

Abstract
PB1-F2 is a virulence factor of influenza A virus (IAV) whose functions remain misunderstood. The different roles of PB1-F2 may be linked to its structural polymorphism and to its propensity to assemble into oligomers and amyloid fibers in the vicinity of the membrane of IAV-infected cells. Here, we monitored the impact of PB1-F2 on the biochemical composition and protein structures of human epithelial pulmonary cells (A549) and monocytic cells (U937) upon IAV infection using synchrotron Fourier-transform infrared (FTIR) and deep UV (DUV) microscopies at the single-cell level. Cells were infected with a wild-type IAV and its PB1-F2 knock-out mutant for analyses at different times post-infection. IR spectra were recorded in each condition and processed to evaluate the change in the component band of the spectra corresponding to the amide I (secondary structure) and the CH stretching region (membrane). The IR spectra analysis revealed that expression of PB1-F2 in U937 cells, but not in A549 cells, results in the presence of a specific β-aggregate signature. Furthermore, the lipid membrane composition of U937 cells expressing PB1-F2 was also altered in a cell type-dependent manner. Using DUV microscopy and taking advantage of the high content of tryptophan residues in the sequence of PB1-F2 (5/90 aa), we showed that the increase of the autofluorescent signal recorded in monocytic cells could be correlated with the IR detection of β-aggregates. Altogether, our results constitute an important step forward in the understanding of the cell type-dependent function of PB1-F2.
Keywords: amyloid, Fourier transform IR (FTIR), influenza virus, membrane, microscopy, oligomerization, fluorescence imaging, synchrotron radiation experiment
Introduction
Influenza A viruses (IAVs)2 are responsible each year for seasonal epidemics resulting in considerable illness, death, and important economic losses (1). IAVs are also major pathogens of animals and cause devastating outbreaks in domestic poultry and massive culling to control the viral spread.
IAVs are members of the Orthomyxoviridae family and their genome is constituted of eight segments of single-stranded negative-sense RNA (2). The viral segment 2 encodes the polymerase subunit PB1 and two additional proteins, N40, a N-truncated version of PB1 devoid of transcriptase activity, and PB1-F2 from an alternative reading frame (3). PB1-F2 is a small protein of 90 amino acids with a strong polymorphism in sequence and length depending on the viral strain. Although PB1-F2 is expressed in its full-length version in 96% of A/H5N1 avian strains (4), less than 10% of A/H1N1 human viruses isolated since 1949 express a functional version of PB1-F2, i.e. of 79 amino acids or more (5, 6). Full-length PB1-F2 was shown to contribute to the virulence of IAVs (7), notably of highly virulent A/H5N1 (8) and of the Spanish flu virus of 1918 (9). Full-length PB1-F2 was also expressed by the pandemic viruses of 1957 and 1968 (6). Nevertheless, the last 2009 pandemic A/H1N1 virus expressed only an 11-amino acid C-terminal-truncated protein as a result of the accumulation of premature stop codon (10). Although PB1-F2 was shown to exacerbate the pathogenicity of IAVs in mouse models (11–14), PB1-F2 attenuates virulence in chicken (15). Such observations are in accordance with the hypothesis that the loss of expression of PB1-F2 in mammals is beneficial for viral fitness, whereas in avian species, PB1-F2 is positively selected to contribute to an optimized spreading of the virus without increased virulence. The question of PB1-F2 function remains unsolved. PB1-F2 was first described as a proapoptotic protein targeting the mitochondria (16–18) and inducing apoptosis by loss of the mitochondrial membrane potential (19). Another proposed function of PB1-F2 is the modulation of innate immune response (20). Several reports have shown that PB1-F2 enhances the inflammatory response in the lungs of IAV-infected mice notably by recruiting a massive number of leukocytes within the airways (14, 21). PB1-F2 also facilitates secondary bacterial pneumonia in mammals (9, 22). PB1-F2 was shown to exacerbate the production of interferon during A/WSN/1933 (H1N1) virus infection (23).
PB1-F2 is disordered in aqueous solution (24) but can switch from a random state to a β-sheet secondary structure in a membrane-mimicking environment (25). Moreover, PB1-F2 has a strong propensity to oligomerize. We previously showed by thioflavin staining that PB1-F2 formed amyloid fibers in vitro using recombinant protein and in IAV-infected cells (25). PB1-F2 is present as unstructured monomers in the early stage of infection (23, 26, 27), and rapidly forms soluble β-oligomers (28, 29) to finally accumulate as β-amyloid aggregates at a later stage of infection (25). Interestingly, other reports referred to the propensity of PB1-F2 to oligomerize and to be implicated in the extracellular activation of the NLRP3 inflammasome pathway (30), which is known to be triggered by protein amyloid aggregates (31). However, the presence of amyloid fibers in different types of IAV target cells during infection remained to be validated by new techniques.
In the present study, we evidenced the presence of PB1-F2 β-aggregates in IAV-infected cells using synchrotron radiation. Fourier transform infrared (FTIR) microscopy is a non-invasive technique for monitoring biochemical changes in situ at the single cell level (32, 33). IR microspectroscopy of cells in culture is a rapidly growing area of research allowing to spectrally distinguish cells in different physiological states, e.g. uninfected and infected (34, 35). Here, FTIR microspectroscopy appeared as a valuable method to study the formation of PB1-F2 fibers and their impact on infected cells due to its ability to detect simultaneously a variety of changes in molecular structure and composition. Moreover, FTIR spectroscopy is a sensitive tool to study protein secondary structure and protein folding, unfolding, and misfolding (36, 37). FTIR approaches were developed on amyloid fibril structure (38) revealing structural differences between native β-sheet proteins and amyloid fibrils (39). FTIR microscopy coupled to a synchrotron source (sFTIR) allows achieving spatial resolution close to the diffraction limit. Thanks to the high signal to noise ratio and spatial resolution offered by synchrotron radiation FTIR, we optimized the signal detection of PB1-F2 fibers in infected cells without the necessity of immunostaining or purification procedures. Synchrotron deep ultraviolet (sDUV) fluorescence microscopy is a powerful tool for studying biological samples and allows localizing aromatic residues such as Trp without any external probe or staining at high spatial resolution (40–42). So, taking advantage of the high frequency of tryptophan (Trp) in PB1-F2, we used sDUV microscopy (below 350 nm (43)) to detect the presence of PB1-F2 aggregates.
In this paper, we used objective and unsupervised methods of multivariable statistics applied to sFTIR spectroscopy and sDUV microscopy at the single cell level to detect PB1-F2 β-aggregated structures in IAV-infected cells. Multivariate statistical analyses in the amide I region of the spectra (corresponding to protein secondary structures) showed a clear and specific IR signature for the β-aggregated structure in a cell-type and time-dependent manner in IAV-infected monocytic cells. We confirmed using sDUV microscopy an increase of the autofluorescent signal in IAV-infected monocytic cells expressing PB1-F2. Moreover, we observed significant differences in the CH region of the spectra (corresponding to membrane) between infected cells expressing or not PB1-F2. Finally, the impact of PB1-F2 on membrane appeared to be cell type-dependent, because loss of membrane fluidity occurred in monocytic cells, whereas perturbation of the CH2/CH3 ratios was observed in epithelial cells.
Experimental Procedures
Cell Culture and Viral Infection
HeLa and 293T cells in Dulbecco's minimum essential medium (Lonza) and the human alveolar epithelial (A549) cells and the Madin-Darby canine kidney cells in Eagle's minimum essential medium (Lonza), supplemented with 10% fetal calf serum (Perbio), 2 mm l-glutamine, 100 IU/ml of penicillin, and 100 μg/ml of streptomycin, were grown as monolayer. The human monocytic (U937) cells were maintained in RPMI 1640 medium (Lonza) supplemented with 10% fetal calf serum (Perbio), 2 mm l-glutamine, 100 IU/ml of penicillin, and 100 μg/ml of streptomycin. All cultures were maintained in a 5% CO2 incubator at 37 °C. For infection assays, all cells were seeded on zinc-selenide (ZnSe) 4-mm diameter IR transparent windows or on 25-mm diameter suprasyl coverslips inserted into 6-well plates for synchrotron FTIR and DUV microscopy, respectively. Cells were allowed to attach overnight before infection. Cells were washed with fetal calf serum-free medium and incubated with virus at a multiplicity of infection of 5 for 1 h at 37 °C. Following this, the infection mixture (fetal calf serum-free medium with excess virus) was replaced by new growth medium and the cells were maintained for 8 or 24 h in a 5% CO2 incubator at 37 °C. Then, ZnSe windows and suprasyl coverslips were collected and infected cells were fixed in 4% formalin for 30 min at room temperature. Cells were rinsed with PBS and once with pure water to eliminate the remaining salts before analysis. Finally, the samples were left to dry in a desiccator at room temperature prior to observation. Mock cells, or “intact cells” received the same treatment as infected cells with the exception of the addition of virus in the infection mixture.
Generation of Recombinant Knocked Out PB1-F2 Mutant Influenza Virus
Influenza A/WSN/1933 (H1N1) wild-type (WT) and the PB1-F2 knock-out (F2) viruses used in this study were produced using the 12-plasmid reverse genetic system as previously described (25). Briefly, 293T cells were transfected with 12 plasmids following the FuGENE protocol (Roche): eight plasmids encoding the viral RNA segments of IAV and four encoding the three subunits of the viral polymerase and the nucleoprotein. The rescued WT and F2 viruses were amplified on Madin-Darby canine kidney cells and titrated by plaque assays. Generation of F2 virus was achieved using the QuikChange mutagenesis kit (Stratagene) at nucleotides coding for the initiation codons of PB1-F2 substituting ATG to ACG (resulting in silent mutations in the PB1 open reading frame). All introduced mutations were confirmed by sequencing.
Synchrotron FTIR Microspectroscopy
Synchrotron FTIR microscopy was performed at the SOLEIL synchrotron (Gif-sur-Yvette, France) using the SMIS beamline (Synchrotron SOLEIL). All spectra were recorded in transmission mode on a Nicolet Continuum XL microscope (Thermo Scientific). The microscope comprises a motorized sample stage and a liquid nitrogen-cooled mercury cadmium telluride (MCT-A) detector (50 μm element size). The microscope operates in confocal mode using a ×32 infinity corrected Schwarzschild objective (NA = 0.65) and a matching ×32 condenser. All IR spectra were recorded using a dual mask aperture of 10 × 10 μm2. Individual spectra were saved in log (1/R) format at 4 cm−1 spectral resolution, with 128 co-added scans encompassing the mid-IR region from 4000 to 800 cm−1. All IR spectra were pre-processed and submitted to multivariate data analysis (The Unscrambler, CAMO Process AS). Second derivatives of the spectral data were assessed (9-point Savitzky-Golay filter) to enhance the spectral resolution of the absorption bands. The second derivative IR spectra were unit vector normalized and analyzed by applying principal component analysis (PCA). The computation of principal components was based on the non-linear iterative projections by alternating least-squares (NIPALS) algorithm. Although the score plots allowed a comparison of IR spectra, the corresponding loading plots revealed the main characteristic absorption bands.
Synchrotron DUV Fluorescence Imaging
The full field synchrotron DUV imaging system is constructed around a Zeiss Axio Observer Z1 (Carl Zeiss) inverted microscope constructed with quartz-only optics. The white beam of DISCO beamline at Synchrotron SOLEIL (43) is monochromatized by an iHR320 (Jobin-Yvon Horiba) before coupling with the entrance of the modified Zeiss Axio Observer Z1, the monochromatic beam was settled at 280 nm to determine the level of tryptophan in cells. A sharp dichroic mirror transmitting only above 300 nm (Omega Optical) reflected the incident light before focalization onto the sample through a Zeiss Ultrafluor ×40 (N.A. 0.6, glycerine immersion) (40, 42). Emission was recorded with a Pixis 1024-BUV (Princeton Instruments) camera after passing through a series of bandpass filters (Semrock). Fluorescence images where typically recorded in few seconds exposure. Power measured on the sample was close to μW/cm2. The whole system is controlled via μManager (44).
Results
Background and Experimental Set-up
To determine whetherthe expression of PB1-F2 drives the formation of amyloid aggregated structures in different IAV susceptible cell types, we infected monocytic cells (U937) and lung epithelial cells (A549) and tried to reveal the presence of such structures using sFTIR and sDUV microspectroscopy (Fig. 1). HeLa cells, which are infectable by IAV but do not allow efficient virus replication, were also integrated in the study (45). Cells were infected with virus engineered by reverse genetic, influenza A/WSN/1933 (H1N1) WT, and its PB1-F2 knock-out (F2) counterpart, fixed at two times post-infection (8 and 24 h) and subjected to comparative analysis. For the relevance of the study, it was crucial to include a virus knocked-out for PB1-F2 expression to distinguish between the observations linked to the infectious context with those specific of PB1-F2 expression. In addition, we had previously confirmed that the deletion of PB1-F2 does not introduce any difference in the growth property of IAV (23). The FTIR spectra of cells and protein have been extensively described in the literature and main spectral peak positions and their assignments are available (Table 1). Because the aim of this study was to evidence the presence of PB1-F2 amyloid aggregates in infected cells, we paid special attention to the amide I region of the spectrum. FTIR is a classical method to study the folding/unfolding and aggregation of proteins in particular thanks to the amide I region (1715–1575 cm−1), which is very sensitive to the protein secondary structure (36, 46). The study of this region of the IR spectrum allows discriminating between different types of β-sheet structure signatures (39). Moreover, as PB1-F2 was shown to aggregate in the vicinity of membranes and to create pores, we evaluated the impact of PB1-F2 in different cell types by analyzing the region of the IR spectrum corresponding to the CH stretching region (2970–2840 cm−1) (47). To this end, FTIR raw spectra in the 4000–800 cm−1 region were recorded in the cytoplasm of infected cells using the Nicolet Continuum XL microscope (Fig. 1B, SMIS beamline). A sufficient number of single-cell spectra were recorded and compiled for each condition to statistically represent and compare the studied populations. The FTIR spectra presenting a large baseline deformation were excluded (Fig. 2, A and B). Second derivative spectra were calculated to enhance the resolution of the absorption bands and visualize the point of maxima for each band studied (Fig. 2C). The amide I and CH stretching regions were independently studied to follow changes in the component bands corresponding to the secondary structure of protein and lipid components, respectively. Preprocessed data were then analyzed by PCA to study the unsupervised variation pattern in the data. PCA enable the decomposition of original data and can be used as a technique that reduces the number of parameters needed to represent the variance in the spectral dataset (48). Thus, PCA can resolve a complete spectral dataset into a few key spectral components to identify and isolate important trends within the dataset. Results were represented as score plots that enable the data to be plotted in a form where each single-cell spectrum is represented as a single point in a relevant subspace (principal components (PC)). This representation permits to indicate easily which parameters (i.e. wavenumber values) were responsible for most of the variance in the data and to estimate sample heterogeneity. Then, a loading plot is used to express the correlation between variables (wavenumber values) and determine to what extent a variable is dominating or influencing the model of study (here IR signature of infected cells) and thus its contribution to each PC.
FIGURE 1.

SOLEIL synchrotron FTIR and DUV set up and experiments. A, A549 or U937 cells were infected with IAV virus expressing (WT) or not PB1-F2 (F2), fixed at various times post-infection and collected prior to observation by IR or DUV microscopy. B, the image shows the Continuum XL microscope (Thermo Fisher Scientific) used for FTIR microspectroscopic analysis performed on the SMIS beamline. C, transmission image representing IAV-infected A549 cells observed with the Continuum XL microscope before recording the IR spectra. Each annotated red cross corresponds to the acquisition of one single cell IR spectrum. D, recorded IR spectra in the 3500–1000 cm−1 region. Spectra were standardized and pre-treated before multivariate statistical analysis. E, the image shows the Telemos full field microscope used to record the Trp fluorescence of IAV-infected cells in the DUV with the DISCO beamline. F, transmission image in bright field microscopy (left image) and DUV fluorescent microscopy (right image) of IAV-infected U937 cells are presented.
TABLE 1.
Band position and assignment for the amide I and lipid region according to the literature
FIGURE 2.

Infrared spectra preprocessing procedure. Top left, raw spectra, top right, filtered spectra (to remove background disturbed spectra), and bottom, 2nd derivative Savitzky-Golay spectra (9 pts, 2 order) and unit vector normalized. A close-up view on CH stretching and the Amide I region are shown in the bottom panel.
sFTIR Detection of PB1-F2 β-Aggregates in IAV-infected Cells
The objective of this study was to investigate whether sFTIR microspectroscopy could reveal the presence of β-aggregated structures in different types of infected cells and at different stages of the infectious cycle. As mentioned above, PCA was performed on the normalized second-derivative spectra of the amide I region to study secondary structures at a single-cell level. Fig. 3 displays the comparison between secondary structures content of mock-, WT-, and F2-infected U937 cells at 8 h post-infection. The score plot presented in Fig. 3A revealed three clusters partially independent using the first two components, PC1 and PC2, that represent 54 and 13% of the total variance, respectively. Following the PC2 axis, the cluster F2 and mock are totally overlapping in contrast to the plots of the WT cluster, which are mainly separated along the PC2 axis, although with a partial overlap. PC2 allows to differentiate between PB1-F2 expressing and non-expressing cells on a basis highlighted by the loading plot presented in Fig. 3B. The PC2 loading plot highlights the vibrational wavenumbers that are important in the separation observed along PC2 in the PC1 versus PC2 scores plot. The main variation in the positive direction of PC2 is due to variables at 1631, 1678, and 1693 cm−1. The peak at 1693 cm−1 is indicative of the antiparallel β-sheet, and commonly observed in in vitro amyloid IR spectra (49). However, associated with the second peak at 1631 cm−1 it is specifically ascribed to aggregated β-sheet structures, such as amyloid aggregates (Table 1) (50). The third peak at 1678 cm−1 could be assigned to β-turns (36, 46). The main variation in the negative direction of PC2, associated to F2 and mock-infected cells spectra, is due to variables at 1660 and 1666 cm−1 related to loop/α-helical structures (50) and internal random coiled segments (50, 51) (disordered structures), respectively. PCA of the spectra recorded at 24 h post-infection leads to the same observation: clear separation along the PC2 axis between PB1-F2 expressing and non-expressing U937 cells and a correlated loading plot showing in the positive direction of PC2 two major peaks at 1630 and 1693 cm−1 assigned to aggregated β-sheet structures (data not shown). Thus, the comparison of spectra corresponding to WT, F2, and mock-infected U937 cells enabled identification of an aggregated β-sheet structure spectral pattern associated to the WT-infected U937 cells spectra as early as 8 h post-infection.
FIGURE 3.
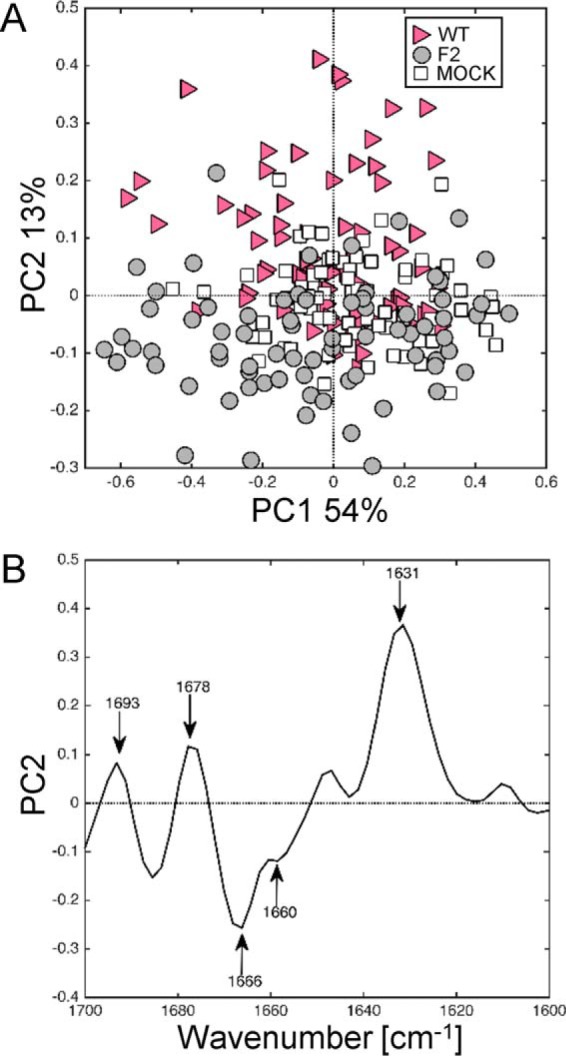
Synchrotron IR microspectroscopy comparison between the secondary structure contents of mock, WT-, and F2-infected U937 cells at 8 h post-infection within the amide I region. A, score plot of PCA from the 1700–1600 cm−1 band IR spectra. The explained variance for PC1 and PC2 is 54 and 13%, respectively. Marked data with the pink triangle correspond to U937 cells infected with WT virus, gray circle to cells infected with F2, and white square to mock-infected cells. B, loading plot linking the variable space and principal component subspace (PC2). PCA score plots show that the WT and F2-mock groups separate along PC2. Compared with mock and F2 groups, the WT group shows higher IR intensity at 1631–1693 cm−1 bands attributed to aggregated β-sheet structures and at the 1678 cm−1 band assigned to turns structures. PC, principal component.
We have previously shown that expression of PB1-F2 in the infectious context lead to the accumulation of β-sheet structures (soluble oligomers) in IAV-infected U937 cells at late stage infection (28). IR spectra of WT-infected cells at 8 and 24 h post-infection and mock-infected cells were submitted to PCA analysis to check if we could observe an accumulation of β-aggregates in a time-dependent manner (Fig. 4). PCA was performed on second derivative spectra of mock- and WT-infected U937 cells at 8 and 24 h post-infection. The score plot presented in Fig. 3A revealed two independent clusters using the first and third component, PC1 and PC3 representing 55 and 11% of the total variance, respectively. The three clusters partially overlap but PC3 in the positive direction clearly differentiates WT- and mock-infected U937 cells. The loading plot presented in Fig. 4B indicates a pattern similar to that observed at 8 h post-infection (Fig. 3B), namely two peaks at 1693 and 1628 cm−1 that could be assigned to aggregated β-structures. This intense peak at 1628 cm−1 associated to a minor peak at 1693 cm−1 is indicative of a highly aggregated β-sheet with a minor peak at 1640–1650 cm−1 indicative of some α-helical and/or random coil structure in the negative direction of PC3 corresponding to mock-infected U937 cells. The minor peak at 1678 cm−1 ascribed to β-turns was observed again in this analysis, but not the two other peaks previously observed at 1666 and 1660 cm−1 in the PCA of WT-F2-mock spectra at 8 h post-infection (Fig. 3B). The comparison of raw spectra (Fig. 4C) highlights the progressive accumulation of β-aggregated structures by a distinct shift in the amide I region of the IR spectrum. More precisely, a clear shift was observed from 1640 to 1628 cm−1, within parallel the appearance of a peak at 1693 cm−1 and the reduction of the peak around 1678 cm−1. This shift was only partial at 8 h post-infection but was totally achieved at 24 h post-infection displaying the specific IR signature assigned to β-aggregated structures with two peaks at 1693 and 1628 cm−1. Thus, thanks to sFTIR microspectroscopy, we have clearly confirmed the presence of β-aggregated structures in IAV-infected U937 cells expressing PB1-F2 in a time-dependent manner and the propensity of PB1-F2 β-sheet structures to accumulate over time.
FIGURE 4.
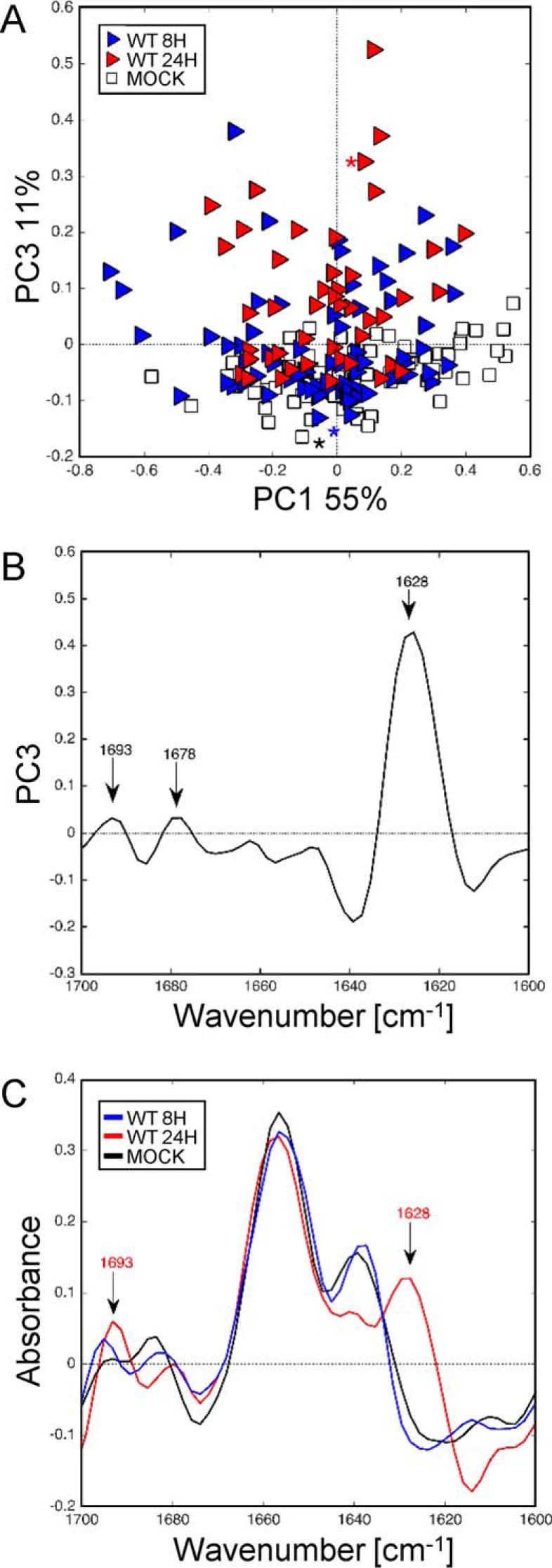
Synchrotron IR microspectroscopy comparison between the secondary structure content of mock and WT-infected U937 cells at 8 and 24 h post-infection within the amide I region. A, score plot of PCA from the 1700–1600 cm−1 band IR spectra. The explained variance for PC1 and PC3 is 55 and 11%, respectively. Marked data with the white square corresponds to mock-infected cells and blue and red triangles correspond to cells infected with WT virus at 8 and 24 h post-infection, respectively. B, loading plot linking the variable space and principal component subspace (PC3). PCA score plots show that the WT and mock groups separate along PC3. Compared with the mock group, WT groups show higher IR intensity at 1628–1693 cm−1 bands attributed to aggregated β-sheet structures. C, marked points second derivative spectra: mock-infected cells (black line) and WT-infected cells at 8 (blue line) and 24 h (red line) post-infection in the range 1700–1600 cm−1. A progressive shift from 1643 cm−1 (unordered β-sheet structure) to 1628 cm−1 was observed in a time-dependent manner for WT-infected cells 2nd derivative spectra compared with mock-infected cells. PC, principal component.
In our previous work, we showed that monomeric PB1-F2 and soluble oligomers of PB1-F2 were detectable at various times post-infection in WT-infected A549 cells (23, 26, 28). However, ThS staining did not allow demonstrating that PB1-F2 forms amyloid fibers in these cells. To explore if FTIR microspectroscopy was suitable to detect the β-aggregated structure, U937, A549, and HeLa cells were infected with the IAV expressing or not PB1-F2 and submitted to the same treatment. The IR spectra recorded for infected A549 and HeLa cells at 8 and 24 h post-infection were submitted to analysis by PCA. The resulting score plot for infected A549 cells shows the samples plotted in a relevant subspace determined by PC1 and PC2 that represented 37 and 18% of the total variance, respectively (Fig. 5A). In contrast to what we observed in infected U937, PCA failed to discriminate between WT and F2-infected A549 cells. However, PCA revealed a clear separation along the PC1 axis between mock-infected cells and IAV-infected cells. PC1 that was permitted to differentiate on a basis highlighted by the corresponding loading plot is presented in Fig. 5B. The main variation on the positive direction of PC1 is due to wavenumbers at 1653 and 1631 cm−1 assigned to α-helical structures and β-sheet structures, respectively. The side band at 1693 was not observed in IAV-infected A549 cells. The same loading plot pattern was observed at 8 and 24 h post-infection even if the intensity of the peak assigned to β-sheet structures increased compared with the peak ascribed to α-helical structures at 24 h post-infection suggesting an accumulation phenomenon. Together, these results collectively suggest that WT infection and PB1-F2 expression does not directly cause the formation of a detectable amount of β-aggregates structures in WT-infected A549 cells, confirming our previous results with classical methods. Moreover, the PCA of the IR spectra recorded in IAV-infected HeLa cells did not discriminate between IAV-infected and non-infected cells (data not shown). Thus, to summarize, spectral changes due to the expression of PB1-F2 in infected cells vary in extent and quality for the different cellular types, and the IR signature specifically corresponding to the β-aggregated feature could only be observed in U937 cells expressing PB1-F2. This observation is in accordance with our previous results and the cell type-dependent function of PB1-F2.
FIGURE 5.
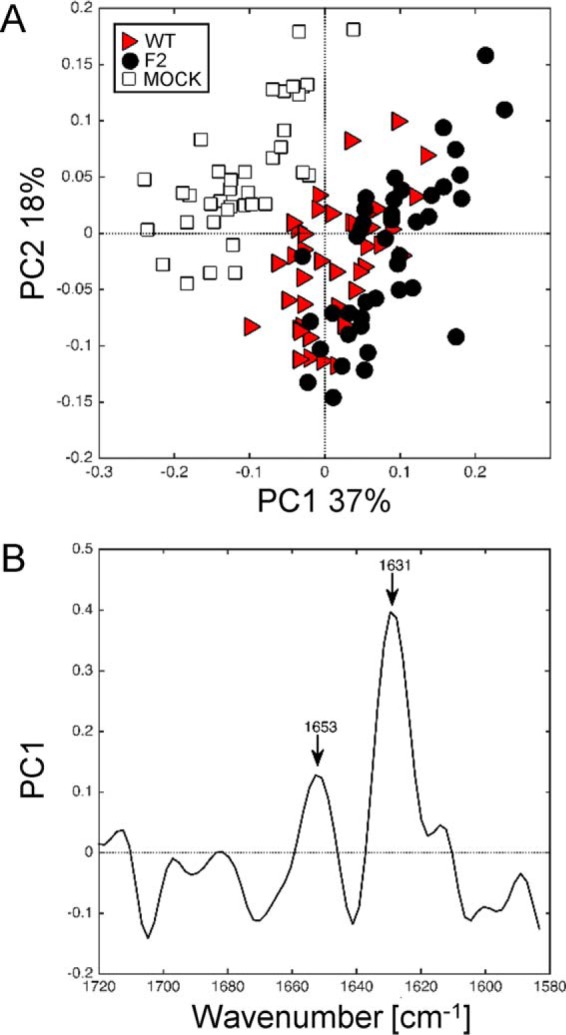
Synchrotron IR microspectroscopy comparison between secondary structure content of mock, WT, and F2-infected A549 cells at 24 h post-infection within the amide I region. A, score plot of PCA from the 1720–1580 cm−1 band IR spectra. The explained variance for PC1 and PC2 is 37 and 18%, respectively. Marked data with a red triangle correspond to A549 cells infected with WT virus, black circle to cells infected with PB1-F2 knocked out virus, and white square to mock-infected cells. B, loading plot linking the variable space and principal component subspace (PC1). PCA score plots show that the WT and F2 groups separate from the mock group along PC1. Compared with the mock group, WT and F2 groups show higher IR intensity at 1631 cm−1 bands attributed to β-sheet structures and at the 1653 cm−1 band assigned to α-helical structures. PC, principal component.
Quantification of Trp Autofluorescence in IAV-infected Cells Assessed with sDUV Microspectroscopy
We confirmed the presence of PB1-F2 β-aggregated structures in WT-infected U937 cells using FTIR microspectroscopy. Thus, taking advantage of the high content of Trp in PB1-F2 (5/90 amino acids), we explored the subcellular localization of PB1-F2 aggregates using the Trp autofluorescent signal. For this purpose, we benefited from the DISCO beamline devoted to synchrotron radiation UV fluorescent microscopy, which allow mapping many compounds without any external probe. The experiments were performed with a full field microscope using a 280-nm excitation and a DUV emission filter (300–380 nm) to follow Trp fluorescence using sDUV microscopy. The fluorescent signal of Trp in infected cells was recorded, quantified, and normalized to the surface area to allow a comparison in the different conditions studied. To be statistically relevant, a minimum of 50 cells for each condition were individually observed. Fig. 6A presents an example of images obtained with A549 and U937 cells at 8 h post-infection by DUV microscopy and its counterpart in the bright field. No relevant difference of fluorescent signal was observed in A549 cells at 8 h post-infection. In contrast, the fluorescence intensity was consistently lower for F2- and mock-infected cells than for WT-infected U937 cells. However, even if intense fluorescent spots were observed, the Trp autofluorescent signal was diffuse in the cytoplasm of the cells and therefore it was not possible to determine specifically the subcellular localization of PB1-F2 β-aggregated structures. Then, ImageJ software (52) was used to quantify the Trp fluorescent intensity of each single cell and calculate the mean of fluorescence normalized to the surface area of the cells, as different cell types were involved in the experiments. Finally, an analysis of variance test was performed to determine the statistical relevance of the data. The results presented in Fig. 6B confirmed a significant increase of fluorescent signal in WT-infected U937 cells compared with F2 and mock-infected cells (p < 0.001). A higher fluorescent signal was also recorded in monocytic cells expressing PB1-F2 at 24 h post-infection (data not shown). In contrast to U937 cells, no relevant difference of fluorescent signal was observed between WT-infected A549 cells compared with F2 and mock-infected cells at 8 and 24 h post-infection. Compared with infected U937 cells, which only exhibited a visible but weak IAV cytopathic effect, WT and F2-infected A549 cells showed a drastic cytopathic effect illustrated in Fig. 7. At 24 h post-infection, the reduction of the surface of the IAV-infected epithelial cells introduced a bias in the quantification of the fluorescent signal and did not allow relevant comparisons. Finally, HeLa cells were used as negative cellular control because they only allowed an incomplete replication of the virus in tissue cell culture. No relevant difference of fluorescent signal and cytopathic effect was observed. Even if we cannot rule out that some aggregates could be formed in IAV-infected A549 or HeLa cells, PB1-F2 preferentially aggregates in U937 monocytic cells. Altogether, our DISCO results are in accordance with the cell-type and time-specific structural behavior of PB1-F2 during the viral cycle and allow to correlate FTIR and DUV microspectroscopy analysis.
FIGURE 6.

Synchrotron deep UV microspectroscopy comparison of fluorescence intensity between mock, WT, and F2-infected cells at 8 h post-infection within the 300–380 nm emission range. A, U937 (top panel) and A549 (bottom panel) cells were infected and observed at 8 h post-infection. For each condition, WT, F2, and mock-infected cells, the fluorescent image obtained by the DUV fluorescent microscope within the 300–380 nm emission range (top) and its corresponding image obtained by transmission microscopy (bottom) are presented. Cells were observed with a ×100 objective. The greyscale was the same for all the fluorescent images. Bars = 20 μm. B, comparison of the fluorescence intensity measured in mock, WT, and F2-infected cells. Fluorescence intensity were quantified using ImageJ software and normalized to the surface area of the cells. Data are expressed as mean ± S.E. by an analysis of variance test. ***, p < 0.001, NS, not significant.
FIGURE 7.

Visualization of cytopathic effect provoked by IAV infection at 24 h post-infection. U937 and A549 IAV-infected cells show a strong cytopathic effect compared with HeLa cells. Cells were observed by transmission microscopy in the bright field using a ×100 objective. Bars = 20 μm.
FTIR Assessment of the Impact of PB1-F2 in IAV-infected Cells in the CH Region
Because we previously observed that PB1-F2 possesses a membrane tropism and the ability to permeabilize artificial membranes (25), we tested whether it was possible to detect a specific lipid IR signature ascribed to the expected damages of PB1-F2 oligomerization in IAV-infected cells. Thus, we focused our attention on the fatty acid region in the spectral range from 2840 and 3000 cm−1 on characteristic vibrations corresponding to the C-H stretching region attributed to lipids. More precisely, this spectral range is attributed to C-H stretching vibrations, i.e. CH2 and CH3 acyl chain symmetric (s) and asymmetric (as) vibrations (51) (Table 1). As previously done within the amide I region, the specific spectral features that cause the distinction between the samples (single cell spectra) were assessed by PCA and presented in the score plots. IR spectra of U937 cells recorded at 8 and 24 h post-infection were submitted to PCA and plotted in a relevant subspace determined by principal component PC1 and PC2 in the resulting score plot (Fig. 8A). IR spectra of WT, F2, and mock-infected U937 cells were separated in two clusters along PC1 with an explained variance of 33%. The three clusters are partially overlapping but PC1 in the negative direction clearly differentiates WT from F2- and mock-infected U937 cells. The corresponding loading plot is presented in Fig. 8B. High loading values at 2951, 2916, 2872, and 2846 cm−1 were positively correlated with F2 and mock clusters, whereas high loading values at 2964, 2931, 2874, and 2858 cm−1 were negatively correlated with WT cluster. The couples of peaks at 2964–2951 and 2931–2916 cm−1 are assigned to asymmetric CH3 and CH2 acyl chain stretching, whereas the weaker peaks at 2874–2872 cm−1 and the peaks at 2858–2846 cm−1 correspond to symmetric CH3 and CH2 stretching, respectively. Examination of raw spectra (marked with an asterisk in Fig. 8A) clearly showed a shift from 2956 to 2954 cm−1 (asCH3), 2922 to 2920 cm−1 (asCH2), 2874 to 2872 cm−1 (sCH3), and 2852 to 2850 cm−1 (sCH2) assigned to modification in the content of long chain-saturated (asymmetric) and unsaturated (symmetric) fatty acids, respectively. This shift was detectable on raw spectra (Fig. 8C) thanks to the high brilliance of the synchrotron source. The shift of the 2874 cm−1 band was not evident in the raw spectra but appeared in the loading plot highlighting the potential of PCA to reveal hidden patterns in the data. Altogether, our FTIR microspectroscopy results showed distinct and reproducible changes in the CH stretching region attributed to change in the membrane composition and a perturbation of membrane fluidity, which could be correlated with the detection of β-aggregated structures in WT-infected U937 cells.
FIGURE 8.
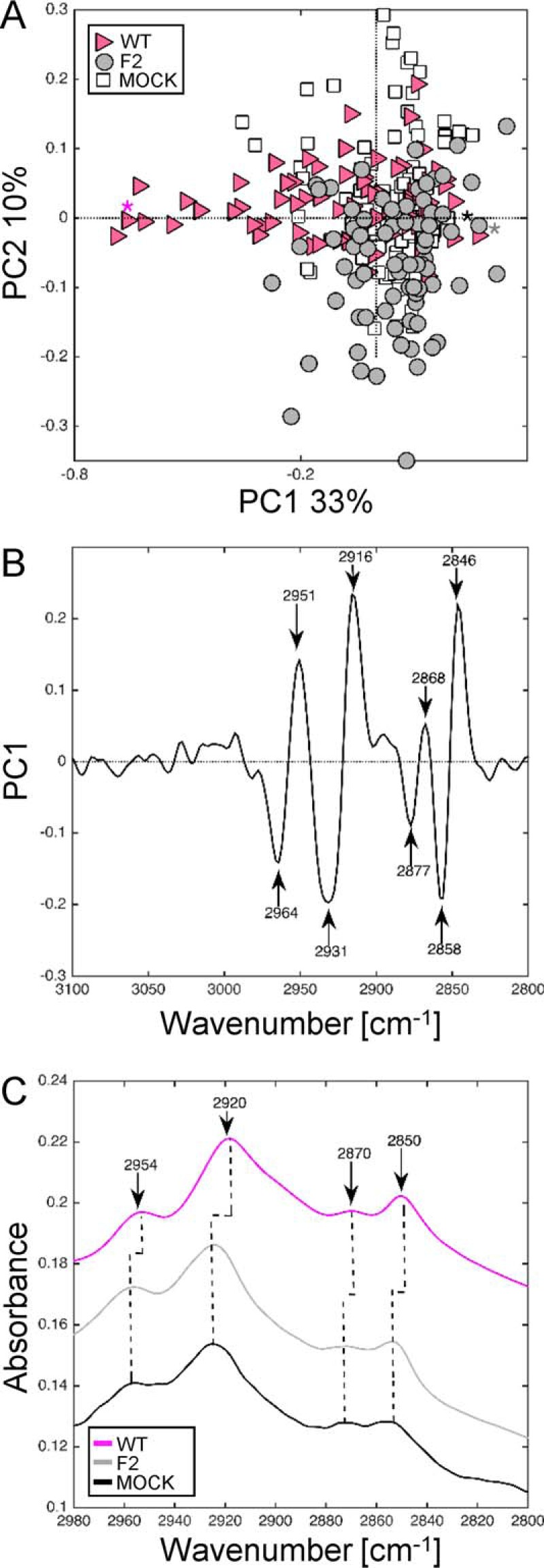
Synchrotron IR microspectroscopy comparison of mock, WT, and F2-infected U937 cells at 8 h post-infection within the CH region. A, score plot of PCA from the 3100–2800 cm−1 band IR spectra. The explained variance for PC1 and PC2 is 33 and 10%, respectively. Marked data with pink triangles correspond to U937 cells infected with WT virus, gray circle to cells infected with F2 virus, and white square to mock-infected cells. B, loading plot linking the variable space and principal component subspace (PC1). PCA score plots show that the WT and F2-mock groups separate along PC1. C, mean IR spectra of mock-infected cells (black line), WT (pink line), and F2-infected cells (gray line) at 8 h post-infection in the 2980–2800 cm−1 range. A shift for the principal peaks corresponding to CH2-CH3 stretching (2954, 2920, and 2850 cm−1) associated to perturbation of membrane fluidity was observed for WT-infected cells spectra compared with mock and F2-infected cells. PC, principal component.
The impact of PB1-F2 expression on membrane compounds of A549-infected cells was assessed by analyzing the same lipid region of the spectra (Fig. 9). IR spectra of A549 cells recorded at 24 h post-infection were submitted to PCA and the resulting score plot showed the samples plotted in a relevant subspace determined by principal components PC1 and PC2 with an explained variance of 87 and 3%, respectively (Fig. 9A). IR spectra of WT-, F2-, and mock-infected A549 cells were separated along PC1 in the negative direction. The corresponding loading plot (Fig. 9B) showed high loading values at 2958, 2924, 2873, and 2846 cm−1 attributed to sCH2/CH3 acyl chains stretching. In contrast to the analysis of IR spectra of infected U937 cells, no shift was observed with the raw spectra of infected A549 cells (Fig. 9C). Interestingly, the band ascribed to stretching vibrations of CH3 acyl chains (sCH3, 2873 cm−1 and asCH3, 2958 cm−1) were more intense for WT than for mock-infected A549 cells. Conversely, the bands assigned to stretching vibrations of CH2 acyl chains (sCH2, 2852 cm−1 and asCH2, 2924 cm−1) were more intense for mock compared with WT-infected A549 cells. This increase of the CH2/CH3 signal intensity ratio has been associated with perturbation of the membranes and correlated with apoptosis feature (53). In our case, this FTIR signature could be assigned to the cytopathic effect generated by IAV infection in epithelial cells.
FIGURE 9.
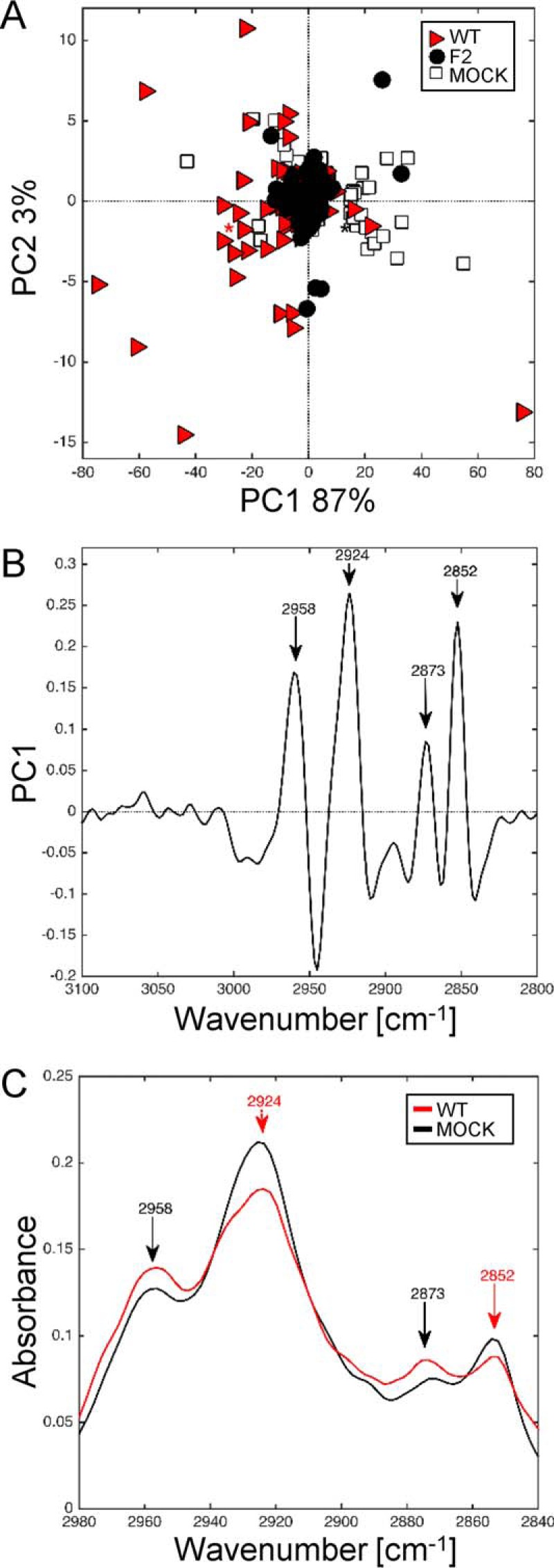
Synchrotron IR microspectroscopy comparison of mock, WT, and F2-infected A549 cells at 24 h post-infection within the CH region. A, score plot of PCA from the 3100–2800 cm−1 band IR spectra. The explained variance for PC1 and PC2 is 87 and 3%, respectively. Marked data with a red triangle correspond to A549 cells infected with WT virus, black circles to cells infected with F2 virus, and white squares to mock-infected cells. B, loading plot linking the variable space and principal component subspace (PC1). PCA score plots show that the WT and F2-mock groups separate along PC1. C, mean IR spectra of mock-infected cells (black line) and WT-infected cells (red line) at 24 h post-infection in the 2980–2840 cm−1 range. The bands assigned to CH3 symmetric/asymmetric (2958 and 2873 cm−1, respectively) were more intense for WT-infected cells than for mock-infected cells. The contrary was observed for bands assigned to CH2 symmetric/asymmetric (2924 and 2852 cm−1, respectively). PC, principal component.
Thus, we showed that PB1-F2 expression differentially affects the membranes in U937 and A549 cells during the infectious cycle. If the effect of PB1-F2 on A549 membranes remains elusive, the loss of membrane fluidity of U937 cells expressing PB1-F2 seems to be clearly correlated with the presence of PB1-F2 β-aggregates.
Discussion
The present work elucidated the structural behavior of PB1-F2 at a single-cell scale in three different cell lines during the viral cycle. To achieve this, combined multimodal synchrotron FTIR and DUV microspectroscopies were used. To our knowledge, this is the first time that such an approach has been used to study the structural behavior of a viral protein in infected cells.
FTIR microspectroscopy is a powerful analytical tool allowing the characterization of biochemical composition of cells and microorganisms. It also allows understanding the molecular basis of their adaptive responses when exposed to stress conditions such as viral infection (54). The use of the bright synchrotron radiation source allows optimizing the signal and provides a very good signal-to-noise ratio and a high spatial resolution to monitor biochemical changes on small-sized biological objects like cells (32, 55). For example, FTIR could rapidly and accurately differentiate between uninfected and virus-infected cells (34). No labels, stains, or dyes are required for FTIR microspectroscopy and it is a non-destructive, sensitive and fast tool. We had previously observed the presence of ThS-positive aggregates in IAV-infected monocytic cells (25). However, we failed to detect a specific ThS positive signal by fluorescent microscopy in WT-infected A549 cells although we proved that oligomerized forms of PB1-F2 were present in such cells using others methods (23, 28). Indeed, ThS staining is subject to the nonspecific signal and does not allow distinguishing between the different oligomerized forms. Thus, a new method and tools were needed to evidence the presence of amyloid aggregates and their impact in different type of cells during the viral cycle. Two spectral regions were studied by FTIR microspectroscopy: the amide I and the CH stretching region.
The amide I band is highly sensitive to protein secondary structures and has been extensively used to measure protein folding, unfolding, and misfolding (36), especially for proteins with a propensity to form β-sheet structures. Generally, β-sheet structures exhibit IR signatures at 1670–1695 and 1630–1640 cm−1, α-helix at 1648–1660 cm−1, and unordered structures at 1640–1657 cm−1 (Table 1). Amyloid fibrils and aggregates present a specific signature ranging from 1615 to 1630 cm−1 that is clearly distinguishable from the IR signature assigned to native β-sheet proteins (39). In WT-infected monocytic cells, an IR signature composed of peaks of high intensity at 1631, 1678, and 1693 cm−1 was observed as early as 8 h post-infection. Although the presence of a single peak at 1693 cm−1 is indicative of anti-parallel β-sheet, the association of this peak with a second peak at 1631 cm−1 is specifically assigned to aggregated β-sheet structures and amyloid fibers (50). The same couple of peaks was found at 24 h post-infection with an increase of the signal suggesting an accumulation of the β-aggregated structures at a later stage of infection. The third peak at 1678 cm−1, present at 8 and 24 h post-infection could be assigned to β-turns and is representative of a global switch from an unordered state to the acquisition of ordered structures. In contrast to monocytic cells, PCA of WT-infected A549 cells did not result in the same specific IR signature ascribed to β-aggregates but revealed two main peaks at 1653 and 1631 cm−1. The peak at 1631 cm−1 could also be associated to the presence of amyloid structures. However, the absence of the second peak at 1693 cm−1 suggested that the β-structures detected were non-aggregated. The recorded signal could be associated to the soluble oligomers that we previously detected in IAV-infected cells (28). The ratio between the peaks at 1653 cm−1/1631 cm−1 decreases at 24 h post-infection arguing for a higher β-sheet and a lower α-helix content in the cells. Unfortunately the separation between WT and F2-infected A549 IR spectra was too weak to associate it unambiguously with the expression of PB1-F2. Indeed, the cytopathic effect induced by IAV in epithelial cells at a late stage of infection could participate to a global enrichment in the β-sheet content of the cells. In a previous study with bacteria, the authors suggested that in dead cells, an increase of the disordered secondary structures could be due to transitions between active (α-helix) and inactive (β-sheet) conformations of the proteins (55). Given that we cannot demonstrate unambiguously that the variance associated to the peak at 1631 cm−1 is only associated to the expression of PB1-F2, we cannot speculate further on the origin of the differences in the spectra. Finally, we took advantage of the high content of Trp residues in the sequence of PB1-F2 to visualize directly by autofluorescence the presence of PB1-F2 aggregates in infected cells and to correlate the quantification of Trp autofluorescence with IR spectra analysis. Thus, we observed a significant increase of the Trp autofluorescence signal only in WT-U937-infected cells. No significant variation of fluorescence was measured in IAV-infected A549 cells at the early stage of infection. We cannot rule out that PB1-F2 partially aggregates in A549 cells but the aggregates signal was too low to be detected at the given spatial resolution. A recent study describes a specific phenomenon of protein amyloids that develop an intrinsic fluorescence during the aggregation process (56). This observation is in accordance with our results: PB1-F2 mainly aggregates in monocytic cells, displaying an increase of fluorescent signal in contrast with the epithelial cells fluorescent signal. Altogether, the results obtained by FTIR and DUV microspectroscopy analyses allowed us to confirm that PB1-F2 is expressed as a monomer at the earlier stage of infection and then oligomerizes to form a preferentially β-aggregated structure that accumulates at later a stage of the viral cycle in U937 cells, whereas PB1-F2 forms β-soluble oligomers in A549 cells but without the final aggregation step.
PB1-F2 was previously shown to oligomerize and create pores in a membrane environment (25). Thus, we focused our attention on the lipid region of the IR-spectra (CH stretching region) to evaluate the impact of PB1-F2 on cell membranes. Synchrotron FTIR microspectroscopy was also shown to be a valuable technique to study modification of the cellular structure (57, 58). The region from 3,010 to 2,800 cm−1 is representative of fatty acids, mostly encountered in cell membranes. Striking differences in cell composition of IAV-infected cells expressing or not PB1-F2 were clearly observed in the CH2/CH3 region but in two different manners: a significant shift of all the asymmetric/symmetric CH2 and CH3 bands for U937 cells, whereas an inversion of the maxima of the asymmetric/symmetric CH2 and CH3 bands was observed for A549 cells. In U937 cells, the correlation loading plot revealed that wavenumbers 2931 and 2858 cm−1 were correlated with PB1-F2 expressing cells, whereas PB1-F2 non-expressing cells (F2 and Mock) were clearly correlated to wavenumbers 2916 and 2846 cm−1. This could be attributed to a downshift of the symmetric CH2 stretching peak. This result demonstrates that the membrane of PB1-F2 expressing cells contained more “trans” unsaturated fatty acids than those of PB1-F2 non-expressing cells suggesting a loss of membrane fluidity (59). It is worth noting that the FTIR signature of PB1-F2 expressing cells correspond to the signature assigned to dead bacteria as shown in a previous study investigating the effect of stress on bacteria and on their membrane fluidity (60). Thus, changes in the CH-stretching region led to the assumption that the expression and aggregation of PB1-F2 may influence the membrane state-of-order via lipid-protein interactions, rendering the lipid biolayer more rigid and thus decreasing the membrane fluidity more generally. Amyloid aggregates are the hallmark of several human diseases and understanding the mechanisms of their cytotoxicity is fundamental. Numerous reports indicated that the proteins and peptides aggregating to form amyloids behave similarly as for their cytotoxic effects (61). The cell susceptibility to face this phenomenon depends on their capacity to accumulate such cytotoxic oligomers and aggregates at their plasma membrane resulting in membrane perturbation and cell damage leading to cell death. In the case of PB1-F2, the presence of lipid membrane influences the oligomerization and aggregation process of the protein and in return PB1-F2 β-aggregates modify the membrane structure and fluidity, which may lead to the permeability of the membrane as previously shown (25). We recently proposed the molecular basis of those membrane/PB1-F2 interactions (62). We showed that PB1-F2 amyloid oligomers are highly cytotoxic and result in membrane disruption and cell death in IAV-infected U937 cells. In contrast, the expression of PB1-F2 in A549 cells did not exacerbate cell death. Nevertheless, a high cytotoxic effect was observed when PB1-F2 amyloid oligomers or pre-fibrils were added to the culture medium. Amorphous structures of PB1-F2 showed no toxicity, confirming that the membrane-lytic activity of PB1-F2 is closely linked to the supramolecular organization of the protein. Thus, the observed shift in IAV-infected immune cells may be the result of cell response to aggregated PB1-F2. It would be worth deciphering such a mechanism to understand the link between PB1-F2 aggregation, membrane perturbation, and enhancement of pathogenicity. Recent work pointed out that the formation of amyloid oligomers and aggregates at the membrane vicinity leads to its structure/function impairment and triggers apoptosis (63). Our results provide the first evidence that the structural behavior of PB1-F2 may be linked to the triggering of apoptosis in immune cells, and not in epithelial cells. Indeed, no shift was observed in PB1-F2 expressing epithelial cells but a specific inversion of the maxima of CH2 and CH3 symmetric/asymmetric stretching peaks. This phenomenon remains poorly documented and difficult to interpret biologically. To summarize, our results show that the impact of PB1-F2 on the cell membrane and the resulting IR signatures of IAV-infected immune or epithelial cells are totally different.
In conclusion, the results obtained in this study are complementary to our previous works and the data provided by the bibliography. Since the first articles focusing on PB1-F2 function (19), PB1-F2 was shown to induce apoptosis in a cell type-specific manner. Indeed monocytes are affected, whereas epithelial cell lines are spared. In contrast, exacerbation of IFN-β expression and the inflammatory response were observed in epithelial cells (23). The different functions of PB1-F2 might be linked to its disordered protein feature, capable to switch from random to α-helical or β-sheet secondary structure, depending on the concentration and localization during infection in a particular cell type (25). In a recent study (20), the authors evidenced that PB1-F2 localizes to the mitochondrial inner membrane space of epithelial cells by interacting with Tom40 and that the PB1-F2 interacting form is a highly ordered oligomer, composed of more than three molecules. On the other hand, another recent study (30) demonstrated that the high molecular weight species of PB1-F2, which might correspond to PB1-F2 fibers and aggregates, activate the NLRP3 inflammasome and induce IL1-β secretion in macrophages. Moreover, the authors showed the rapid phagocytosis of PB1-F2 aggregates and their incorporation in the lysosomal pathway suggesting a release from dying infected cells that may lead to the recruitment of more inflammatory immune cells and thus constitute a feedforward inflammatory circuit. Thus, our data provide new insight into the structural behavior of PB1-F2 and contribute to understand the role of PB1-F2 in IAV pathogenesis. Moreover, an increasing number of disordered proteins are described in a wide range of pathogens including viruses and our approach by FTIR microspectroscopy could serve as a new method to detect such aggregates as a marker of exacerbated pathogenicity.
Author Contributions
C. C., F. J., B. D., and R. L. G. conceived and designed the experiments. C. C., F. J., R. L. G., and O. L. performed the experiments. C. C., F. J., and R. L. G. analyzed the data. M. R. and F. J. contributed reagents/materials/analysis tools. C. C. and F. J. wrote the paper.
Acknowledgments
We thank Stéphane Biacchesi (VIM, INRA, Jouy-en-Josas) and Martine Morzel (UMR1324, INRA, Dijon) for critical reading of the manuscript.
This work was supported in part by Synchrotron SOLEIL, SMIS, and DISCO beamlines proposals 20110735 and 20120904. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding was received for this study. The authors declare that they have no conflicts of interest with the content of this article.
- IAV
- influenza A virus
- sDUV
- Synchrotron deep ultraviolet
- PCA
- principal component analysis.
References
- 1. World Health Organization (2014) Global Influenza Surveillance and Response System (GISRS), http://www.who.int/influenza/gisrs_laboratory/en
- 2. Kawaoka Y. (ed) (2006) Influenza Virology: Current Topics, Caister Academic Press, Wymondham [Google Scholar]
- 3. Wise H. M., Foeglein A., Sun J., Dalton R. M., Patel S., Howard W., Anderson E. C., Barclay W. S., and Digard P. (2009) A complicated message: identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 83, 8021–8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pasricha G., Mishra A. C., and Chakrabarti A. K. (2013) Comprehensive global amino acid sequence analysis of PB1F2 protein of influenza A H5N1 viruses and the influenza A virus subtypes responsible for the 20th-century pandemics. Influenza Other Respir. Viruses 7, 497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zell R., Krumbholz A., Eitner A., Krieg R., Halbhuber K. J., and Wutzler P. (2007) Prevalence of PB1-F2 of influenza A viruses. J. Gen. Virol. 88, 536–546 [DOI] [PubMed] [Google Scholar]
- 6. Chakrabarti A. K., and Pasricha G. (2013) An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses. Virology 440, 97–104 [DOI] [PubMed] [Google Scholar]
- 7. Schrauwen E. J., de Graaf M., Herfst S., Rimmelzwaan G. F., Osterhaus A. D., and Fouchier R. A. (2014) Determinants of virulence of influenza A virus. Eur. J. Clin. Microbiol. Infect. Dis. 33, 479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmolke M., Manicassamy B., Pena L., Sutton T., Hai R., Varga Z. T., Hale B. G., Steel J., Pérez D. R., and García-Sastre A. (2011) Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog. 7, e1002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McAuley J. L., Hornung F., Boyd K. L., Smith A. M., McKeon R., Bennink J., Yewdell J. W., and McCullers J. A. (2007) Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2, 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trifonov V., Racaniello V., and Rabadan R. (2009) The contribution of the PB1-F2 protein to the fitness of influenza A viruses and its recent evolution in the 2009 influenza A (H1N1) pandemic virus. PLoS Curr. 1, RRN1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zamarin D., Ortigoza M. B., and Palese P. (2006) Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol. 80, 7976–7983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alymova I. V., Green A. M., van de Velde N., McAuley J. L., Boyd K. L., Ghoneim H. E., and McCullers J. A. (2011) Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 85, 12324–12333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alymova I. V., Samarasinghe A., Vogel P., Green A. M., Weinlich R., and McCullers J. A. (2014) A novel cytotoxic sequence contributes to influenza A viral protein PB1-F2 pathogenicity and predisposition to secondary bacterial infection. J. Virol. 88, 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Le Goffic R., Leymarie O., Chevalier C., Rebours E., Da Costa B., Vidic J., Descamps D., Sallenave J. M., Rauch M., Samson M., and Delmas B. (2011) Transcriptomic analysis of host immune and cell death responses associated with the influenza A virus PB1-F2 protein. PLoS Pathog. 7, e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leymarie O., Embury-Hyatt C., Chevalier C., Jouneau L., Moroldo M., Da Costa B., Berhane Y., Delmas B., Weingartl H. M., and Le Goffic R. (2014) PB1-F2 attenuates virulence of highly pathogenic avian H5N1 influenza virus in chickens. PLoS ONE 9, e100679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen C. J., Chen G. W., Wang C. H., Huang C. H., Wang Y. C., and Shih S. R. (2010) Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 84, 10051–10062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gibbs J. S., Malide D., Hornung F., Bennink J. R., and Yewdell J. W. (2003) The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 77, 7214–7224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamada H., Chounan R., Higashi Y., Kurihara N., and Kido H. (2004) Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 578, 331–336 [DOI] [PubMed] [Google Scholar]
- 19. Chen W., Calvo P. A., Malide D., Gibbs J., Schubert U., Bacik I., Basta S., O'Neill R., Schickli J., Palese P., Henklein P., Bennink J. R., and Yewdell J. W. (2001) A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7, 1306–1312 [DOI] [PubMed] [Google Scholar]
- 20. Yoshizumi T., Ichinohe T., Sasaki O., Otera H., Kawabata S., Mihara K., and Koshiba T. (2014) Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 5, 4713. [DOI] [PubMed] [Google Scholar]
- 21. Leymarie O., Jouvion G., Hervé P. L., Chevalier C., Lorin V., Lecardonnel J., Da Costa B., Delmas B., Escriou N., and Le Goffic R. (2013) Kinetic characterization of PB1-F2-mediated immunopathology during highly pathogenic avian H5N1 influenza virus infection. PLoS ONE 8, e57894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McAuley J. L., Chipuk J. E., Boyd K. L., Van De Velde N., Green D. R., and McCullers J. A. (2010) PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 6, e1001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le Goffic R., Bouguyon E., Chevalier C., Vidic J., Da Costa B., Leymarie O., Bourdieu C., Decamps L., Dhorne-Pollet S., and Delmas B. (2010) Influenza A virus protein PB1-F2 exacerbates IFN-β expression of human respiratory epithelial cells. J. Immunol. 185, 4812–4823 [DOI] [PubMed] [Google Scholar]
- 24. Bruns K., Studtrucker N., Sharma A., Fossen T., Mitzner D., Eissmann A., Tessmer U., Röder R., Henklein P., Wray V., and Schubert U. (2007) Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza A virus protein. J. Biol. Chem. 282, 353–363 [DOI] [PubMed] [Google Scholar]
- 25. Chevalier C., Al Bazzal A., Vidic J., Février V., Bourdieu C., Bouguyon E., Le Goffic R., Vautherot J. F., Bernard J., Moudjou M., Noinville S., Chich J. F., Da Costa B., Rezaei H., and Delmas B. (2010) PB1-F2 influenza A virus protein adopts a beta-sheet conformation and forms amyloid fibers in membrane environments. J. Biol. Chem. 285, 13233–13243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miodek A., Sauriat-Dorizon H., Chevalier C., Delmas B., Vidic J., and Korri-Youssoufi H. (2014) Direct electrochemical detection of PB1-F2 protein of influenza A virus in infected cells. Biosens. Bioelectron 59, 6–13 [DOI] [PubMed] [Google Scholar]
- 27. Vidic J., Chevalier C., Le Goffic R., Miodek A., Bourdieu C., Richard C.-A., Moudjou M., and Delmas B. (2013) Surface plasmon resonance immunosensor for detection of PB1-F2 influenza A virus protein in infected biological samples. J. Anal. Bioanal. Tech. S7, 006 [Google Scholar]
- 28. Miodek A., Vidic J., Sauriat-Dorizon H., Richard C. A., Le Goffic R., Korri-Youssoufi H., and Chevalier C. (2014) Electrochemical detection of the oligomerization of PB1-F2 influenza A virus protein in infected cells. Anal. Chem. 86, 9098–9105 [DOI] [PubMed] [Google Scholar]
- 29. Vidic J., Le Goffic R., Miodek A., Bourdieu C., Richard C.-A., Moudjou M., Delmas B., and Chevalier C. (2013) Detection of soluble oligomers formed by PB1-F2 influenza A virus protein in vitro. J. Anal. Bioanal. Tech. 4, 169 [Google Scholar]
- 30. McAuley J. L., Tate M. D., MacKenzie-Kludas C. J., Pinar A., Zeng W., Stutz A., Latz E., Brown L. E., and Mansell A. (2013) Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 9, e1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gustot A., Gallea J. I., Sarroukh R., Celej M. S., Ruysschaert J. M., and Raussens V. (2015) Amyloid fibrils are the molecular trigger of inflammation in Parkinson's disease. Biochem. J. 471, 323–333 [DOI] [PubMed] [Google Scholar]
- 32. Jamin N., Dumas P., Moncuit J., Fridman W. H., Teillaud J. L., Carr G. L., and Williams G. P. (1998) Highly resolved chemical imaging of living cells by using synchrotron infrared microspectrometry. Proc. Natl. Acad. Sci. U.S.A. 95, 4837–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller L. M., and Dumas P. (2010) From structure to cellular mechanism with infrared microspectroscopy. Curr. Opin. Struct. Biol. 20, 649–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hastings G., Krug P., Wang R., Guo J., Lamichhane H. P., Tang T., Hsu Y. S., Ward J., Katz D., and Hilliard J. (2009) Viral infection of cells in culture detected using infrared microscopy. Analyst 134, 1462–1471 [DOI] [PubMed] [Google Scholar]
- 35. Kretlow A., Wang Q., Kneipp J., Lasch P., Beekes M., Miller L., and Naumann D. (2006) FTIR-microspectroscopy of prion-infected nervous tissue. Biochim. Biophys. Acta 1758, 948–959 [DOI] [PubMed] [Google Scholar]
- 36. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 37. Barth A., and Zscherp C. (2002) What vibrations tell us about proteins. Q. Rev. Biophys. 35, 369–430 [DOI] [PubMed] [Google Scholar]
- 38. Hiramatsu H., and Kitagawa T. (2005) FT-IR approaches on amyloid fibril structure. Biochim. Biophys. Acta 1753, 100–107 [DOI] [PubMed] [Google Scholar]
- 39. Zandomeneghi G., Krebs M. R., McCammon M. G., and Fändrich M. (2004) FTIR reveals structural differences between native beta-sheet proteins and amyloid fibrils. Protein Sci. 13, 3314–3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jamme F., Villette S., Giuliani A., Rouam V., Wien F., Lagarde B., and Réfrégiers M. (2010) Synchrotron UV fluorescence microscopy uncovers new probes in cells and tissues. Microsc. Microanal. 16, 507–514 [DOI] [PubMed] [Google Scholar]
- 41. Batard E., Jamme F., Villette S., Jacqueline C., de la Cochetière M. F., Caillon J., and Réfrégiers M. (2011) Diffusion of ofloxacin in the endocarditis vegetation assessed with synchrotron radiation UV fluorescence microspectroscopy. PLoS ONE 6, e19440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jamme F., Kascakova S., Villette S., Allouche F., Pallu S., Rouam V., and Réfrégiers M. (2013) Deep UV autofluorescence microscopy for cell biology and tissue histology. Biol. Cell 105, 277–288 [DOI] [PubMed] [Google Scholar]
- 43. Giuliani A., Jamme F., Rouam V., Wien F., Giorgetta J. L., Lagarde B., Chubar O., Bac S., Yao I., Rey S., Herbeaux C., Marlats J. L., Zerbib D., Polack F., and Réfrégiers M. (2009) DISCO: a low-energy multipurpose beamline at synchrotron SOLEIL. J. Synchrotron Radiat. 16, 835–841 [DOI] [PubMed] [Google Scholar]
- 44. Edelstein A., Amodaj N., Hoover K., Vale R., and Stuurman N. (2010) Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol. Chapter 14, Unit 14.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takizawa T., Matsukawa S., Higuchi Y., Nakamura S., Nakanishi Y., and Fukuda R. (1993) Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. J. Gen. Virol. 74, 2347–2355 [DOI] [PubMed] [Google Scholar]
- 46. Goormaghtigh E., Ruysschaert J. M., and Raussens V. (2006) Evaluation of the information content in infrared spectra for protein secondary structure determination. Biophys. J. 90, 2946–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arrondo J. L., and Goñi F. M. (1998) Infrared studies of protein-induced perturbation of lipids in lipoproteins and membranes. Chem. Phys. Lipids 96, 53–68 [DOI] [PubMed] [Google Scholar]
- 48. Wood B. R., Chernenko T., Matthäus C., Diem M., Chong C., Bernhard U., Jene C., Brandli A. A., McNaughton D., Tobin M. J., Trounson A., and Lacham-Kaplan O. (2008) Shedding new light on the molecular architecture of oocytes using a combination of synchrotron Fourier transform-infrared and Raman spectroscopic mapping. Anal. Chem. 80, 9065–9072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rak M., Del Bigio M. R., Mai S., Westaway D., and Gough K. (2007) Dense-core and diffuse Aβ plaques in TgCRND8 mice studied with synchrotron FTIR microspectroscopy. Biopolymers 87, 207–217 [DOI] [PubMed] [Google Scholar]
- 50. Perisic N., Afseth N. K., Ofstad R., and Kohler A. (2011) Monitoring protein structural changes and hydration in bovine meat tissue due to salt substitutes by Fourier transform infrared (FTIR) microspectroscopy. J. Agric. Food Chem. 59, 10052–10061 [DOI] [PubMed] [Google Scholar]
- 51. Movasaghi Z., Rehman S., and Rehman I. U. (2008) Fourier transform infrared (FTIR) spectroscopy of biological tissues. Appl. Spectrosc. Rev. 43, 134–179 [Google Scholar]
- 52. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blankenberg F. G., Katsikis P. D., Storrs R. W., Beaulieu C., Spielman D., Chen J. Y., Naumovski L., and Tait J. F. (1997) Quantitative analysis of apoptotic cell death using proton nuclear magnetic resonance spectroscopy. Blood 89, 3778–3786 [PubMed] [Google Scholar]
- 54. Pénicaud C., Landaud S., Jamme F., Talbot P., Bouix M., Ghorbal S., and Fonseca F. (2014) Physiological and biochemical responses of Yarrowia lipolytica to dehydration induced by air-drying and freezing. PLoS ONE 9, e111138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Saulou C., Jamme F., Girbal L., Maranges C., Fourquaux I., Cocaign-Bousquet M., Dumas P., and Mercier-Bonin M. (2013) Synchrotron FTIR microspectroscopy of Escherichia coli at single-cell scale under silver-induced stress conditions. Anal. Bioanal. Chem. 405, 2685–2697 [DOI] [PubMed] [Google Scholar]
- 56. Chan F. T., Kaminski Schierle G. S., Kumita J. R., Bertoncini C. W., Dobson C. M., and Kaminski C. F. (2013) Protein amyloids develop an intrinsic fluorescence signature during aggregation. Analyst 138, 2156–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Romeo M., Mohlenhoff B., Jennings M., and Diem M. (2006) Infrared micro-spectroscopic studies of epithelial cells. Biochim. Biophys. Acta 1758, 915–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jamme F., Vindigni J. D., Méchin V., Cherifi T., Chardot T., and Froissard M. (2013) Single cell synchrotron FT-IR microspectroscopy reveals a link between neutral lipid and storage carbohydrate fluxes in S. cerevisiae. PLoS ONE 8, e74421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Loffhagen N., Hartig C., Geyer W., Voyevoda M., and Harms H. (2007) Competition between cis, trans, and cyclopropane fatty acid formation and its impact on membrane fluidity. Eng. Life Sci. 7, 67–74 [Google Scholar]
- 60. Moen B., Janbu A. O., Langsrud S., Langsrud O., Hobman J. L., Constantinidou C., Kohler A., and Rudi K. (2009) Global responses of Escherichia coli to adverse conditions determined by microarrays and FT-IR spectroscopy. Can. J. Microbiol. 55, 714–728 [DOI] [PubMed] [Google Scholar]
- 61. Bucciantini M., and Cecchi C. (2010) Biological membranes as protein aggregation matrices and targets of amyloid toxicity. Methods Mol. Biol. 648, 231–243 [DOI] [PubMed] [Google Scholar]
- 62. Vidic J., Richard C. A., Péchoux C., Da Costa B., Bertho N., Mazerat S., Delmas B., and Chevalier C. (2016) Amyloid assemblies of influenza A virus PB1-F2 protein damage membrane and induce cytotoxicity. J. Biol. Chem. 291, 739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bucciantini M., Nosi D., Forzan M., Russo E., Calamai M., Pieri L., Formigli L., Quercioli F., Soria S., Pavone F., Savistchenko J., Melki R., and Stefani M. (2012) Toxic effects of amyloid fibrils on cell membranes: the importance of ganglioside GM1. FASEB J. 26, 818–831 [DOI] [PubMed] [Google Scholar]