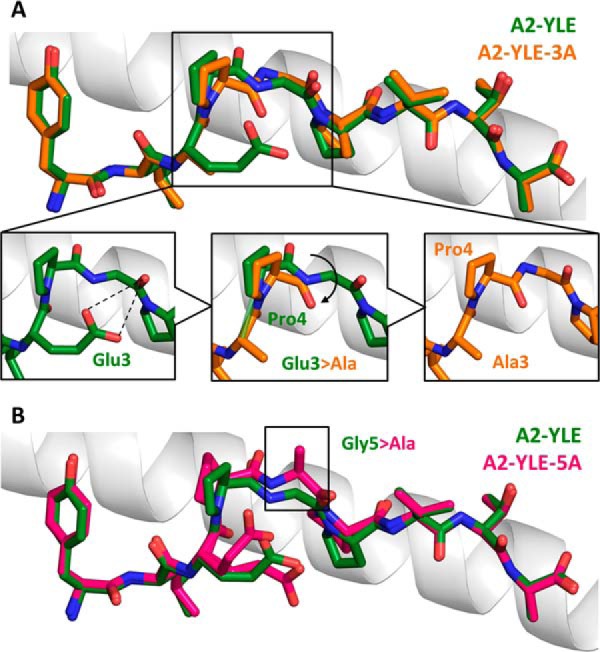
Conformational comparison of YLE, YLE-3A, and A2-YLE-5A peptides presented by HLA-A*0201. A, YLE (dark green sticks) and YLE-3A (orange sticks) peptide alignment by superimposition of HLA-A*0201 α1 helix (gray schematic). Boxed residues indicate the mutation of Glu3 into an alanine. The insets show how the Glu3 → Ala substitution causes a shift in position (black arrow) of neighbor residue Pro4 in the A2-YLE-3A structure compared with the A2-YLE structure. B, YLE (dark green sticks) and YLE-5A (pink sticks) peptide alignment by superimposition of HLA-A*0201 α1 helix (gray schematic). Boxed residues indicate the mutation of glycine 5 into an alanine.
Alanine Substitutions at YLE Peptide Residues 3 and 5 Abrogate T-cell Activation
To determine the effect of gp100280–288 altered peptide ligands on the activation of T-cells, we examined the ability of these mutants to induce MIP-1β, TNFα production and specific cytotoxicity (Fig. 6). These are key effector functions of activated CD8+ T-cells, which can be measured over a wide range of peptide concentrations. Human primary CD8+ T-cells were transduced with a lentiviral construct carrying the gp100 TCR and enriched for high and equal levels of TCR expression. Staining with TCRVβ mAb showed that ∼40% of total CD8+ T-cells stained as positive for gp100 TCR expression by flow cytometry in three independent donors (data not shown). Transduced CD8+ T-cells were stimulated with peptide-pulsed HLA-A*0201+ T2 target cells, across a range of different concentrations of gp100280–288 altered peptide ligands. Antigen-specific responses of gp100 TCR-engineered T-cells were validated at the level of production of MIP-1β and specific lysis of pulsed targets. Non-transduced CD8+ T-cells were used to control for nonspecific activation through the endogenous TCR; T-cells transduced with the MEL5 TCR (specific for the HLA-A*0201 restricted cancer epitope ELA from the Melan-A/MART-1 protein) were used as an irrelevant control in all experiments (data not shown). Peptide titration experiments showed marked differences in the ability to sensitize target cells for MIP-1β production by CD8+ gp100-specific T-cells (Fig. 6A). In particular, target cells pulsed with YLE and YLE-9V were recognized more efficiently than those pulsed with YLE-1A, YLE-8A, YLE-4A, and YLE-7A. No MIP-1β production was measured with YLE-3A and YLE-5A peptide ligands, even at higher peptide concentrations. TNFα production was measured by ELISA from the same supernatants (Fig. 6C), and low levels of this cytokine were only detected when cells were pulsed with YLE, YLE-9V, YLE-8A, or YLE-6A. Fig. 6B shows specific lysis of target cells pulsed with the same range of peptides and measured by a 51Cr release assay. Similar to the MIP-1β response curves, the specific lysis induced by these altered gp100280–288 ligands was variable. Most importantly, no cytotoxic T lymphocyte-mediated lysis was observed when peptide YLE-3A or YLE-5A was used. Taken together, these data are consistent with the molecular analysis demonstrating that the structural and biophysical alterations induced by peptide modifications translate directly to the effects that we observed upon T-cell recognition.
FIGURE 6.

Production of MIP-1β and TNFα and cytotoxicity by gp100 TCR-transduced CD8+ T-cells in response to stimulation with YLE peptide ligands. A, gp100 TCR-transduced CD8+ T-cells were stimulated with peptide-pulsed target cells as indicated. Supernatant was assayed for MIP-1β by ELISA. B, gp100 TCR-transduced CD8+ T-cells were tested in a standard 4-h 51Cr release assay against peptide-pulsed targets. C, gp100 TCR-transduced CD8+ T-cells were stimulated with peptide-pulsed target cells (10−5 m peptide) as indicated. Supernatant was assayed for TNFα by ELISA. ELA (Melan-A/MART-1) peptide was used as an irrelevant control in all experiments. Results of one donors (of three) are shown. Error bars, S.E.
Discussion
TCRs specific for cancer epitopes are generally characterized by low binding affinities (binding KD values in the high micromolar range) (35). This lower binding affinity is thought to be a result of negative selection of T-cells that bear TCRs with higher affinity for self-ligands in the thymus. Because TCR affinity plays an important role in T-cell activation, the TCR affinity gap between anti-pathogen and anti-cancer T-cells leaves the latter at a distinct disadvantage and makes it more difficult to break self-tolerance to such antigens. One approach to enhance the T-cell response to tumor antigen-derived peptides has been to immunize patients with altered peptide ligands that differ from the native sequence by a single or multiple amino acid residues. However, such “heteroclitic” peptides with even single amino acid substitutions that are predicted to only contact the HLA can have unpredictable, yet important, effects on TCR engagement. To date, only a few x-ray structures of TCRs bound to cognate tumor antigens have been determined (18, 36–38). Given the growing evidence that plasticity at the TCR·pHLA interface can influence immune recognition (39), structural and biophysical studies should be taken into account when attempting to design altered peptide ligands with improved immunogenicity.
We solved the first structure of a naturally occurring αβTCR in complex with a gp100 HLA-A*0201-restricted melanoma epitope. Overall, the PMEL17 TCR bound with a typical diagonal orientation over the central peptide residues and mainly contacted residues 4, 7, and 8 of the YLE peptide, which protruded out of the HLA-A*0201 binding groove. It is important to underscore that the PMEL17 TCR was characterized by a binding affinity (KD) of 7.6 μm. This value falls at the very high end of the affinity range described so far for cancer TCRs (11, 35). These results suggest that T-cells bearing TCRs with reasonable affinity for some tumor-associated antigens may escape central tolerance, opening the door to further TCR engineering for medical applications (40).
We also provide insight into the role of each residue in gp100280–288 during TCR recognition by performing an alanine scan mutagenesis with two different gp100280–288-specific αβTCRs. With regard to HLA anchor-modified “heteroclitic” peptides, previous studies have shown that even highly immunogenic designer peptides (e.g. ELA epitope from Melan-A/MART-1 protein) do not necessarily induce a better clinical response (13, 41, 42). Fortunately, this is not the case for the gp100 YLE-9V peptide, which has been successfully adopted in clinical trials (15). These observations are consistent with our in vitro findings, in that the A2-YLE-9V bound with similar affinity to PMEL17 TCR and gp100 TCR compared with the native peptide. Interestingly, both the PMEL17 TCR (TRAV21 TRBV7-3) and gp100 TCR (TRAV17 TRBV19) were most sensitive to mutations at position 3 or 5 of the native gp100280–288 peptide sequence despite these TCRs being constructed from completely different Vα and Vβ genes. A previous study of gp100 altered peptide ligands also showed the YLE-3A mutant to be a null agonist when tested on gp100-specific TCR-transfected human T-cells (17). Our results provide a molecular explanation for this finding.
We show that PMEL17 TCR non-responsiveness to A2-YLE-3A was caused by an unexpected molecular switch in the peptide, repositioning the Pro4 residue, which was at the center of a sizeable network of interactions (both van der Waals contacts and hydrogen bonds) in the PMEL17·A2-YLE-9V structure. Position 3 in HLA-A*0201 restricted peptides is known to be a secondary anchor residue (43), in that it supports the exposed peptide bulge that is normally involved in TCR binding. Interestingly, mutation in position 3 in the YLE peptide did not alter the conformation of the peptide backbone itself but resulted in a “knock-on” effect on the neighboring residue Pro4 that completely abolished TCR binding and T-cell recognition. We have recently described a similar molecular switch in an HIV-1-derived peptide, with important implications for the immune control of HIV infection and patterns of viral escape mutants (44). Additionally, we have demonstrated the existence of a novel mode of flexible peptide presentation in a diabetes model, showing the dynamic nature of the region surrounding the HLA F-pocket (39, 45). Taken together, these studies highlight that the peptide-HLA interaction is more plastic and dynamic than previously appreciated, with obvious implications for immune recognition, epitope prediction, and structural modeling.
Overall, our results represent the first structural insight into TCR recognition of an important tumor antigen, targeted by many clinical therapies. These data reveal that two very different TCRs share a similar pattern of specificity, demonstrated by their nearly identical sensitivity to different peptide modifications. Finally, we show that even changes in a single peptide residue that are not heavily engaged by a TCR can have important, knock-on effects on other residues in an HLA-bound peptide that can dramatically alter T-cell recognition. Such “transmitted” structural changes need to be taken into consideration when designing improved peptides for cancer vaccination.
Author Contributions
V. B., G. D., A. B., G. D., A. F., A. T., P. J. R., and D. K. C. performed experiments and analyzed the data. V. B., A. K. S., and D. K. C. wrote the manuscript. A. K. S. and D. K. C. conceived and directed the study. A. K. S. and D. K. C. funded the study. All authors contributed to discussions.
Acknowledgment
We thank the staff at Diamond Light Source for providing facilities and support.
The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 5EU3, 5EU4, 5EU5, and 5EU6) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- pHLA
- peptide human leukocyte antigen
- TCR
- T-cell receptor
- gp100
- glycoprotein 100
- SPR
- surface plasmon resonance
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- ITC
- isothermal titration calorimetry.
References
- 1. Boon T., Coulie P. G., Van den Eynde B. J., and van der Bruggen P. (2006) Human T cell responses against melanoma. Annu. Rev. Immunol. 24, 175–208 [DOI] [PubMed] [Google Scholar]
- 2. Raposo G., and Marks M. (2007) Melanosomes: dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 8, 786–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wagner S. N., Wagner C., Schultewolter T., and Goos M. (1997) Analysis of Pmel17/gp100 expression in primary human tissue specimens: implications for melanoma immuno- and gene-therapy. Cancer Immunol. Immunother. 44, 239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawakami Y., Eliyahu S., Jennings C., Sakaguchi K., Kang X., Southwood S., Robbins P. F., Sette A., Appella E., and Rosenberg S. A. (1995) Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J. Immunol. 154, 3961–3968 [PubMed] [Google Scholar]
- 5. Kawakami Y., Eliyahu S., Sakaguchi K., Robbins P. F., Rivoltini L., Yannelli J. R., Appella E., and Rosenberg S. A. (1994) Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J. Exp. Med. 180, 347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salgaller M. L., Marincola F. M., Cormier J. N., and Rosenberg S. A. (1996) Immunization against epitopes in the human melanoma antigen gp100 following patient immunization with synthetic peptides. Cancer Res. 56, 4749–4757 [PubMed] [Google Scholar]
- 7. Rosenberg S. A., Yang J. C., Schwartzentruber D. J., Hwu P., Marincola F. M., Topalian S. L., Restifo N. P., Dudley M. E., Schwarz S. L., Spiess P. J., Wunderlich J. R., Parkhurst M. R., Kawakami Y., Seipp C. A., Einhorn J. H., and White D. E. (1998) Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 4, 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawakami Y., Eliyahu S., Delgado C. H., Robbins P. F., Sakaguchi K., Appella E., Yannelli J. R., Adema G. J., Miki T., and Rosenberg S. A. (1994) Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc. Natl. Acad. Sci. U.S.A. 91, 6458–6462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Skipper J. C., Gulden P. H., Hendrickson R. C., Harthun N., Caldwell J. A., Shabanowitz J., Engelhard V. H., Hunt D. F., and Slingluff C. L. (1999) Mass-spectrometric evaluation of HLA-A*0201-associated peptides identifies dominant naturally processed forms of CTL epitopes from MART-1 and gp100. Int. J. Cancer 82, 669–677 [DOI] [PubMed] [Google Scholar]
- 10. Parkhurst M. R., Salgaller M. L., Southwood S., Robbins P. F., Sette A., Rosenberg S. A., and Kawakami Y. (1996) Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J. Immunol. 157, 2539–2548 [PubMed] [Google Scholar]
- 11. Aleksic M., Liddy N., Molloy P. E., Pumphrey N., Vuidepot A., Chang K.-M., and Jakobsen B. K. (2012) Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur. J. Immunol. 42, 3174–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cole D. K., Pumphrey N. J., Boulter J. M., Sami M., Bell J. I., Gostick E., Price D. A., Gao G. F., Sewell A. K., and Jakobsen B. K. (2007) Human TCR-binding affinity is governed by MHC class restriction. J. Immunol. 178, 5727–5734 [DOI] [PubMed] [Google Scholar]
- 13. Cole D. K., Edwards E. S. J., Wynn K. K., Clement M., Miles J. J., Ladell K., Ekeruche J., Gostick E., Adams K. J., Skowera A., Peakman M., Wooldridge L., Price D. A., and Sewell A. K. (2010) Modification of MHC anchor residues generates heteroclitic peptides that alter TCR binding and T cell recognition. J. Immunol. 185, 2600–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miles K. M., Miles J. J., Madura F., Sewell A. K., and Cole D. K. (2011) Real time detection of peptide-MHC dissociation reveals that improvement of primary MHC-binding residues can have a minimal, or no, effect on stability. Mol. Immunol. 48, 728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lesterhuis W. J., Schreibelt G., Scharenborg N. M., Brouwer H. M., Gerritsen M.-J., Croockewit S., Croockewit ra, Coulie P. G., Torensma R., Adema G. J., Figdor C. G., de Vries I. J., and Punt C. J. (2011) Wild-type and modified gp100 peptide-pulsed dendritic cell vaccination of advanced melanoma patients can lead to long-term clinical responses independent of the peptide used. Cancer Immunol. Immunother. 60, 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaft N., Willemsen R. A., de Vries J., Lankiewicz B., Essers B. W., Gratama J.-W., Figdor C. G., Bolhuis R. L., Debets R., and Adema G. J. (2003) Peptide fine specificity of anti-glycoprotein 100 CTL is preserved following transfer of engineered TCR α β genes into primary human T lymphocytes. J. Immunol. 170, 2186–2194 [DOI] [PubMed] [Google Scholar]
- 17. Schaft N., Coccoris M., Drexhage J., Knoop C., de Vries I. J., Adema G. J., and Debets R. (2013) An altered gp100 peptide ligand with decreased binding by TCR and CD8α dissects T cell cytotoxicity from production of cytokines and activation of NFAT. Front. Immunol. 4, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Madura F., Rizkallah P. J., Holland C. J., Fuller A., Bulek A., Godkin A. J., Schauenburg A. J., Cole D. K., and Sewell A. K. (2015) Structural basis for ineffective T-cell responses to MHC anchor residue-improved “heteroclitic” peptides. Eur. J. Immunol. 45, 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boulter J. M., Glick M., Todorov P. T., Baston E., Sami M., Rizkallah P., and Jakobsen B. K. (2003) Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 16, 707–711 [DOI] [PubMed] [Google Scholar]
- 20. Garboczi D. N., Utz U., Ghosh P., Seth A., Kim J., VanTienhoven E. A., Biddison W. E., and Wiley D. C. (1996) Assembly, specific binding, and crystallization of a human TCR-αβ with an antigenic Tax peptide from human T lymphotropic virus type 1 and the class I MHC molecule HLA-A2. J. Immunol. 157, 5403–5410 [PubMed] [Google Scholar]
- 21. Wyer J. R., Willcox B. E., Gao G. F., Gerth U. C., Davis S. J., Bell J. I., van der Merwe P. A., and Jakobsen B. K. (1999) T cell receptor and coreceptor CD8 αα bind peptide-MHC independently and with distinct kinetics. Immunity 10, 219–225 [DOI] [PubMed] [Google Scholar]
- 22. Bulek A. M., Madura F., Fuller A., Holland C. J., Schauenburg A. J. A., Sewell A. K., Rizkallah P. J., and Cole D. K. (2012) TCR/pMHC optimized protein crystallization Screen. J. Immunol. Methods 382, 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 [Google Scholar]
- 24. Collaborative Computational Project, Number 4 (1994) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 25. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 28. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 29. Armstrong K. M., and Baker B. M. (2007) A comprehensive calorimetric investigation of an entropically driven T cell receptor-peptide/major histocompatibility complex interaction. Biophys. J. 93, 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parker K. C., Bednarek M. A., Hull L. K., Utz U., Cunningham B., Zweerink H. J., Biddison W. E., and Coligan J. E. (1992) Sequence motifs important for peptide binding to the human MHC class I molecule, HLA-A2. J. Immunol. 149, 3580–3587 [PubMed] [Google Scholar]
- 31. Tickle I. J., Laskowski R. A., and Moss D. S. (2000) Rfree and the rfree ratio. II. Calculation Of the expected values and variances of cross-validation statistics in macromolecular least-squares refinement. Acta Crystallogr. D Biol. Crystallogr. 56, 442–450 [DOI] [PubMed] [Google Scholar]
- 32. Rudolph M. G., Stanfield R. L., and Wilson I. A. (2006) How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466 [DOI] [PubMed] [Google Scholar]
- 33. Tynan F. E., Burrows S. R., Buckle A. M., Clements C. S., Borg N. A., Miles J. J., Beddoe T., Whisstock J. C., Wilce M. C., Silins S. L., Burrows J. M., Kjer-Nielsen L., Kostenko L., Purcell A. W., McCluskey J., and Rossjohn J. (2005) T cell receptor recognition of a “super-bulged” major histocompatibility complex class I-bound peptide. Nat. Immunol. 6, 1114–1122 [DOI] [PubMed] [Google Scholar]
- 34. Armstrong K. M., Insaidoo F. K., and Baker B. M. (2008) Thermodynamics of T-cell receptor-peptide/MHC interactions: progress and opportunities. J. Mol. Recognit. 21, 275–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bridgeman J. S., Sewell A. K., Miles J. J., Price D. A., and Cole D. K. (2012) Structural and biophysical determinants of αβ T-cell antigen recognition. Immunology 135, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Borbulevych O. Y., Santhanagopolan S. M., Hossain M., and Baker B. M. (2011) TCRs used in cancer gene therapy cross-react with MART-1/Melan-A tumor antigens via distinct mechanisms. J. Immunol. 187, 2453–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deng L., Langley R. J., Brown P. H., Xu G., Teng L., Wang Q., Gonzales M. I., Callender G. G., Nishimura M. I., Topalian S. L., and Mariuzza R. A. (2007) Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat. Immunol. 8, 398–408 [DOI] [PubMed] [Google Scholar]
- 38. Chen J.-L., Stewart-Jones G., Bossi G., Lissin N. M., Wooldridge L., Choi E. M. L., Held G., Dunbar P. R., Esnouf R. M., Sami M., Boulter J. M., Rizkallah P., Renner C., Sewell A., van der Merwe P. A., et al. (2005) Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med. 201, 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borbulevych O. Y., Piepenbrink K. H., Gloor B. E., Scott D. R., Sommese R. F., Cole D. K., Sewell A. K., and Baker B. M. (2009) T cell receptor cross-reactivity directed by antigen-dependent tuning of peptide-MHC molecular flexibility. Immunity 31, 885–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liddy N., Bossi G., Adams K. J., Lissina A., Mahon T. M., Hassan N. J., Gavarret J., Bianchi F. C., Pumphrey N. J., Ladell K., Gostick E., Sewell A. K., Lissin N. M., Harwood N. E., Molloy P. E., et al. (2012) Monoclonal TCR-redirected tumor cell killing. Nat. Med. 18, 980–987 [DOI] [PubMed] [Google Scholar]
- 41. Speiser D. E., Baumgaertner P., Voelter V., Devevre E., Barbey C., Rufer N., and Romero P. (2008) Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc. Natl. Acad. Sci. U.S.A. 105, 3849–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Romero P., Valmori D., Pittet M. J., Zippelius A., Rimoldi D., Lévy F., Dutoit V., Ayyoub M., Rubio-Godoy V., Michielin O., Guillaume P., Batard P., Luescher I. F., Lejeune F., Liénard D., et al. (2002) Antigenicity and immunogenicity of Melan-A/MART-1 derived peptides as targets for tumor reactive CTL in human melanoma. Immunol. Rev. 188, 81–96 [DOI] [PubMed] [Google Scholar]
- 43. Ruppert J., Sidney J., Celis E., Kubo R. T., Grey H. M., and Sette A. (1993) Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell 74, 929–937 [DOI] [PubMed] [Google Scholar]
- 44. Kløverpris H. N., Cole D. K., Fuller A., Carlson J., Beck K., Schauenburg A. J., Rizkallah P. J., Buus S., Sewell A. K., and Goulder P. (2015) A molecular switch in immunodominant HIV-1-specific CD8 T-cell epitopes shapes differential HLA-restricted escape. Retrovirology 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Motozono C., Pearson J. A., De Leenheer E., Rizkallah P. J., Beck K., Trimby A., Sewell A. K., Wong F. S., and Cole D. K. (2015) Distortion of the MHC class I binding groove to accommodate an insulin-derived 10-mer peptide. J. Biol. Chem. 290, 18924–18933 [DOI] [PMC free article] [PubMed] [Google Scholar]