

Abstract
The β-arrestins (βarrs) are versatile, multifunctional adapter proteins that are best known for their ability to desensitize G protein-coupled receptors (GPCRs), but also regulate a diverse array of cellular functions. To signal in such a complex fashion, βarrs adopt multiple conformations and are regulated at multiple levels to differentially activate downstream pathways. Recent structural studies have demonstrated that βarrs have a conserved structure and activation mechanism, with plasticity of their structural fold, allowing them to adopt a wide array of conformations. Novel roles for βarrs continue to be identified, demonstrating the importance of these dynamic regulators of cellular signaling.
Keywords: 7-helix receptor, arrestin, G protein-coupled receptor (GPCR), receptor desensitization, receptor endocytosis, signaling
Introduction
β-Arrestins (βarrs)2 are ubiquitously expressed proteins that were first described for their role in desensitizing G protein-coupled receptors (GPCRs) (1). We now appreciate that these proteins are multifunctional adapter proteins that regulate a vast array of cellular functions. βarrs were identified through their sequence homology to visual arrestin (arrestin-1), so named because of its ability to “arrest” rhodopsin signaling in the retina (2). There are two βarr isoforms, β-arrestin1 and β-arrestin2 (also denoted as arrestin-2 and arrestin-3, respectively). Both are expressed ubiquitously and share 78% sequence homology (3). βarrs are highly conserved across species, with ∼50% sequence homology between vertebrates and invertebrates. The other arrestins are expressed in the eye: arrestin-1 (visual arrestin) and arrestin-4 (cone arrestin) (4). There are other proteins, termed α-arrestins or arrestin domain-containing proteins, that share the arrestin structural fold and are involved in receptor endocytosis, although the full breadth of their functions is still emerging (5). Similar to arrestin's function in the visual system, βarrs were first identified for their capacity to desensitize β2 adrenergic receptor (β2AR) G protein signaling following agonist stimulation (1). Through a number of investigations, it became apparent that the two βarr isoforms shared the capability to interact with activated GPCRs, but that they differed in terms of their expression patterns, their specificity for different GPCRs, and their functional effects (6). We now appreciate that the βarrs regulate a diverse array of cellular processes including MAPK signaling, receptor transactivation, receptor trafficking, and transcriptional regulation in addition to the canonical roles of GPCR desensitization and internalization (7, 8). These studies have revealed the current spectrum of βarr-mediated cell processes downstream of GPCRs (Fig. 1).
FIGURE 1.
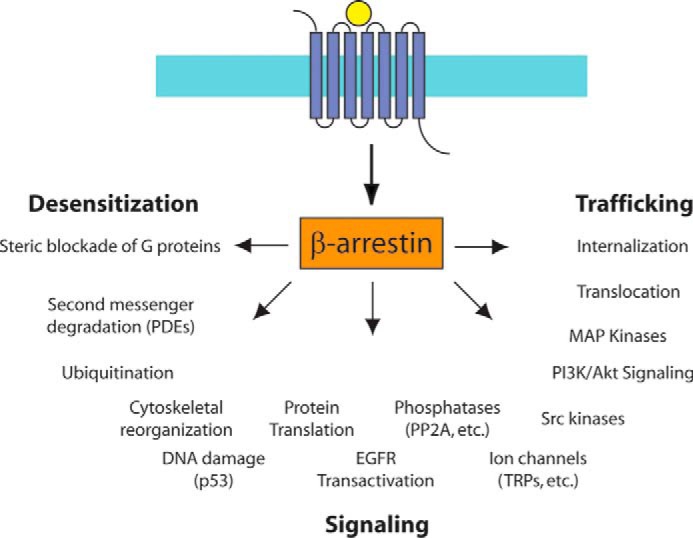
The spectrum of βarr-mediated signaling. βarrs regulate a wide array of pathways downstream of GPCRs (see text). PDEs, phosphodiesterases; EGFR, EGF receptor; PP2A, protein phosphatase 2A; TRP, transient receptor potential.
Distinct and Overlapping Roles for the βarrs
βarr1 and βarr2 knockouts are phenotypically normal and produce viable progeny, but these mice display abnormal responses to physiologic stresses (9, 10). This suggests a compensatory ability for each isoform. Nevertheless, important differences between βarr isoforms are present (11). Although both accumulate in the cytoplasm following overexpression, βarr1, but not βarr2, accumulates in the nucleus. Although both βarr1 (418 amino acids) and βarr2 (410 amino acids) have nuclear localization sequences on their N termini, only βarr2 has a nuclear export sequence located on its C terminus, thereby accounting for differences in nucleocytoplasmic shuttling (12). βarr1 and βarr2 scaffold to different signaling pathways; however, this is often cell type- and receptor-specific. βarr2, but not βarr1, is known to be necessary for creating a signaling that activates JNKs (13). βarr1 and βarr2 can “reciprocally regulate” signaling at certain receptors; that is, one isoform increases pathway-specific signaling, whereas the other isoform inhibits signaling. Reciprocal regulation is observed in the type 1 angiotensin II receptor (AT1R), where siRNA knockdown of βarr2 attenuates ERK signaling, whereas knockdown of βarr1 potentiates ERK signaling (14). However, at other receptors such as the β2AR and the type 1 parathyroid hormone receptor (PTH1R), knockdown of either βarr1 or βarr2 decreases ERK signaling (15, 16). In addition, growing evidence suggests that the kinetics of βarr-mediated signaling is tissue-dependent (17). Adding to the complexity, the functions of βarrs appear to be strongly influenced by their cellular environment, such as the presence or absence of critical signaling partners such as G protein receptor kinases (GRKs) (18).
βarr Post-translational Modifications
Post-translational modifications are critical to βarr signaling and trafficking. βarr1 and βarr2 are constitutively phosphorylated, and both require C-terminal dephosphorylation for targeting internalized receptors to clathrin. The phosphorylation site differs between βarr isoforms (Ser412 for βarr1, Ser361 and Thr383 for βarr2) (19). However, dephosphorylation of βarrs is not required for desensitization of G protein signaling. Differential trafficking of βarr isoforms often controls the kinetics of desensitization, as well as if or when a receptor is recycled back to the membrane surface. Dephosphorylation of βarrs following receptor activation is necessary for full functionality, including receptor internalization and βarr-mediated MAPK signaling. Covalent modification of βarr with ubiquitin (ubiquitination) results in sustained βarr·GPCR complexes and prolonged MAPK activity. Ubiquitination of a GPCR is necessary for receptor degradation, and ubiquitination of βarrs is necessary for GPCR internalization (20). Different patterns of βarr ubiquitination (especially at Lys11 and Lys12) result in changes in receptor trafficking (see below) and the ability to scaffold signalosomes (21). Other reported modifications that regulate βarr function are S-nitrosylation (22) and SUMOylation (23), and it is likely that βarrs are modified in other, yet unexplored, ways that impact their functions.
Desensitization
Receptor desensitization is the process by which repeated stimulation of a GPCR results in a decreased response over seconds to minutes. This is in contrast to down-regulation, the process underlying decreased signaling that occurs over hours. Receptor-dependent activation of heterotrimeric G proteins induces dissociation of Gα and Gβγ subunits, promoting their interactions with effector proteins that lead to downstream signaling. Desensitization of GPCR signaling requires a coordinated response by GRKs and βarrs (24). The first functional effect noted in the arrestin family was the desensitization of G protein-mediated signaling by rhodopsin (2). G protein signaling inhibition was soon recognized as a function of βarrs in tissues outside of the visual system, and inhibition of G protein-mediated signaling was the primary function assigned to βarrs until the mid-1990s. βarrs are thought to quench G protein signaling by sterically inhibiting the G protein interaction at the second (ICL2) and third (ICL3) intracellular loops of a GPCR (6, 25). This steric hindrance uncouples GPCRs from the G protein signal transduction process, which results in desensitization of second messenger pathways (26).
Phosphorylation of the cytoplasmic elements of GPCRs is critical for βarr recruitment and receptor desensitization (2, 24, 27, 28). GPCR phosphorylation can be targeted directly to intracellular regions of the ligand-bound receptor complex (homologous desensitization) or to multiple GPCRs throughout the cell (heterologous desensitization). Heterologous desensitization is often mediated by PKA or PKC (29). In homologous desensitization, phosphorylation of the GPCR intracellular residues is predominately mediated by GRKs (28). There are seven GRK isoforms: GRK1 and GRK7 are confined to the visual system, GRK2, GRK3, GRK5, and GRK6 are ubiquitously expressed, and GRK4 is expressed primarily in the reproductive tract (30). Importantly, phosphorylation of GPCRs appears to be absolutely required for desensitization. Elimination of intracellular phosphorylation, either by using phosphorylation-deficient receptor mutants or by co-transfecting a dominant-negative GRK, abolishes βarr recruitment, desensitization,and internalization (27, 31). This process occurs sequentially, as βarr binding requires both ligand-induced conformational change in the GPCR and GPCR phosphorylation (32). Because GRK-mediated phosphorylation of receptors is often the rate-limiting aspect of receptor desensitization, it can dominate the kinetics of βarr binding to receptors in intact cells. Heterogeneity in the phosphorylation sites is a second source of complexity, because GRK-mediated phosphorylation occurs not only at the C-terminal tail of the receptor (e.g. rhodopsin and the β2AR) but also at many other intracellular sites, most notably ICL3 (e.g. α2 adrenergic receptor (33) and M2 muscarinic receptor (34)).
Trafficking
For a number of GPCRs, βarrs function as adapters to target receptors to clathrin-coated pits through its scaffolding of AP-2 and clathrin (35). Many, but not all (36–38), GPCRs appear to require βarrs for internalization. The recruitment of βarr2 for its receptors can be modified by mutation of selected “receptor discriminator” residues (39). Receptors that follow the clathrin-dependent endocytic pathway are internalized in clathrin-coated pits in a dynamin-dependent fashion (40). βarrs scaffold multiple protein regulators including ARF6 (41) and n-ethylmaleimide-sensitive fusion protein (42), which are implicated in βarr-mediated receptor internalization. Once internalized, the receptor continues to tubulovesicular early endosomes. Here, receptors are sorted to either recycling endosomes, which return GPCRs to the plasma membrane, or multivesicular late endosomes, which traffic receptors to lysosomes for degradation (7). Some GPCRs internalize in the absence of βarrs, but require them for recycling (43).
GPCRs that traffic through the clathrin-dependent endocytic pathway can be divided into two groups, class A and B, based on the characteristics of agonist-dependent βarr binding (44). βarrs facilitate the desensitization and internalization of both receptor classes. Class A receptors, such as the β2 adrenergic receptor, bind βarr2 with greater affinity than βarr1. Class B receptors, such as the V2 vasopressin receptor, bind βarr2 and βarr1 with approximately equal affinities. In class A interactions, receptors internalized in membrane vesicles remain at the cellular surface, and βarrs dissociate from the receptor at or near the plasma membrane. In class B interactions, βarrs form a long-lived complex with the receptor and traffic into endosomes. Class A receptors are associated with transient βarr ubiquitination, and class B receptors are associated with stable βarr ubiquitination. Notably, class A patterns can be switched to class B by covalently linking ubiquitin to βarr or by switching the C terminus of the receptor to that of a class B receptor (20). Differential phosphorylation by GRKs and other kinases also regulates the receptor-βarr interaction (45). These changes in receptor and βarr post-translational modifications appear to be ligand-dependent, as different ligands binding to the same receptor can result in class A or B patterns. In addition to ligand-stimulated receptors, constitutively active receptors that internalize in the absence of ligand appear to rely on βarrs for trafficking (46). βarrs can also traffic receptors to distinct areas of the cell, such as βarr translocation of Smoothened (Smo) during Hedgehog pathway activation. Once formed, this βarr·Smo complex localizes to the primary cilia, where the complex activates Gli transcription factors (47).
Signaling
It is now appreciated that in addition to regulating receptor-stimulated G protein signaling, βarrs are also capable of initiating distinct signaling patterns (48). These signaling patterns are often both spatially and temporally distinct from G protein-mediated signaling, and result in unique cellular, physiological, and pathophysiological consequences. In addition to differential trafficking, βarrs also scaffold MAPKs, including ERK1/2. Both G proteins and βarrs mediate ERK1/2 activation, but through distinct mechanisms. Recruitment of βarrs sterically inhibits G protein interaction with the active receptor, thus quenching the rapid G protein-mediated phase of ERK activation. Sometimes G protein-mediated ERK activation can also include a slow phase (49), so kinetics alone cannot distinguish between G protein- and βarr-mediated ERK signaling. Separately, βarr scaffolds Raf-1, MEK1, and ERK, thus serving to sequester ERK in the cytosol (50). Seclusion of phosphorylated ERK1/2 in the cytosol precludes ERK-mediated transcription and prolongs ERK signaling. Similarly, βarr2 scaffolds JNK1/2 with its upstream kinases MKK4 and MKK7, which phosphorylate different residues in its activation loop (13). Activation of p38 signaling cascades is also βarr-dependent, although a direct scaffolding complex of βarr and p38 has not been elucidated (51, 52). Modified βarrs have also been reported to signal to kinases independently of GPCRs (53).
Ubiquitination is now appreciated to regulate not only protein degradation, but also protein signaling. In addition to being ubiquitinated themselves, βarrs act as adapters for multiple E3 ubiquitin ligases. Complexes containing βarr and E3 ligases are essential for mediating aspects of ubiquitin-dependent signaling. For example, βarrs are critically involved in ubiquitination of receptors, acting on late endosomal populations as a lysosomal degradation signal for the receptor. More broadly, βarrs act as adapters for several E3 ligases that catalyze ubiquitination, such as Mdm2. Mdm2 ubiquitination of βarr2 is required for clathrin-mediated internalization of the β2AR (54), whereas the E3 ligase AIP4 is necessary for sorting of CXCR4 to early endosomes and then lysosomes (55). Endosomal sorting of CXCR4 also requires βarr1 interaction with STAM-1, part of the endosomal sorting complex required for transport (ESCRT-0) machinery (56). βarrs are also regulated by deubiquitinating enzymes such as the ubiquitin-specific protease USP33 (57, 58), thus providing a mechanism for coordinating receptor recycling and resensitization. Interestingly, evidence suggests that receptor post-translational modification can influence later signaling events, either by catalyzing additional post-translational modifications or by controlling downstream signaling pathways (55, 59).
A number of other signaling pathways have been demonstrated to be regulated by βarrs. The transactivation of EGF receptor by GPCRs can be regulated by βarrs, through the activation of a transmembrane matrix metalloprotease that cleaves membrane-bound EGF ligand (60). βarr2 can inhibit NF-κB signaling through stabilization of IκBa (61). βarr1 can directly influence epigenetic modifications through nuclear interaction with histone acetylases and deacetylases that influence chromatin structure (62). There are now even examples of βarr-mediated G protein signaling. βarrs promote G protein signaling by the type 1 parathyroid hormone (63) and V2 vasopressin receptor (64) from endosomes, an effect that is lost with βarr knockdown. The β2AR has also been noted to maintain an active conformation that can signal through G proteins to generate cAMP from endosomes (65). These findings suggest that βarr trafficking of receptors to endosomes results in a receptor that is still capable of activating G proteins. This signaling appears to be mediated by a complex of receptor·βarr·G protein (63), direct evidence of which would fully overturn the classic paradigm of βarrs as “arresting” G protein signaling.
βarr-biased Agonism
Following the discovery of βarr-mediated signaling came the observation that some ligands are capable of selectively signaling through βarrs while blocking signaling through G proteins. This is an example of biased agonism, also referred to as functional selectivity, which is the ability of certain agonists to signal through different pathways of a GPCR with different efficacies (66). Strongly biased agonists activate one pathway while completely blocking signaling through others, whereas partially biased agonists may strongly signal through one pathway while weakly signaling through another. Biased agonism between different G proteins has been appreciated for 30 years (67), and the discovery of βarr-biased agonism resulted in renewed interest in this area (66). Biased agonism changes the classical models of receptor theory associated with single active and inactive receptor conformations to one with multiple receptor conformations. Although balanced ligands stabilize the conformations that are competent for signaling to all downstream pathways, biased ligands stabilize only those conformations that are capable of promoting a subset of downstream signaling effectors. For example, ligands can show bias for either G protein-mediated (G protein-biased) or βarr-mediated (βarr-biased) signaling (Fig. 2). This is necessarily an oversimplification, as recruitment of βarrs requires the activity of GRKs, and hence G protein- and βarr-biased ligands will also be biased with respect to GRK recruitment and receptor phosphorylation.
FIGURE 2.
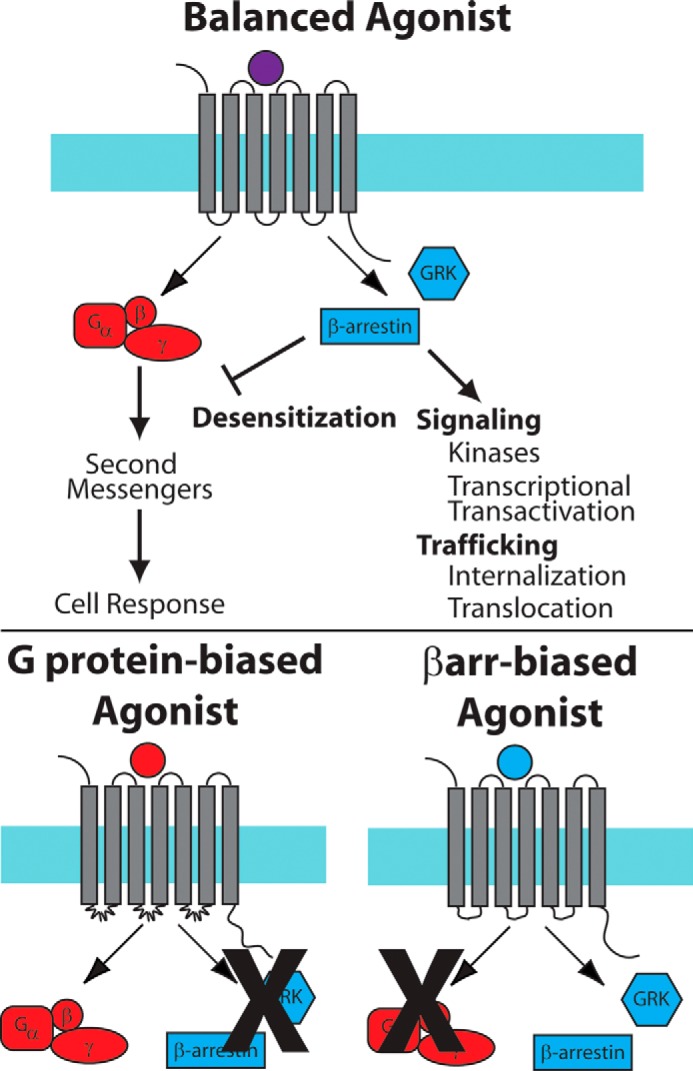
Balanced and biased signaling by GPCRs. Top panel, in balanced signaling, both G protein-mediated and βarr-mediated signaling pathways are activated by the ligand·receptor complex. Bottom panel, in G protein- or βarr-biased signaling, one of the pathways is activated while the other pathway is blocked.
Bias adds a layer of complexity to the traditional definition of ligand action. For example, a βarr-biased AT1R agonist has markedly different physiologic effects from the endogenous agonist angiotensin II (68). Although angiotensin II causes vasoconstriction, cardiac hypertrophy, and increased cardiac contractility, the βarr-biased agonist causes vasodilation and does not cause cardiac hypertrophy, but still increases cardiac contractility via βarr-mediated phosphorylation of tropomyosin and other contractile proteins (69). A number of other G protein- and βarr-biased agonists targeting a variety of receptors are currently being tested in early phase clinical trials, including those of the μ-opioid receptor (70) and apelin receptor (71).
Although many biased agonists have been identified serendipitously, drug development of biased agonists requires an approach for quantifying the degree of ligand bias. Classical parameters of receptor signaling such as maximal effects (Emax) and potencies (EC50) cannot account for differences in receptor reserve and amplification of different signaling pathways (72). In assays with significant amplification, such as second messenger assays, e.g. cyclic AMP formation, both full and partial agonists can reach the same maximal response, whereas in assays with little amplification, such as assays that monitor recruitment of βarr to a receptor, partial agonists have significantly lower maximal responses than full agonists. Multiple approaches have been developed that all address the issue of differential amplification between signaling assays (72–74). As an example, bias factors (72) yield an estimate of bias equivalent to other approaches, and when combined with dissociation constants obtained from a binding experiment, also provide an estimate of relative efficacy. All of these approaches yield similar estimates for bias (75), although relative errors can be significantly higher depending on the assumptions made in the analysis (76).
The Signaling Barcode: A Model for Allosteric Regulation of βarrs
Numerous studies have suggested that βarrs can adopt multiple conformations that differentially regulate distinct cellular signaling events. Regulation of these unique βarr conformations is controlled at a number of levels, through interactions with the ligand·receptor complex, different post-translational modifications of both the receptor and βarrs, and the presence of other cofactors that are cell type-dependent. These different mechanisms for βarr regulation have been integrated in the “signaling barcode” model for receptor·βarr signaling (17, 77) (Fig. 3). Binding of βarr to distinct receptor C-terminal phosphorylation patterns (“barcodes”) generated by different kinases results in different conformations of receptor-bound βarrs. These different βarr conformations are capable of activating distinct downstream signaling events, such as endocytosis, desensitization, or kinase activation. Although it is an attractive hypothesis, there are still only limited data to support it. At the M3 muscarinic receptor, differential phosphorylation of the receptor C terminus was noted in response to different ligands and in different tissues (presumably due to differential expression of GRKs and other kinases) (78). At CXCR4, unique serines are phosphorylated by PKA, GRK2, and GRK6, with different effects on ERK1/2 phosphorylation and calcium influx (79). At the β2AR, a βarr-biased ligand resulted in phosphorylation of distinct sites by GRK2 and GRK6 when compared with a balanced agonist, with different effects on receptor endocytosis and signaling through MAPKs (80). Important questions that need to be addressed in further developing the barcode model are how differential recruitment of kinases to the receptor influences receptor phosphorylation, how the receptor allosterically induces conformational changes in the structures of βarrs, and the means by which specific post-translational modifications of βarrs directly influence βarr conformation and subsequent downstream signaling.
FIGURE 3.

Regulation of βarrs by GPCR signaling barcodes. A–C, in the signaling barcode model, a receptor activated by ligand (A) recruits kinases and other enzymes that generate a signaling barcode (B) on the C-terminal tail of the receptor. This results in the recruitment of βarr and activation of effector molecules (C). D, changes to the barcode result in differential effector coupling by βarrs (shown are the clathrin adapter AP-2 and ERK MAPK). 7TMR, seven-transmembrane class of receptors; Ub, ubiquitin.
A Highly Conserved Structure and Activation Mechanism
The arrestins display high structural conservation as they share features critical for their biological activity, although with some notable differences. Arrestin-1 has N- and C- terminal β-sheet domains with a series of buried polar residues (“polar core”) in the N-domain stabilized by an extended C-terminal tail that locks the molecule into an inactive state (81) (Fig. 4A). βarr1 has an additional cationic amphipathic helix that could serve as a reversible membrane anchor (82). The receptor-binding surface of βarr2 does not form a contiguous β-sheet, consistent with increased flexibility and possibly responsible for its reduced selectivity for activated receptors (83). Arrestin-4 has differences in the concave surfaces of the β-sheets involved in receptor binding and the loop between β-strands 1 and 2 (84). Notably, arrestin-1 was crystallized as a tetramer (a dimer of dimers) and was noted to form dimers and tetramers in solution (although different from those observed in the crystal), although only monomeric arrestin-1 can bind to activated rhodopsin (85). βarr1 and βarr2 self-associate and form heterodimers, which is enhanced by binding to inositol hexakisphosphate (86). The significance of βarr multimerization is unclear, but it may regulate the subcellular distribution of βarrs (86).
FIGURE 4.
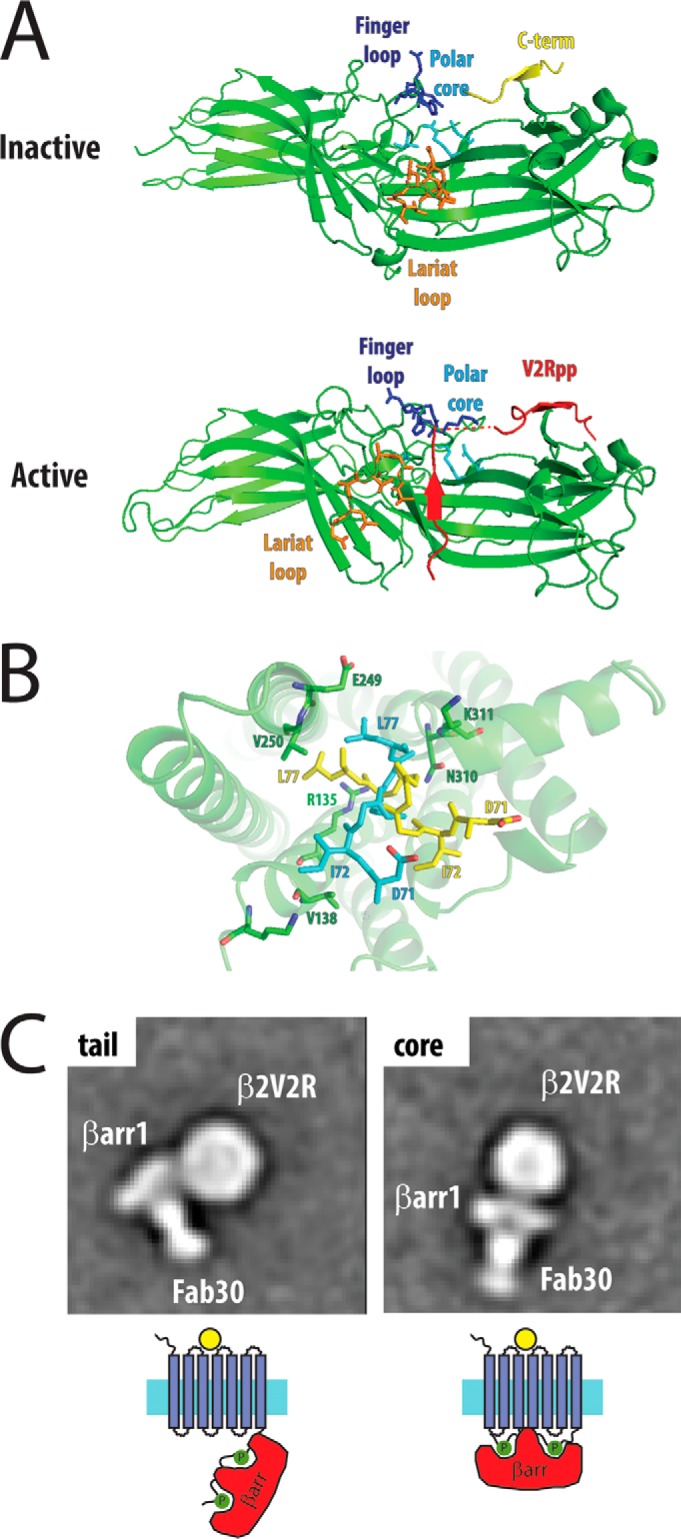
Structural mechanisms for βarr activation and signaling. A, βarractivation occurs through disruption of the polar core (“phosphate sensor”) by the phosphorylated C terminus of the receptor, thereby allowing specific motifs in βarr (“activation sensor,” including the finger and lariat loops) to bind to the ligand-activated receptor (inactive structure, Protein Data Bank (PDB) 1G4M; active structure, PDB 4JQI). B, alternative models for the finger loop interaction from the rhodopsin·finger loop peptide structure (yellow, PDB 4PXF) and the rhodopsin·arrestin-1 structure (cyan, PDB 4ZWJ) with the active receptor (green). C, single particle electron microscopy identifies distinct conformations of β2AR·βarr, with a tail conformation with interactions between the C-terminal tail of the receptor with βarr (phosphate sensor only) and a core conformation with interactions between the transmembrane domains and βarrs (activation sensor and phosphate sensor). EM images courtesy of Thomas Cahill.
A number of studies led to a model for arrestin binding to the receptor via two sensors: a “phosphate sensor” that interacts with the phosphorylated receptor C terminus and an “activation sensor” that interacts with the active conformation of the GPCR induced by agonist (87) (Fig. 4A). This model was largely confirmed by the structure of βarr1 bound to a C-terminal phosphopeptide from the vasopressin 2 receptor (V2R), stabilized by a synthetic antibody fragment (88). The polar core acts as the phosphate sensor: the phosphorylated receptor C terminus displaces the arrestin C terminus and interacts with a number of positively charged residues in the polar core (Fig. 4A). The disruption of the polar core is associated with a significant twist of the N- and C-terminal domains relative to one another. This results in exposure of regions of the protein that act as an activation sensor, most notably the interdomain hinge and the finger, middle, and lariat loops, structural changes that have been observed in earlier biophysical studies (89–93).
Structure of Receptor·Arrestin Complexes
The recent crystal structure of rhodopsin bound to arrestin-1 by serial femtosecond x-ray laser crystallography largely validates this mechanism for arrestin activation (94) (Fig. 4B). In this structure, there are three arrestin-rhodopsin interfaces: the finger loop of arrestin-1, which interacts with TM7 and TM8 of rhodopsin, the interdomain hinge, which forms a cleft that accommodates ICL2 of rhodopsin, and the β-strand, which follows the finger loop and interacts with TM5, TM6, and ICL3. A notable difference is in the conformation of the finger loop when compared with a rhodopsin·arrestin-1 finger loop peptide complex: in the rhodopsin·arrestin-1 structure, a helical structure for the finger loop was refined (Fig. 4B, yellow sticks), whereas in the rhodopsin·peptide complex, a reverse turn structure was observed (Fig. 4B, cyan sticks) (95). The rhodopsin·arrestin-1 structure has the advantage of having the entire arrestin-1 molecule in the structure, and a previous NMR structure has demonstrated a helical conformation of the finger loop (96). However, the rhodopsin·peptide structure was of significantly higher resolution with better electron density in the finger loop region when compared with the rhodopsin·arrestin-1 structure. Therefore, the precise conformation of the finger loop bound to the receptor is currently ambiguous, although both structures demonstrate that arrestin binding results in interactions with highly conserved motifs in the receptor, including the Arg135 of the E(D)RY motif in TM3 and Lys311 of the NPXXY motif at the end of TM6 (the motifs that form the ionic lock in the inactive receptor). This region is similar to the binding crevice that the Gα C terminus uses to bind to the receptor (97), demonstrating that GPCRs share a common binding interface for interacting with G proteins and βarrs.
Crystallography is limited to obtaining protein structures that are stable and sufficiently ordered to produce protein crystals. A complementary technique that has allowed the low-resolution visualization of large protein complexes is EM. Single-particle negative-stain EM has allowed the visualization of different modes of βarr1 binding to the β2AR (98). For these studies, the β2AR·βarr1 complex was stabilized with a synthetic antibody fragment that binds the active βarr conformation. By combining hydrogen/deuterium exchange MS, biochemical experiments, and single particle EM analysis, two distinct conformations of the receptor·βarr complex were identified (Fig. 4C). In the first conformation, βarr binds to the phosphorylated receptor C terminus only; in the second conformation, βarr1 is tightly bound to the receptor through transmembrane core interactions (via the activation sensor) and through the C terminus (via the phosphate sensor). These conformations may represent steps in a multi-step binding process of βarrs to GPCRs or may represent distinct states that are associated with differential signaling.
Signal Transduction to Effectors
Recent structural studies have also addressed the question of how βarrs transmit signals encoded in the receptor to effector molecules. The βarrs can interact with downstream effectors in different modes. For example, βarr1 can bind between blades 1 and 2 of the clathrin β-propeller via an intrinsically disordered clathrin-binding box, but can also interact with a binding pocket formed by blades 4 and 5 of clathrin via an 8-amino acid splice loop found only in the long βarr1 isoform (99). Further insights into the allosteric regulation of βarr signaling have recently been provided by an NMR study that used 19F probes in βarr1 to probe changes in its structure induced by different phosphopeptides derived from the V2R C terminus (100). Although all the phosphopeptides interacted with the phosphate sensor to induce changes in the finger and middle loops, there were also distinct phospho-interaction patterns that were related to the spacing of the multiple βarr phospho-binding sites. These distinct patterns may serve as a structural model for the signaling barcode, by which changes in a GPCR phosphorylation pattern are translated to distinct conformations of βarr that can be “read” by downstream effectors.
Future Directions
Over the past two decades, our understanding of the biology of βarrs has expanded, to the point where we now appreciate that these ubiquitous molecules are involved in virtually every aspect of cell biology. This is a trait that they share with their signaling partners, G protein-coupled receptors, whose over 800 members in the human genome regulate nearly every aspect of physiology. The βarrs are versatile, regulating receptor desensitization, trafficking, and signaling through their ability to interact with a vast array of binding partners. There are still a number of unresolved questions that need to be addressed regarding βarr function. From a structural perspective, it will be important to determine the different biological roles of distinct GPCR·βarr conformations and how those are regulated via the barcode or other signaling mechanisms. It will also be important to obtain structures, via either crystallography or electron microscopy, of GPCRs with βarrs and effectors to fully appreciate how specific signaling modes are encoded. From a pharmacologic perspective, the development of more biased agonists, both as tool compounds to dissect receptor pharmacology and as potential therapeutic agents, will continue to lead to novel insights into how biological information is processed by the cell.
Acknowledgments
We thank Thomas Cahill, Sudha Shenoy, and Robert Lefkowitz for helpful comments and critical review.
This work was supported by National Institutes of Health Grants HL114643 (to S. R.) and 5T32GM007171 (to J. S. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- βarr
- β-arrestin
- GPCR
- G protein-coupled receptor
- β2AR
- β2 adrenergic receptor
- GRK
- G protein receptor kinase
- ICL
- intracellular loop
- TM
- transmembrane.
References
- 1. Lohse M. J., Benovic J. L., Codina J., Caron M. G., and Lefkowitz R. J. (1990) β-Arrestin: a protein that regulates β-adrenergic receptor function. Science 248, 1547–1550 [DOI] [PubMed] [Google Scholar]
- 2. Wilden U., Wüst E., Weyand I., and Kühn H. (1986) Rapid affinity purification of retinal arrestin (48 kDa protein) via its light-dependent binding to phosphorylated rhodopsin. FEBS Lett. 207, 292–295 [DOI] [PubMed] [Google Scholar]
- 3. Attramadal H., Arriza J. L., Aoki C., Dawson T. M., Codina J., Kwatra M. M., Snyder S. H., Caron M. G., and Lefkowitz R. J. (1992) β-Arrestin2, a novel member of the arrestin/β-arrestin gene family. J. Biol. Chem. 267, 17882–17890 [PubMed] [Google Scholar]
- 4. Craft C. M., Whitmore D. H., and Wiechmann A. F. (1994) Cone arrestin identified by targeting expression of a functional family. J. Biol. Chem. 269, 4613–4619 [PubMed] [Google Scholar]
- 5. Patwari P., and Lee R. T. (2012) An expanded family of arrestins regulate metabolism. Trends Endocrinol. Metab. 23, 216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeGraff J. L., Gurevich V. V., and Benovic J. L. (2002) The third intracellular loop of α2-adrenergic receptors determines subtype specificity of arrestin interaction. J. Biol. Chem. 277, 43247–43252 [DOI] [PubMed] [Google Scholar]
- 7. Kang D. S., Tian X., and Benovic J. L. (2014) Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 27, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeWire S. M., Ahn S., Lefkowitz R. J., and Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 9. Conner D. A., Mathier M. A., Mortensen R. M., Christe M., Vatner S. F., Seidman C. E., and Seidman J. G. (1997) β-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to β-adrenergic stimulation. Circ. Res. 81, 1021–1026 [DOI] [PubMed] [Google Scholar]
- 10. Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., and Lin F. T. (1999) Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 [DOI] [PubMed] [Google Scholar]
- 11. Kohout T. A., Lin F. S., Perry S. J., Conner D. A., and Lefkowitz R. J. (2001) β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. U.S.A. 98, 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scott M. G., Le Rouzic E., Périanin A., Pierotti V., Enslen H., Benichou S., Marullo S., and Benmerah A. (2002) Differential nucleocytoplasmic shuttling of β-arrestins: characterization of a leucine-rich nuclear export signal in β-arrestin2. J. Biol. Chem. 277, 37693–37701 [DOI] [PubMed] [Google Scholar]
- 13. Kook S., Zhan X., Kaoud T. S., Dalby K. N., Gurevich V. V., and Gurevich E. V. (2013) Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J. Biol. Chem. 288, 37332–37342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahn S., Wei H., Garrison T. R., and Lefkowitz R. J. (2004) Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by β-arrestins 1 and 2. J. Biol. Chem. 279, 7807–7811 [DOI] [PubMed] [Google Scholar]
- 15. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., and Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 16. Gesty-Palmer D., Chen M., Reiter E., Ahn S., Nelson C. D., Wang S., Eckhardt A. E., Cowan C. L., Spurney R. F., Luttrell L. M., and Lefkowitz R. J. (2006) Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 281, 10856–10864 [DOI] [PubMed] [Google Scholar]
- 17. Tobin A. B., Butcher A. J., and Kong K. C. (2008) Location, location, location … site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 29, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bychkov E., Zurkovsky L., Garret M. B., Ahmed M. R., and Gurevich E. V. (2012) Distinct cellular and subcellular distributions of G protein-coupled receptor kinase and arrestin isoforms in the striatum. PLoS ONE 7, e48912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin F. T., Krueger K. M., Kendall H. E., Daaka Y., Fredericks Z. L., Pitcher J. A., and Lefkowitz R. J. (1997) Clathrin-mediated endocytosis of the β-adrenergic receptor is regulated by phosphorylation/dephosphorylation of β-arrestin1. J. Biol. Chem. 272, 31051–31057 [DOI] [PubMed] [Google Scholar]
- 20. Shenoy S. K., and Lefkowitz R. J. (2003) Trafficking patterns of β-arrestin and G protein-coupled receptors determined by the kinetics of β-arrestin deubiquitination. J. Biol. Chem. 278, 14498–14506 [DOI] [PubMed] [Google Scholar]
- 21. Shenoy S. K., and Lefkowitz R. J. (2005) Receptor-specific ubiquitination of β-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J. Biol. Chem. 280, 15315–15324 [DOI] [PubMed] [Google Scholar]
- 22. Ozawa K., Whalen E. J., Nelson C. D., Mu Y., Hess D. T., Lefkowitz R. J., and Stamler J. S. (2008) S-nitrosylation of β-arrestin regulates β-adrenergic receptor trafficking. Mol. Cell 31, 395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wyatt D., Malik R., Vesecky A. C., and Marchese A. (2011) Small ubiquitin-like modifier modification of arrestin-3 regulates receptor trafficking. J. Biol. Chem. 286, 3884–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Benovic J. L., Kühn H., Weyand I., Codina J., Caron M. G., and Lefkowitz R. J. (1987) Functional desensitization of the isolated β-adrenergic receptor by the β-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. U.S.A. 84, 8879–8882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marion S., Oakley R. H., Kim K. M., Caron M. G., and Barak L. S. (2006) A β-arrestin binding determinant common to the second intracellular loops of rhodopsin family G protein-coupled receptors. J. Biol. Chem. 281, 2932–2938 [DOI] [PubMed] [Google Scholar]
- 26. Ferguson S. S., Downey W. E. 3rd, Colapietro A. M., Barak L. S., Ménard L., and Caron M. G. (1996) Role of β-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271, 363–366 [DOI] [PubMed] [Google Scholar]
- 27. Bouvier M., Hausdorff W. P., De Blasi A., O'Dowd B. F., Kobilka B. K., Caron M. G., and Lefkowitz R. J. (1988) Removal of phosphorylation sites from the β2-adrenergic receptor delays onset of agonist-promoted desensitization. Nature 333, 370–373 [DOI] [PubMed] [Google Scholar]
- 28. Benovic J. L., Strasser R. H., Caron M. G., and Lefkowitz R. J. (1986) β-Adrenergic receptor kinase: identification of a novel protein kinase that phosphorylates the agonist-occupied form of the receptor. Proc. Natl. Acad. Sci. U.S.A. 83, 2797–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pitcher J., Lohse M. J., Codina J., Caron M. G., and Lefkowitz R. J. (1992) Desensitization of the isolated β2-adrenergic receptor by β-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry 31, 3193–3197 [DOI] [PubMed] [Google Scholar]
- 30. Premont R. T., and Gainetdinov R. R. (2007) Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69, 511–534 [DOI] [PubMed] [Google Scholar]
- 31. Kong G., Penn R., and Benovic J. L. (1994) A β-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the β2-adrenergic receptor. J. Biol. Chem. 269, 13084–13087 [PubMed] [Google Scholar]
- 32. Krasel C., Bünemann M., Lorenz K., and Lohse M. J. (2005) β-Arrestin binding to the β2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J. Biol. Chem. 280, 9528–9535 [DOI] [PubMed] [Google Scholar]
- 33. Liggett S. B., Ostrowski J., Chesnut L. C., Kurose H., Raymond J. R., Caron M. G., and Lefkowitz R. J. (1992) Sites in the third intracellular loop of the α2A-adrenergic receptor confer short term agonist-promoted desensitization. Evidence for a receptor kinase-mediated mechanism. J. Biol. Chem. 267, 4740–4746 [PubMed] [Google Scholar]
- 34. Pals-Rylaarsdam R., and Hosey M. M. (1997) Two homologous phosphorylation domains differentially contribute to desensitization and internalization of the m2 muscarinic acetylcholine receptor. J. Biol. Chem. 272, 14152–14158 [DOI] [PubMed] [Google Scholar]
- 35. Kang D. S., Tian X., and Benovic J. L. (2013) β-Arrestins and G protein-coupled receptor trafficking. Methods Enzymol. 521, 91–108 [DOI] [PubMed] [Google Scholar]
- 36. Snyder J. C., Rochelle L. K., Lyerly H. K., Caron M. G., and Barak L. S. (2013) Constitutive internalization of the leucine-rich G protein-coupled receptor-5 (LGR5) to the trans-Golgi network. J. Biol. Chem. 288, 10286–10297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee K. B., Pals-Rylaarsdam R., Benovic J. L., and Hosey M. M. (1998) Arrestin-independent internalization of the m1, m3, and m4 subtypes of muscarinic cholinergic receptors. J. Biol. Chem. 273, 12967–12972 [DOI] [PubMed] [Google Scholar]
- 38. Bhatnagar A., Willins D. L., Gray J. A., Woods J., Benovic J. L., and Roth B. L. (2001) The dynamin-dependent, arrestin-independent internalization of 5-hydroxytryptamine 2A (5-HT2A) serotonin receptors reveals differential sorting of arrestins and 5-HT2A receptors during endocytosis. J. Biol. Chem. 276, 8269–8277 [DOI] [PubMed] [Google Scholar]
- 39. Gimenez L. E., Vishnivetskiy S. A., Baameur F., and Gurevich V. V. (2012) Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J. Biol. Chem. 287, 29495–29505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laporte S. A., Oakley R. H., Zhang J., Holt J. A., Ferguson S. S., Caron M. G., and Barak L. S. (1999) The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. U.S.A. 96, 3712–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Claing A., Chen W., Miller W. E., Vitale N., Moss J., Premont R. T., and Lefkowitz R. J. (2001) β-Arrestin-mediated ADP-ribosylation factor 6 activation and β2-adrenergic receptor endocytosis. J. Biol. Chem. 276, 42509–42513 [DOI] [PubMed] [Google Scholar]
- 42. McDonald P. H., Cote N. L., Lin F. T., Premont R. T., Pitcher J. A., and Lefkowitz R. J. (1999) Identification of NSF as a β-arrestin1-binding protein. Implications for β2-adrenergic receptor regulation. J. Biol. Chem. 274, 10677–10680 [DOI] [PubMed] [Google Scholar]
- 43. Vines C. M., Revankar C. M., Maestas D. C., LaRusch L. L., Cimino D. F., Kohout T. A., Lefkowitz R. J., and Prossnitz E. R. (2003) N-Formyl peptide receptors internalize but do not recycle in the absence of arrestins. J. Biol. Chem. 278, 41581–41584 [DOI] [PubMed] [Google Scholar]
- 44. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., and Barak L. S. (2000) Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 [DOI] [PubMed] [Google Scholar]
- 45. Zidar D. A., Violin J. D., Whalen E. J., and Lefkowitz R. J. (2009) Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. U.S.A. 106, 9649–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barak L. S., Oakley R. H., Laporte S. A., and Caron M. G. (2001) Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. U.S.A. 98, 93–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kovacs J. J., Whalen E. J., Liu R., Xiao K., Kim J., Chen M., Wang J., Chen W., and Lefkowitz R. J. (2008) β-Arrestin-mediated localization of smoothened to the primary cilium. Science 320, 1777–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luttrell L. M., Ferguson S. S., Daaka Y., Miller W. E., Maudsley S., Della Rocca G. J., Lin F., Kawakatsu H., Owada K., Luttrell D. K., Caron M. G., and Lefkowitz R. J. (1999) β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661 [DOI] [PubMed] [Google Scholar]
- 49. Luo J., Busillo J. M., and Benovic J. L. (2008) M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol. Pharmacol. 74, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., and Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sun Y., Cheng Z., Ma L., and Pei G. (2002) β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 277, 49212–49219 [DOI] [PubMed] [Google Scholar]
- 52. Bruchas M. R., Macey T. A., Lowe J. D., and Chavkin C. (2006) Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 281, 18081–18089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Breitman M., Kook S., Gimenez L. E., Lizama B. N., Palazzo M. C., Gurevich E. V., and Gurevich V. V. (2012) Silent scaffolds: inhibition of c-Jun N-terminal kinase 3 activity in cell by dominant-negative arrestin-3 mutant. J. Biol. Chem. 287, 19653–19664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shenoy S. K., McDonald P. H., Kohout T. A., and Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 55. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., and Benovic J. L. (2003) The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell 5, 709–722 [DOI] [PubMed] [Google Scholar]
- 56. Malik R., and Marchese A. (2010) Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol. Biol. Cell 21, 2529–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shenoy S. K., Modi A. S., Shukla A. K., Xiao K., Berthouze M., Ahn S., Wilkinson K. D., Miller W. E., and Lefkowitz R. J. (2009) β-Arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc. Natl. Acad. Sci. U.S.A. 106, 6650–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Berthouze M., Venkataramanan V., Li Y., and Shenoy S. K. (2009) The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 28, 1684–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. von Zastrow M. (2003) Mechanisms regulating membrane trafficking of G protein-coupled receptors in the endocytic pathway. Life Sci. 74, 217–224 [DOI] [PubMed] [Google Scholar]
- 60. Noma T., Lemaire A., Naga Prasad S. V., Barki-Harrington L., Tilley D. G., Chen J., Le Corvoisier P., Violin J. D., Wei H., Lefkowitz R. J., and Rockman H. A. (2007) β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest. 117, 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gao H., Sun Y., Wu Y., Luan B., Wang Y., Qu B., and Pei G. (2004) Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol. Cell 14, 303–317 [DOI] [PubMed] [Google Scholar]
- 62. Kang J., Shi Y., Xiang B., Qu B., Su W., Zhu M., Zhang M., Bao G., Wang F., Zhang X., Yang R., Fan F., Chen X., Pei G., and Ma L. (2005) A nuclear function of β-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell 123, 833–847 [DOI] [PubMed] [Google Scholar]
- 63. Wehbi V. L., Stevenson H. P., Feinstein T. N., Calero G., Romero G., and Vilardaga J. P. (2013) Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gβγ complex. Proc. Natl. Acad. Sci. U.S.A. 110, 1530–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Feinstein T. N., Yui N., Webber M. J., Wehbi V. L., Stevenson H. P., King J. D. Jr., Hallows K. R., Brown D., Bouley R., and Vilardaga J. P. (2013) Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J. Biol. Chem. 288, 27849–27860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G., Sunahara R. K., El-Samad H., Huang B., and von Zastrow M. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rajagopal S., Rajagopal K., and Lefkowitz R. J. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kenakin T. P. (1985) The quantification of relative efficacy of agonists. J. Pharmacol. Methods 13, 281–308 [DOI] [PubMed] [Google Scholar]
- 68. Boerrigter G., Soergel D. G., Violin J. D., Lark M. W., and Burnett J. C. Jr. (2012) TRV120027, a novel β-arrestin biased ligand at the angiotensin II type I receptor, unloads the heart and maintains renal function when added to furosemide in experimental heart failure. Circ. Heart Fail. 5, 627–634 [DOI] [PubMed] [Google Scholar]
- 69. Tarigopula M., Davis R. T. 3rd, Mungai P. T., Ryba D. M., Wieczorek D. F., Cowan C. L., Violin J. D., Wolska B. M., and Solaro R. J. (2015) Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc. Res. 107, 226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Soergel D. G., Subach R. A., Burnham N., Lark M. W., James I. E., Sadler B. M., Skobieranda F., Violin J. D., and Webster L. R. (2014) Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155, 1829–1835 [DOI] [PubMed] [Google Scholar]
- 71. Brame A. L., Maguire J. J., Yang P., Dyson A., Torella R., Cheriyan J., Singer M., Glen R. C., Wilkinson I. B., and Davenport A. P. (2015) Design, characterization, and first-in-human study of the vascular actions of a novel biased apelin receptor agonist. Hypertension 65, 834–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rajagopal S., Ahn S., Rominger D. H., Gowen-MacDonald W., Lam C. M., Dewire S. M., Violin J. D., and Lefkowitz R. J. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 80, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., and Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tran J. A., Chang A., Matsui M., and Ehlert F. J. (2009) Estimation of relative microscopic affinity constants of agonists for the active state of the receptor in functional studies on M2 and M3 muscarinic receptors. Mol. Pharmacol. 75, 381–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brust T. F., Hayes M. P., Roman D. L., Burris K. D., and Watts V. J. (2015) Bias analyses of preclinical and clinical D2 dopamine ligands: studies with immediate and complex signaling pathways. J. Pharmacol. Exp. Ther. 352, 480–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Onaran H. O., Rajagopal S., and Costa T. (2014) What is biased efficacy? Defining the relationship between intrinsic efficacy and free energy coupling. Trends Pharmacol. Sci. 35, 639–647 [DOI] [PubMed] [Google Scholar]
- 77. Kim J., Ahn S., Ren X. R., Whalen E. J., Reiter E., Wei H., and Lefkowitz R. J. (2005) Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Butcher A. J., Prihandoko R., Kong K. C., McWilliams P., Edwards J. M., Bottrill A., Mistry S., and Tobin A. B. (2011) Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 286, 11506–11518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Busillo J. M., Armando S., Sengupta R., Meucci O., Bouvier M., and Benovic J. L. (2010) Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J. Biol. Chem. 285, 7805–7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., and Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hirsch J. A., Schubert C., Gurevich V. V., and Sigler P. B. (1999) The 2.8 Å crystal structure of visual arrestin: a model for arrestin's regulation. Cell 97, 257–269 [DOI] [PubMed] [Google Scholar]
- 82. Han M., Gurevich V. V., Vishnivetskiy S. A., Sigler P. B., and Schubert C. (2001) Crystal structure of β-arrestin at 1.9 Å: possible mechanism of receptor binding and membrane Translocation. Structure 9, 869–880 [DOI] [PubMed] [Google Scholar]
- 83. Zhan X., Gimenez L. E., Gurevich V. V., and Spiller B. W. (2011) Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 406, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sutton R. B., Vishnivetskiy S. A., Robert J., Hanson S. M., Raman D., Knox B. E., Kono M., Navarro J., and Gurevich V. V. (2005) Crystal structure of cone arrestin at 2.3 Å: evolution of receptor specificity. J. Mol. Biol. 354, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 85. Hanson S. M., Van Eps N., Francis D. J., Altenbach C., Vishnivetskiy S. A., Arshavsky V. Y., Klug C. S., Hubbell W. L., and Gurevich V. V. (2007) Structure and function of the visual arrestin oligomer. EMBO J. 26, 1726–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Milano S. K., Kim Y. M., Stefano F. P., Benovic J. L., and Brenner C. (2006) Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J. Biol. Chem. 281, 9812–9823 [DOI] [PubMed] [Google Scholar]
- 87. Gurevich V. V., and Benovic J. L. (1993) Visual arrestin interaction with rhodopsin: sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J. Biol. Chem. 268, 11628–11638 [PubMed] [Google Scholar]
- 88. Shukla A. K., Manglik A., Kruse A. C., Xiao K., Reis R. I., Tseng W. C., Staus D. P., Hilger D., Uysal S., Huang L. Y., Paduch M., Tripathi-Shukla P., Koide A., Koide S., Weis W. I., et al. (2013) Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Granzin J., Stadler A., Cousin A., Schlesinger R., and Batra-Safferling R. (2015) Structural evidence for the role of polar core residue Arg175 in arrestin activation. Sci. Rep. 5, 15808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nobles K. N., Guan Z., Xiao K., Oas T. G., and Lefkowitz R. J. (2007) The active conformation of β-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of β-arrestins1 and -2. J. Biol. Chem. 282, 21370–21381 [DOI] [PubMed] [Google Scholar]
- 91. Kim Y. J., Hofmann K. P., Ernst O. P., Scheerer P., Choe H. W., and Sommer M. E. (2013) Crystal structure of pre-activated arrestin p44. Nature 497, 142–146 [DOI] [PubMed] [Google Scholar]
- 92. Hanson S. M., Francis D. J., Vishnivetskiy S. A., Kolobova E. A., Hubbell W. L., Klug C. S., and Gurevich V. V. (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim M., Vishnivetskiy S. A., Van Eps N., Alexander N. S., Cleghorn W. M., Zhan X., Hanson S. M., Morizumi T., Ernst O. P., Meiler J., Gurevich V. V., and Hubbell W. L. (2012) Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. U.S.A. 109, 18407–18412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kang Y., Zhou X. E., Gao X., He Y., Liu W., Ishchenko A., Barty A., White T. A., Yefanov O., Han G. W., Xu Q., de Waal P. W., Ke J., Tan M. H., Zhang C., et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Szczepek M., Beyrière F., Hofmann K. P., Elgeti M., Kazmin R., Rose A., Bartl F. J., von Stetten D., Heck M., Sommer M. E., Hildebrand P. W., and Scheerer P. (2014) Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat. Commun. 5, 4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Feuerstein S. E., Pulvermüller A., Hartmann R., Granzin J., Stoldt M., Henklein P., Ernst O. P., Heck M., Willbold D., and Koenig B. W. (2009) Helix formation in arrestin accompanies recognition of photoactivated rhodopsin. Biochemistry 48, 10733–10742 [DOI] [PubMed] [Google Scholar]
- 97. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shukla A. K., Westfield G. H., Xiao K., Reis R. I., Huang L. Y., Tripathi-Shukla P., Qian J., Li S., Blanc A., Oleskie A. N., Dosey A. M., Su M., Liang C. R., Gu L. L., Shan J. M., et al. (2014) Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kang D. S., Kern R. C., Puthenveedu M. A., von Zastrow M., Williams J. C., and Benovic J. L. (2009) Structure of an arrestin2-clathrin complex reveals a novel clathrin binding domain that modulates receptor trafficking. J. Biol. Chem. 284, 29860–29872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yang F., Yu X., Liu C., Qu C. X., Gong Z., Liu H. D., Li F. H., Wang H. M., He D. F., Yi F., Song C., Tian C. L., Xiao K. H., Wang J. Y., and Sun J. P. (2015) Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F-NMR. Nat. Commun. 6, 8202. [DOI] [PMC free article] [PubMed] [Google Scholar]