

Abstract
Tissue transglutaminase (tTG) is an acyltransferase/GTP-binding protein that contributes to the development of various diseases. In human cancer cells, tTG activates signaling pathways that promote cell growth and survival, whereas in other disorders (i.e. neurodegeneration), overexpression of tTG enhances cell death. Therefore, it is important to understand how tTG is differentially regulated and functioning to promote diametrically distinct cellular outcomes. Previous structural studies revealed that tTG adopts either a nucleotide-bound closed conformation or a transamidation-competent open conformation. Here we provide evidence showing that these different conformational states determine whether tTG promotes, or is detrimental to, cell survival, with the open conformation of the protein being responsible for inducing cell death. First, we demonstrate that a nucleotide binding-defective form of tTG, which has previously been shown to induce cell death, assumes an open conformation in solution as assessed by an enhanced sensitivity to trypsin digestion and by small angle x-ray scattering (SAXS) analysis. We next identify two pairs of intramolecular hydrogen bonds that, based on existing x-ray structures, are predicted to form between the most C-terminal β-barrel domain and the catalytic core domain of tTG. By disrupting these hydrogen bonds, we are able to generate forms of tTG that constitutively assume an open conformation and induce apoptosis. These findings provide important insights into how tTG participates in the pathogenesis of neurodegenerative diseases, particularly with regard to the actions of a C-terminal truncated form of tTG (TG-Short) that has been linked to such disorders and induces apoptosis by assuming an open-like conformation.
Keywords: apoptosis, cell death, conformational change, protein cross-linking, small-angle x-ray scattering (SAXS), transglutaminase
Introduction
tTG2 is a member of a family of enzymes that are best known for their ability to catalyze a calcium (Ca2+)-dependent transamidation reaction that results in the formation of an amide bond between a glutamine residue in one protein and a primary amino group in another protein or polyamine (1). It is also capable of binding and hydrolyzing GTP, similar to other GTP-binding proteins (2, 3). The activities of tTG have been implicated in the regulation of several physiological processes including cellular differentiation, endocytosis, wound healing, and cell adhesion (3–8). However, aberrant tTG expression is also a hallmark of several human diseases (9–17). For example, increases in tTG expression are frequently observed in the most malignant and high grade brain and breast cancers, where it has been show to play important roles in promoting cell growth, survival, and therapy resistance (9–13). Interestingly and perhaps counterintuitively, tTG levels are also up-regulated in Alzheimer, Parkinson, and Huntington diseases, where its enzymatic transamidation activity is believed to contribute to the protein aggregation events and cytotoxicity that are characteristic of these neurodegenerative disorders (14–17). Therefore, understanding the mechanisms through which tTG influences cellular events that give rise to cell survival in some circumstances, and cell death in others, is of great interest, especially as targeting tTG has been proposed as a strategy to treat cancer and neurodegenerative disorders (10, 11, 18–20).
Given the wide range of roles played by tTG in normal and disease state contexts, it is not surprising to find that both the levels of tTG expression and activation are tightly regulated. tTG is typically expressed at low levels in most normal cell types as well as in low grade, non-aggressive cancer cells, whereas its expression can be up-regulated by prolonged growth factor stimulation or upon the induction of cellular differentiation and in response to cell stresses (4, 9, 10, 12, 21). Once expressed, the transamidation and GTP binding activities of tTG are subjected to further regulation (1, 3, 17, 22, 23). Structural studies have shown that the binding of GTP or GDP to tTG causes it to adopt a “closed” conformation that prevents it from catalyzing transamidation (i.e. protein cross-linking) as an acyltransferase (24). In the closed conformation, the C terminus of tTG folds over onto itself and blocks substrate access to the transamidation active site located within the catalytic core domain (3). However, under conditions that result in increases in intracellular Ca2+ concentrations, such as commonly occurs after cell stresses (25), tTG has a weakened binding affinity for GTP or GDP (3, 22, 23), causing the C terminus of tTG to move away from the active site (26). Therefore, this “open” conformation of tTG is enzymatically active and capable of catalyzing protein cross-linking reactions.
There have been reasons to suspect that the two distinct conformational states of tTG are responsible for the profoundly different cellular effects attributed to the protein (3, 22, 27). For example, the guanine nucleotide-bound state of tTG has been implicated in cellular transformation (10, 27, 28). The ectopic expression of wild type (WT) tTG (tTG WT), which would be expected to be bound to GTP, in NIH3T3 fibroblasts protects them from serum starvation-induced apoptosis by stimulating the activation of PI 3-kinase (28). Moreover, our laboratory has also recently shown that GTP-bound tTG contributes to the aggressiveness of high grade brain tumors by binding to the E3 ubiquitin ligase c-Cbl and interfering with its ability to properly target the epidermal growth factor receptor (EGFR) for lysosomal degradation (10). The build-up of EGFRs on the surfaces of brain tumor cells promotes their growth and resistance to chemotherapy and radiation. Importantly, tTG can only activate PI 3-kinase and bind c-Cbl when it is capable of binding GTP, thus suggesting that the guanine nucleotide-bound closed conformation is necessary for these effects (10, 28).
However, when tTG is incapable of binding GTP (i.e. as an outcome of a point-mutation), it has an adverse effect on cells (22, 27, 29). Perhaps the best example of this came from a study that showed ectopically expressing a GTP binding-defective form of tTG (tTG R580K), which presumably adopts an open conformation, induces normal as well as cancer cell lines to undergo apoptosis (22). Although this outcome was initially suspected to be due to the up-regulated enzymatic transamidation activity exhibited by the tTG R580K mutant, it was subsequently shown not to be the case. Specifically, changing an essential cysteine residue in the active site to a valine (30) within the tTG R580K background, thus generating a form of tTG that is incapable of catalyzing protein cross-linking, still induced cell death (22). Consistent with these findings, an alternatively spliced form of tTG called tTG-Short, which is often detected in the brains of Alzheimer disease patients (31–33) and exhibits greatly reduced transamidation activity (34), has also been shown to induce cell death (32, 34). The fact that tTG-Short lacks a significant portion of the C terminus (33, 34) such that it is incapable of effectively binding guanine nucleotide, would suggest that it adopts an open conformational state.
When taken together, these different findings support the idea that the different conformational states of tTG (i.e. the closed versus open states) represent the underlying basis for its markedly distinct functions on cell survival. To test this idea, we set out to gain a more detailed understanding of the molecular basis by which tTG assumes these two distinct conformations. To this end we show that the truncated splice variant of tTG, tTG-Short (33), and a GTP binding-defective form of tTG, tTG R580K (22), are cytotoxic and indeed adopt conformations that resemble the open state in solution, as assessed by SAXS analysis, whereas, tTG WT assumes the closed conformation. We then set out to identify important intramolecular interactions that help to maintain tTG WT in a closed conformational state. We discovered two pairs of hydrogen bonds that formed between the C-terminal 15 amino acids of tTG and its catalytic core domain, which were essential for maintaining tTG in the closed conformation. When these interactions were disrupted by site-directed mutagenesis, tTG assumed the open conformation and induced cell death. Thus, these findings demonstrate that it is the ability of tTG to assume the open conformation for extended periods of time that is severely detrimental to cell survival.
Experimental Procedures
Reagents
The Myc and HA antibodies were from Covance, and the 5-(biotinamido) pentylamine (BPA) and horseradish peroxidase-conjugated streptavidin were from Pierce. The tTG and actin antibodies, 4′,6-diamidino-2-phenylindole (DAPI), GTP, BODIPY FL GTPγS, trypsin, retinoic acid (RA), monodansylcadaverine, Coomassie Brilliant Blue Dye, and pre-cast 4–20% Tris glycine acrylamide gels were obtained from Thermo Fisher Scientific. The Z-DON was from Zeidira, and the polyvinylidene difluoride (PVDF) transfer membrane and the Western Lightning Plus ECL reagent were from PerkinElmer Life Sciences. The mouse and rabbit IgG horseradish peroxidase-conjugated secondary antibodies were from Cell Signaling. The Oregon green 488-conjugated and Rhodamine Red-conjugated secondary antibodies as well as all the cell culture reagents including Dulbecco's modified Eagle's medium (DMEM), calf serum (CS), and Lipofectamine were purchased from Invitrogen.
Constructs and Site-directed Mutagenesis
The Myc-tagged and HA-tagged pcDNA3 mammalian expression constructs and the His-tagged pET-28a bacterial expression constructs encoding tTG WT, tTG-Short, tTG R580K, and tTG R580K/C277V were generated as previously described (22, 34). The additional tTG mutants used in the study were prepared by using the QuikChange Site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. For each mutant generated, a PCR reaction was performed using a pair of oligonucleotide primers containing the desired mutation and an expression plasmid encoding tTG WT as the template. The PCR reactions were digested with Dpn1 restriction enzyme to linearize the template plasmid DNA before being transformed into DH5α-competent cells. All mutations were confirmed by DNA sequencing.
Recombinant Proteins
The expression and purification of recombinant tTG WT, tTG-Short, and the various tTG mutants were carried-out as previously described (22).
BODIPY-GTPγS Binding Assays
The purified recombinant tTG proteins (600 nm final concentration) were added to nucleotide-binding reaction buffer (50 mm Tris-HCl, pH 7.4, 2 mm DTT, and 1 mm EDTA), and fluorescent measurements were made using a Varian Eclipse spectrofluorimeter. The excitation and emission wavelengths were set at 504 and 520, respectively. BODIPY FL GTPγS (1 mm final concentration) was added to the reaction incubations, and changes in fluorescence were measured. After maximal binding of BODIPY FL GTPγS to tTG proteins was attained, an excess of unlabeled GTP (30 mm final concentration) was added to each reaction to compete off the BODIPY FL GTPγS bound to the recombinant tTG proteins.
Trypsin Digestion Assays
The purified recombinant tTG proteins (3.5 μg) were added to trypsin digestion reaction buffer (20 mm Tris-HCl, pH 7.4, 300 mm NaCl, and 10% glycerol) containing 80 ng of trypsin. The reactions were carried out on ice for 2 h, at which point Laemmli sample buffer was immediately added to each reaction incubation and boiled. The samples were then resolved by SDS-PAGE, and the gels were stained with Coomassie Brilliant Blue dye to visualize the proteins.
SAXS Data Collection and Analysis
SAXS data collection was carried out at the Cornell High Energy Synchrotron Source (CHESS) on beamline F2 at an electron energy of 10 keV. Protein samples were prepared in 20 mm HEPES buffer, pH 7.4, containing 300 mm NaCl, 2% glycerol, and 0.5 mm tris(2-carboxyethyl)phosphine at concentrations between 1 and 10 mg/ml. Ten data frames were collected for each sample as well as for the corresponding control buffer solution. The data were corrected for background scattering, averaged, and scaled using the program BioXtas (35). Guinier plots were used to estimate Rg values, and Kratky plots were analyzed to assess the folded state of the proteins and overall data quality. Only data showing no signs of radiation damage were used for further analysis. Due to concentration-dependent protein aggregation, only samples with concentrations between 1 and 2.5 mg/ml were used to compose the scattering curves in the low-q range. Data processing was carried out using GNOM (36, 37) and CRYSOL (38). An indirect Fourier transform (IFT) method, as implemented in the GNOM program, was used to determine the intraparticle distance distribution function, p(r), Dmax, the radius of gyration of the protein, Rg, the integral properties of p(r), and the extrapolated forward scattering intensity, I (q = 0), from the experimental scattering data profiles. Using the p(r) function as the target, SAXS-based reconstructions were carried out with DAMMIN (39). Ten independent models of each were calculated and averaged using DAMAVER (40). The envelopes of the reconstructed shapes were superposed onto their respective models with SUBCOMB (41). Further optimization of the positioning and orientation of the proteins was performed using SASREF (42), where the components of the monomer/dimer equilibrium of tTG (PDB codes 1KV3 and 2Q3Z) were treated as rigid bodies and the modeling of domains to the SAXS data were performed through a simulated annealing protocol. SASREF simulations were computed and compared visually. CRYSOL was used to calculate the theoretical scattering curves for models in which the subunits were manually positioned.
Cell Culture and Preparation of Whole Cell Lysates
NIH3T3 fibroblasts were grown in DMEM containing 10% CS. To express the different tTG proteins in cells, Myc-tagged and HA-tagged pcDNA3 constructs encoding tTG WT, tTG-Short, and the various mutant forms of tTG were introduced into cells using Lipofectamine according to the manufacture's instructions. For RA treatment, plates of NIH3T3 fibroblasts were cultured in DMEM containing 2% CS supplemented without or with 1.0 μm RA for the indicated lengths of time. Preparing whole cell lysates involved rinsing the cells with phosphate-buffered saline (PBS) and then lysing them with cell lysis buffer (25 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1 mm DTT, 1 mm Na3VO4, 10 mm β-glycerol phosphate, and 1 μg/ml each of leupeptin and aprotinin). The Bio-Rad DC protein assay was used to determine the protein concentrations of the whole cell lysates.
Apoptotic Assays
Cells ectopically expressing either the vector alone or the indicated combinations of HA-tagged and Myc-tagged tTG proteins were cultured in DMEM medium supplemented without (serum-starved) or with 2% CS, 50 μm monodansylcadaverine, or 5 μm Z-DON, as indicated. Approximately 36 h later the cells were fixed with 3.7% formaldehyde, permeabilized with 0.2% Triton X-100, blocked in PBS containing 10% bovine serum albumin (BSA), and then incubated with the Myc and HA antibodies. Oregon green 488-conjugated and Rhodamine Red-conjugated secondary antibodies were used to detect the Myc and HA antibodies. The cells were also stained with DAPI (1 μg/ml) to label nuclei before being visualized using a Zeiss Axioscop with a 40× objective lens. Transfectants undergoing apoptosis were identified by nuclear condensation and blebbing. Images of cells were taken using a Sensicam qe camera (Cooke Co.) and processed using IPLab software (BD Biosciences).
Immunoblot Analysis
Whole cell lysates (40 μg of each) were resolved by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked in 10% BSA diluted in TBST (20 mm Tris, pH 7.4, 130 mm NaCl, and 0.02% Tween 20) for 1 h and then incubated in primary antibodies diluted in TBST overnight at 4 °C. Horseradish peroxidase-conjugated secondary antibodies were used to detect the primary antibodies followed by exposure to ECL reagent.
Transamidation Activity Assays
Cell extracts (10 μg of each) collected from NIH3T3 fibroblasts transiently expressing either the vector alone or a Myc-tagged form of tTG WT, tTG-Short, or one of the tTG mutants were incubated in a buffer containing 10 mm DTT, 10 mm CaCl2, and 50 μm BPA for 10 min. The reactions were stopped by the addition of Laemmli sample buffer followed by boiling. Each sample was resolved by SDS-PAGE, transferred to PVDF membranes, and blocked in BBST (100 mm boric acid, 20 mm sodium borate, 0.01% SDS, 0.01% Tween 20, and 80 mm NaCl) containing 20% BSA. The membranes were incubated with HRP-conjugated streptavidin diluted in BBST containing 10% BSA for 45 min followed by extensive washing with BBST. The proteins that incorporated BPA were visualized on x-ray film after exposing the membranes to ECL reagent.
Results
tTG Can Mediate Distinct Effects on Cell Viability
How tTG promotes cell growth and survival in certain contexts (i.e. cancer) and cell death in others (i.e. neurodegenerative diseases) has become a pervasive question in the field (3, 27). One possibility is that the opposing cellular actions attributed to tTG are due to its ability to adopt two markedly distinct conformational states (24, 26). tTG WT is composed of four major domains: an N-terminal β-sandwich domain, a catalytic core domain, and two β-barrel domains (Fig. 1A, top panel). Previous x-ray crystallographic studies (24) revealed that when tTG WT is bound to GTP or GDP, it adopts a closed conformation where the C-terminal β-barrels fold over and cover its catalytic core domain (Fig. 1B, left panel, PDB code 1KV3). In this closed state substrate access to the transamidation active site is blocked, preventing tTG from functioning as an acyltransferase capable of catalyzing the cross-linking of proteins (3). However, when tTG WT is not bound to nucleotide (GTP or GDP) but is covalently modified with a substrate-mimetic gluten peptide, it adopts a much more extended or open conformation (Fig. 1B, middle panel, PDB code 2Q3Z) (26). In this case, the C-terminal β-barrel domains of tTG have moved away from the catalytic core domain, allowing substrate access to the transamidation active site. Therefore, the open conformation represents the enzymatically active state of tTG.
FIGURE 1.
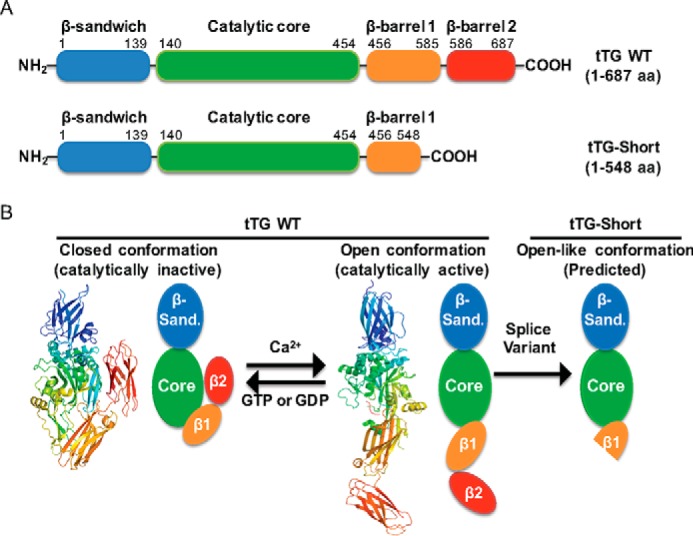
tTG adopts two different conformational states. A, linear representations of tTG WT (top) and tTG-Short (bottom). tTG WT is composed of four domains including an N-terminal β-sandwich (blue) and a catalytic core (green) followed by β-barrel 1 (orange) and β-barrel 2 (red) at its C terminus. tTG-Short is an alternative splice variant of tTG that lacks approximately the last third of β-barrel 1 and all of β-barrel 2. The numbers indicate the amino acids that encode each domain. B, the x-ray crystal structures and simplified diagrams show the closed (catalytically inactive) and open (catalytically active) conformations of tTG WT as well as the open-like conformation that tTG-Short is suspected to assume. The x-ray crystal structure of the closed conformation of tTG WT (far left) represents the GDP-bound form of the protein (PDB code 1KV3), whereas the structure shown for the open state conformation (center) represents tTG WT covalently modified with a substrate-mimetic gluten peptide (PDB code 2Q3Z). GDP and the gluten peptide are shown in sticks and colored in blue and red, respectively. The simplified diagrams show how the different domains in tTG WT are arranged in the closed (far left) versus open (middle) conformations as well as are used to highlight the suspected conformation of tTG-Short (far right). The various domains in tTG are labeled as follows; the N-terminal β-sandwich (β-sand.), the catalytic core (Core), and the two C-terminal β-barrels (β1 and β2).
To investigate whether the conformational changes that tTG can undergo are indeed responsible for its ability to mediate diametrically opposing cellular outcomes, we started by determining the effects of ectopically expressing Myc-tagged forms of tTG WT and tTG-Short, a splice variant of tTG that is truncated from the C terminus (Fig. 1A, bottom panel), in NIH3T3 fibroblasts. These non-transformed, immortalized cells express undetectable levels of tTG (Fig. 2A, lane labeled Untreated), thus enabling an unambiguous assessment of the activity and function of any ectopically expressed tTG isoform or mutant. However, it is worth noting that expression of the full-length form of tTG (tTG WT), but not its shorter counterpart (tTG-Short), can be up-regulated in NIH3T3 fibroblasts by treatment with the differentiation factor RA (Fig. 2A, lanes labeled RA Treatment), highlighting that the mechanisms regulating the expression of these two tTG isoforms are unique. Immunoblot analysis was then performed to determine the relative amounts of Myc-tagged tTG WT and Myc-tagged tTG-Short that are expressed in NIH3T3 fibroblasts as a function of time. Fig. 2B (lanes labeled tTG WT) shows that Myc-tagged tTG WT was readily detectable 24 h after the cells were transfected, and its levels remained high for the duration of the experiment. This was in sharp contrast to Myc-tagged tTG-Short expression, which was maximal at 24 h and then rapidly declined (Fig. 2B, lanes labeled tTG-Short).
FIGURE 2.

tTG WT and tTG-Short differently influence cell survival. A, cell lysates collected from NIH3T3 fibroblasts that were either treated with RA for the indicated lengths of time (left panels) or ectopically expressing the vector alone, Myc-tagged tTG WT, or Myc-tagged tTG-Short for ∼36 h (right panels) were immunoblotted (IB) with tTG and actin antibodies. B, cell lysates collected from NIH3T3 fibroblasts ectopically expressing either the vector alone for 36 h or Myc-tagged forms of either tTG WT and tTG-Short for the indicated lengths of time were immunoblotted with Myc and actin antibodies. C, NIH3T3 cells ectopically expressing the vector alone, Myc-tagged tTG WT, and Myc-tagged tTG-Short were either cultured in medium containing 2% CS or serum-starved (SS) for ∼36 h and fixed. Immunofluorescence (IF) was performed on the cells using a Myc antibody to detect the transfectants. The cells were also stained with DAPI to label nuclei. Representative images of the transfectants and their nuclei are shown. Arrows highlight the nuclei of a given transfectant, whereas asterisks indicate condensed/blebbed (apoptotic) nuclei. D, the experiment shown in C was performed three separate times, and the percent apoptosis for each condition was determined by calculating the ratio of apoptotic to non-apoptotic cells. The data shown represent the means ± S.E. E, cell lysates collected from NIH3T3 cells ectopically expressing either the vector alone for 48 h or the indicated combinations of Myc-tagged forms of tTG WT and tTG-Short for 24 and 48 h were immunoblotted with Myc and actin antibodies. F, cells ectopically expressing the vector alone, Myc-tagged tTG WT, HA-tagged tTG-Short, or Myc-tagged tTG WT and HA-tagged tTG-Short were cultured in medium containing 2% CS for ∼36 h and fixed. Immunofluorescence was performed on the cells using Myc and HA antibodies to detect the transfectants. The cells were also stained with DAPI to label nuclei. The percent of cells with condensed/blebbed (apoptotic) nuclei for each condition was determined by calculating the ratio of apoptotic to non-apoptotic cells. The experiment was performed three separate times, and the data shown represent mean ± S.E.
We then examined the ability of the tTG isoforms to influence cell viability. Serum starvation is a stress known to induce cell death (9, 22, 28, 34). Thus, we took NIH3T3 fibroblasts ectopically expressing the vector alone or Myc-tagged forms of tTG WT or tTG-Short and placed them in medium supplemented without (serum-starved) or with 2% CS for 36 h (a time point where Myc-tagged tTG-Short levels were clearly declining). The cells were stained with a Myc antibody to identify the transfectants, and DAPI was used to label nuclei. The cells expressing vector alone underwent serum starvation-induced cell death, as readout by the appearance of cells with condensed and/or blebbed nuclei (Fig. 2C, column labeled Vector, SS, and Fig. 2D, compare the first two bars). Expression of Myc-tagged tTG WT reduced the amount of cell death caused by serum starvation by ∼6-fold compared with the control cells, i.e. expressing vector alone (Fig. 2D, compare the second and fourth bars), whereas Myc-tagged tTG-Short caused an ∼2-fold increase (Fig. 2D, compare the second and sixth bars). Even when cultured in medium containing serum, the Myc-tagged tTG-Short expressing cells appeared small and rounded (Fig. 2C, column labeled tTG-Short, 2% CS) and underwent cell death at a rate similar to that for cells expressing vector alone that had been serum-starved (Fig. 2D, compare the second and fifth bars). Thus, the time-dependent reduction in tTG-Short expression is most likely the outcome of negative selection due to its cytotoxic effects.
We then determined how the ability of tTG-Short to promote cell death was affected by tTG WT. NIH3T3 cells ectopically expressing Myc-tagged forms of tTG WT, tTG-Short, or tTG WT and tTG-Short were cultured in medium containing 2% CS for different lengths of time and lysed. Immunoblot analysis carried-out on the cell extracts again showed that tTG-Short expression was rapidly reduced to undetectable levels by 48 h (Fig. 2E, lane labeled tTG-Short, 48 h). However, when expressed together with tTG WT, the decrease in tTG-Short levels was slowed such that it was still detectable after 48 h of expression (Fig. 2E, compare the fifth and seventh lanes). Consistent with these findings, the expression of tTG WT consistently reduced the amount of cell death caused by tTG-Short by half (Fig. 2F, compare the third and fourth bars), further supporting the idea that these two forms of tTG have antagonistic effects on cell viability.
We generated purified recombinant forms of tTG WT and tTG-Short (Fig. 3A, lanes labeled tTG WT and tTG-Short) and assayed their ability to bind the fluorescent GTP analog, BODIPY-GTP, and to catalyze transamidation (cross-linking) reactions. As previously shown (22, 23), tTG WT was able to rapidly bind BODIPY-GTP, and this binding could be reversed by the addition of excess unlabeled GTP (Fig. 3B, blue line). tTG WT also catalyzed the Ca2+-dependent incorporation of BPA into lysate proteins (Fig. 3C, lane labeled tTG WT), demonstrating that it is fully functional. On the other hand, tTG-Short failed to bind BODIPY-GTP (Fig. 3B, green line) or catalyze transamidation activity to any appreciable degree (Fig. 3C, lane labeled tTG-Short).
FIGURE 3.

GTP binding-defective forms of tTG induce cell death. A, the indicated recombinant forms of tTG were expressed, purified, and then resolved by SDS-PAGE. The gels were stained with Coomassie Blue to visualize the proteins. A protein molecular mass (M.W.) marker was included on the gel to show the sizes of the recombinant proteins. B, the recombinant tTG proteins (600 nm) were subjected to fluorescent measurements using a spectrofluorimeter. BODIPY FL GTPγS was added to the reactions (labeled on the graph as BODIPY-GTP Added), and changes in fluorescence were determined. At the indicated times, an excess of unlabeled GTP was added to the reactions (labeled on the graph as GTP Added). These experiments were performed at least three separate times, each yielding similar results. C, cell lysates collected from NIH3T3 fibroblasts ectopically expressing the indicated forms of Myc-tagged tTG were assayed for their enzymatic transamidation activity as read out by the incorporation of BPA into cell lysate proteins (top panel). The same lysates were also immunoblotted (IB) with Myc and actin antibodies (bottom two panels). D, cell lysates collected from NIH3T3 fibroblasts ectopically expressing the vector-alone for 36 h and either Myc-tagged tTG WT or Myc-tagged tTG R580K for the indicated lengths of time were immunoblotted with Myc and actin antibodies. E, NIH3T3 cells ectopically expressing the vector alone or one of the indicated Myc-tagged tTG proteins were cultured in medium containing 10% CS for ∼36 h and fixed. Immunofluorescence was performed on the cells using a Myc antibody to detect the transfectants, and the cells were also stained with DAPI to label nuclei. Cells undergoing apoptosis were identified by nuclear condensation/blebbing. The experiment was performed three separate times, and the percent apoptosis for each condition was determined by calculating the ratio of apoptotic to non-apoptotic cells. The data shown represent the mean ± S.E. F, NIH3T3 cells ectopically expressing the vector alone or one of the indicated Myc-tagged tTG proteins were cultured in medium containing 2% CS without (−) or with either monodansylcadaverine or Z-DON for ∼36 h and fixed. Immunofluorescence was performed on the cells using a Myc antibody to detect the transfectants, and the cells were also stained with DAPI to label nuclei. Cells undergoing apoptosis were identified by nuclear condensation/blebbing. The experiment was performed three separate times, and the percent apoptosis for each condition was determined by calculating the ratio of apoptotic to non-apoptotic cells. The data shown represent mean ± S.E.
Mutant forms of tTG that are defective in GTP binding have been reported to have deleterious effects on cells (22). One such mutant, tTG R580K, in which an arginine residue essential for binding GTP is changed to a lysine, was generated in Escherichia coli as a recombinant protein (Fig. 3A, lane labeled tTG R580K) and shown to have completely lost its affinity for BODIPY-GTP (Fig. 3B, purple line). When ectopically expressed in actively growing NIH3T3 fibroblasts (i.e. the cells were cultured in DMEM with 10% CS), a time-dependent selection against Myc-tagged tTG R580K expression was observed (Fig. 3D, lanes labeled tTG R580K). Nearly 25% of the cells expressing this GTP binding-defective form of tTG for 36 h were undergoing apoptosis (Fig. 3E, compare the first and the fourth bars), similar to what was observed in cells expressing Myc-tagged tTG-Short (Fig. 3E, compare the third and fourth bars).
Given that a loss of nucleotide binding capability enhances the transamidation activity exhibited by the tTG R580K mutant, compared with tTG WT (Fig. 3C, compare the lanes labeled tTG R580K and tTG WT), we wanted to verify that the apoptotic-inducing effect of tTG R580K was not a result of its uncontrolled cross-linking of proteins that are essential for maintaining cell viability. Thus, an additional mutation was made in the background of tTG R580K that inhibited its enzymatic transamidation activity by changing an essential active site cysteine (30) to valine (Fig. 3A, lane labeled tTG R580K/C277V). The tTG R580K/C277V double-mutant lacks both GTP binding (Fig. 3B, red line) and transamidation activity (Fig. 3C, lane labeled tTG R580K/C277V). When introduced into NIH3T3 fibroblasts, it induced cell death to a similar extent as tTG R580K (Fig. 3E, compare the fourth and fifth bars). Likewise, treating cells expressing tTG-Short or tTG R580K with the transamidation inhibitors monodansylcadaverine and Z-DON did not significantly alter the extent of cell death caused by these tTG constructs (Fig. 3F, compare the fourth through sixth bars and the seventh through ninth bars), suggesting that the excessive transamidation activity associated with tTG R580K is not required for its ability to kill cells.
The tTG R580K Mutant Adopts an Open Conformational State
We suspected that the nucleotide binding-defective form of tTG (i.e. tTG R580K) would assume an open conformation. To confirm whether this is the case, we first performed limited trypsin proteolysis on the purified recombinant forms of tTG WT and tTG R580K and then resolved the samples by SDS-PAGE. Although recombinant tTG WT is largely insensitive to trypsin cleavage (Fig. 4A, compare the first two lanes), tTG R580K showed increased sensitivity to protease digestion (Fig. 4A, compare the third and fourth lanes). These results supported the idea that tTG WT primarily adopts a closed, more compact conformation that is insensitive to proteolysis and that tTG R580K adopts an open conformation that is more accessible for trypsin digestion.
FIGURE 4.

tTG R580K and tTG-Short adopt open conformations in solution. A, purified recombinant forms of tTG WT and tTG R580K (3.5 μg of each) were incubated without or with trypsin for 2 h before being resolved by SDS-PAGE and stained with Coomassie Blue to visualize the proteins. Note that the tTG R580K mutant is more susceptible to trypsin digestion than tTG WT. B, SAXS analysis was performed on recombinant tTG WT. A monomeric model derived from the crystal structure of GDP-bound wild type tTG (PDB code 1KV3) was fitted into the calculated SAXS envelope, with the N-terminal β-sandwich and the catalytic core domains colored in blue and the two C-terminal β-barrel domains colored in red (left panel). A schematic representation shows the superimposition of the fitted model (blue and red) onto the structure of wild type tTG bound to GDP (PDB code 1KV3, yellow) (middle panel). The experimental scattering profiles from SAXS are shown as blue dots, and the scattering profile for the GDP-bound wild type tTG structure (PDB code 1KV3) is shown as a green line (right panel). C, SAXS analysis was performed on recombinant tTG R580K. Two monomeric models derived from a substrate-bound tTG WT crystal structure (PDB code 2Q3Z) were fitted into the calculated SAXS envelope for tTG R580K in a head-to-tail fashion. The N-terminal β-sandwich and the catalytic core domains of the fitted models are colored in blue, and the two C-terminal β-barrel domains are colored in red (left panel). A schematic representation shows the superimposition of one of the monomers in the fitted model (blue and red) onto the structure of the substrate-bound tTG (PDB code 2Q3Z, yellow) (middle panel). The experimental scattering profiles from SAXS are shown as blue dots, and the scattering profile for the substrate-bound tTG WT structure (PDB code 2Q3Z) is shown as a green line (right panel). D, SAXS analysis was performed on recombinant tTG-Short. Two monomeric models derived from a substrate-bound tTG WT crystal structure (PDB code 2Q3Z) were fitted into the calculated SAXS envelope for tTG-Short in a head-to-tail fashion. One monomer is colored in red and the other in blue (left panel). A schematic representation shows one of the monomers in the fitted model. The N-terminal β-sandwich is colored in blue, and the catalytic core domain and the portion of β-barrel that is present in tTG-Short are colored red (middle panel). The experimental scattering profiles from SAXS are shown as blue dots, and the scattering profile for the substrate-bound tTG WT structure (PDB code 2Q3Z) is shown as a green line (right panel).
SAXS analyses were then performed on these two forms of tTG to gain additional information about their overall size and shape in solution, similar to what we have done in the past (43). The Rg values for tTG WT and tTG R580K were 31.6 Å and 42.5 Å, respectively (Fig. 4, B and C, panels on the right). Molecular mass calculations using scattering profiles from SAXS data for tTG WT showed that it exists as a monomer in solution with a calculated molecular mass of 86.1 kDa, whereas tTG R580K exists as a homodimer with a calculated molecular mass of 153.4 kDa. After applying an unbiased approach for generating all possible models using available tTG structures and testing which of these best reflects the experimental scattering curve, we found that a monomer of a closed conformation of tTG (PDB code 1KV3) best fits the SAXS envelope calculated for tTG WT, with a χ2 value of 0.9 (Fig. 4B, left), whereas a dimer in an open conformation (PDB code 2Q3Z) fits well for the envelope calculated for the tTG R580K mutant, with a low χ2 value of 0.5 (Fig. 4C, left).
SAXS was also performed on tTG-Short. The Rg value for tTG-Short was 35.2 Å (Fig. 4D, panel on the right), and the molecular mass calculations using scattering profiles from the SAXS data showed that it exists as a homodimer with a calculated molecular mass of 119.5 kDa. The model for tTG-Short that best fits all the criteria resembles an open-state conformation, with a χ2 value of 0.7 (Fig. 4D, left).
Identification of Intramolecular Interactions That Are Critical for Stabilizing tTG in Its Closed Conformation
The open conformation that tTG R580K assumes supported the idea that this conformational state is highly toxic to cells. To directly examine this possibility, we set out to determine the intramolecular interactions that enable tTG WT to adopt the closed conformation. The x-ray structures for tTG WT show that the second β-barrel located at the C terminus makes close contacts with the catalytic core domain when tTG is in its closed configuration. These contacts should be lost in the open conformation due to the movement of the C-terminal β-barrels away from the catalytic core domain (see Fig. 1B, compare the left and middle structural depictions). Moreover, tTG-Short, which adopts a conformation resembling the open state and induces cell death, is missing nearly half of the first C-terminal β-barrel and the entire second β-barrel in tTG WT (see Fig. 1B, right), further supporting the idea that the intramolecular interactions occurring between the second C-terminal β-barrel domain and the catalytic core may play an important role in stabilizing tTG WT in its closed conformation.
We, therefore, set out to directly test this idea. Because forms of tTG that assume the open conformation (i.e. tTG R580K and tTG-Short) consistently induce cell death, we used this as a readout to indicate whether a given tTG truncation mutant was in the open or closed state. The first tTG truncation mutant that was generated lacked only the C-terminal 15 amino acids (amino acids 673–687) and was thus designated tTG-1–672 (Fig. 5A). We ectopically expressed a Myc-tagged version of this mutant along with Myc-tagged tTG WT, tTG-Short, and tTG R580K in actively growing NIH3T3 fibroblasts (Fig. 5B) and then evaluated the ability of each of these forms of tTG to induce cell death. Interestingly, after ∼36 h of expression, Myc-tagged tTG-1–672 caused >25% of the cells to spontaneously undergo apoptosis, similar to the extents of cell death caused by tTG-Short and the tTG R580K mutant (Fig. 5C, compare the third, fourth, and fifth bars).
FIGURE 5.

Identification of a region in the C terminus of tTG that is crucial for its ability to assume the closed conformation. Linear representations showing the relative lengths of Myc-tagged forms of tTG WT, tTG-Short, and a tTG 1–672. The numbers shown indicate amino acids. B, cell lysates collected from NIH3T3 fibroblasts ectopically expressing the vector alone or one of the indicated Myc-tagged tTG proteins for ∼24 h were immunoblotted (IB) with Myc and actin antibodies. C, NIH3T3 cells ectopically expressing the vector alone or one of the indicated Myc-tagged tTG proteins were cultured in medium containing 10% CS for ∼36 h and fixed. Immunofluorescence was performed on the cells using a Myc antibody to detect the transfectants, and the cells were also stained with DAPI to label nuclei. Cells undergoing apoptosis were identified by nuclear condensation/blebbing. The experiment was performed three separate times, and the percent apoptosis for each condition was determined by calculating the ratio of apoptotic to non-apoptotic cells. The data shown represent the mean ± S.E. D, the C-terminal 15 amino acids of tTG (residues 673–687) are highlighted in purple in the x-ray crystal structure of tTG bound to GDP (top panel). Two pairs of hydrogen bonds formed between residues Asn-681 and Asp-434 and between residues Lys-677 and Trp-254 are shown in magenta with participating residues shown in sticks (bottom panels). The distances of each interaction are shown.
Given that there appears to be key residues present within the C-terminal 15 amino acids of tTG WT that are required to maintain the closed conformation, we focused our attention on this region, i.e. corresponding to the last β-sheet within the second β-barrel domain (Fig. 5D, top panel, area highlighted in purple). Close examination of the x-ray structure for GDP-bound tTG revealed that two pairs of hydrogen bonds are formed between Lys-677 in the β-barrel 2 domain and Trp-254 in the catalytic core domain (Fig. 5D, middle) as well as between Asn-681 in the β-barrel 2 domain and Asp-434 in the catalytic core domain (Fig. 5D, bottom panels). The distance between each of these pairs of hydrogen bonds was calculated to be ∼3.0 Å.
To determine whether the formation of these hydrogen bonds contributes to the stabilization of the closed conformation of tTG, we changed each of the four participating residues to alanine. We hypothesized that if these two pairs of hydrogen bonds play an important role in stabilizing the closed conformation, then substitutions that block their formation should cause tTG to assume an open state. We first examined whether the enzymatic transamidation activity and GTP binding capability of tTG were affected by these substitutions. NIH3T3 fibroblasts ectopically expressing either Myc-tagged tTG WT or one of the hydrogen bond point mutants of tTG were lysed, and then the transamidation activity associated with each of the cell extracts was assayed. The W245A substitution completely abolished transamidation activity, whereas the D434A substitution greatly reduced it (Fig. 6A, compare the second and third lanes to the first lane). Both Trp-254 and Asp-434 are localized in the catalytic core domain and are within close proximity to the active site cysteine residue (i.e. Cys-277) (30). Thus, substitutions at these two sites may alter local structures within the catalytic domain that potentially perturb enzymatic activity. However, the substitutions made at the more distant C-terminal residues (i.e. tTG K677A and N681A) had a more modest effect on enzymatic activity, and interestingly, the tTG N681A mutant reproducibly showed slightly elevated transamidation activity compared with tTG WT (Fig. 6A, compared the fourth and fifth lanes to the first lane). This indicates that the tTG N681A mutant might be especially effective at adopting the open state conformation, making the catalytic active site more accessible for substrate binding and thus exhibiting enhanced enzymatic activity similar to tTG R580K (see Fig. 3C).
FIGURE 6.

The identification of key residues in tTG that are essential for its ability to adopt the closed conformation. A, cell lysates collected from NIH3T3 cells ectopically expressing the indicated forms of Myc-tagged tTG were assayed for their enzymatic transamidation activity (top panel) as well as immunoblotted (IB) with Myc and actin antibodies (bottom two panels). The lines indicate where a portion of the blot was removed. B, the indicated recombinant forms of tTG were expressed, purified, and then resolved by SDS-PAGE. The gels were stained with Coomassie Blue to visualize the proteins. A protein molecular mass (M.W.) marker was included on the gel to show the sizes of the recombinant proteins. C, purified recombinant forms of tTG WT, tTG W254A, and tTG N681A (600 nm) were incubated with BODIPY-GTP (indicated on the graph as BODIPY-GTP Added), and the resulting changes in fluorescence were determined using a spectrofluorimeter. These experiments were performed at least three separate times, each yielding similar results. D, purified recombinant forms of tTG WT, tTG R580K, tTG W254A, and tTG N681A (3.5 μg of each) were incubated without or with trypsin for 2 h before being resolved by SDS-PAGE and then stained with Coomassie Blue to visualize the proteins. Note that only tTG WT was not efficiently digested by trypsin. E, SAXS was performed on purified recombinant tTG W254A. Two monomeric models derived from a substrate-bound tTG WT crystal structure (PDB code 2Q3Z) were fitted into the calculated SAXS envelope for tTG-Short in a head-to-tail fashion. The N-terminal β-sandwich and the catalytic core domains of the fitted models are colored in blue, and the two C-terminal β-barrel domains are colored in red (left panel). A schematic representation shows the superimposition of one of the monomers in the fitted model (blue and red) (right panel). The experimental scattering profiles from SAXS are shown as blue dots, and the scattering profile for the substrate-bound tTG WT structure (PDB code 2Q3Z) is shown as a green line (bottom panel).
We also assayed the nucleotide binding capabilities of the different tTG point mutants using BODIPY-GTP. Whereas attempts to generate recombinant forms of tTG D434A and tTG K677A mutants were not successful, most likely because these proteins are unstable and degrade rapidly, we were able to generate the recombinant tTG W254A and tTG N681A mutants (Fig. 6B, lanes labeled tTG W254A and tTG N681A). Consistent with our earlier findings, tTG WT has a high affinity for BODIPY-GTP, as indicated by the significant enhancement in the BODIPY fluorescence (Fig. 6C, blue line). However, both the tTG W254A (red line) and tTG N681A (green line) mutants showed little ability to bind BODIPY-GTP.
We next examined whether these mutants adopt the open conformation. First, we assayed their sensitivity to trypsin proteolysis. Fig. 6D shows that, like the GTP binding-defective tTG R580K mutant, both the tTG W254A and N681A mutants showed increased sensitivity to trypsin digestion compared with tTG WT, suggesting that they adopt the open conformation. We then performed SAXS analysis on tTG W254A and tTG N681A. Although we were not able to obtain useable data for the tTG N681A mutant, the tTG W254A mutant showed a reliable scattering profile. Molecular mass calculations using scattering profiles from the SAXS data showed that tTG W254A exists as a homodimer in solution with a calculated molecular mass of 168 kDa (Fig. 6E, bottom panel). The Rg value for tTG W254A was 47.5 Å, and a dimer of the tTG W254A molecules in the open conformation fit the SAXS envelope well, with a χ2 value of 0.5 (Fig. 6E, top panel).
We then examined whether disrupting the intramolecular interactions that occur between the second C-terminal β-barrel domain in tTG WT and its catalytic core domain, so as to drive tTG WT to the open conformation, was sufficient to induce cell death. Fig. 7, A and B, shows that this was indeed the case, as the ectopic expression of any of these tTG point mutants was toxic to cells, whereas tTG WT, which adopts the closed conformation, was not.
FIGURE 7.

Forms of tTG that assume an open conformation are consistently cytotoxic. A, cell lysates collected from NIH3T3 cells ectopically expressing the vector alone or the indicated forms of Myc-tagged tTG were immunoblotted (IB) with Myc and actin antibodies. B, NIH3T3 fibroblasts ectopically expressing the same Myc-tagged tTG proteins shown in A were cultured in medium containing 10% CS for ∼36 h and fixed. Immunofluorescence was performed on the cells using a Myc antibody to detect the transfectants. The cells were also stained with DAPI to label nuclei. Apoptotic cells were identified by nuclear condensation/blebbing. The experiment was performed three separate times, and the percent apoptosis for each condition was determined by calculating the ratio of apoptotic to non-apoptotic cells. The data shown represent the mean ± S.E.
Discussion
The overexpression and deregulation of tTG have been linked to a number of pathological conditions and disease states (3, 17, 20). Perhaps foremost among these has been the overexpression of tTG in a variety of cancers and particularly in the most aggressive forms of the disease, where it has been implicated in cancer cell survival and invasiveness (9–13). However, what has been somewhat puzzling is that tTG has also been reported to contribute to cellular apoptosis and to have important roles in neurodegenerative disorders including Alzheimer, Parkinson, and Huntington diseases (3, 14–17, 22, 29, 34). These seemingly contradictory functions have often led to confusion in the field as well as to various speculative ideas regarding the specific conditions in which tTG plays a positive role in cell survival versus exerting a damaging cell death-inducing activity (3, 27).
An initial clue that helped to shed some light on the discrepant functions of tTG came from studies implicating a splice variant of the protein that lacks the C-terminal 138 amino acids and is thus called tTG-Short in neurodegeneration (31–34). Indeed, we previously showed that tTG-Short was capable of inducing an apoptotic response and was poorly tolerated by most cells, such that they exert a selective pressure against its expression (34). There initially appeared to be two plausible possibilities for the apoptotic activity of tTG-Short, these being that either the induction of cell death was due to an excess transamidation (protein-cross-linking) capability because tTG-Short shows only very weak binding of GTP and thus cannot effectively regulate this enzymatic activity or because of the propensity of tTG-Short to undergo aggregation in cells. However, we were subsequently able to rule out both of these possibilities. In particular, we showed that introducing transamidation-defective mutations within the tTG-Short background did not eliminate its apoptotic activity (34) and subsequently demonstrated that a different class of tTG point mutants that was incapable of binding GTP and showed no tendency to undergo higher order aggregation could nonetheless induce apoptosis similar to tTG-Short (22).
Collectively, these findings seemed to suggest that the expression of tTG-Short as well as GTP binding-defective point mutants of tTG exert some type of dominant-inhibitory effect. The question then was how might such a dominant-negative function be mediated. An intriguing possibility came from comparisons of the high resolution x-ray crystal structure for guanine nucleotide-bound tTG (24) versus that for tTG containing an inhibitor within its transamidation active site (26). Specifically, guanine nucleotide-bound tTG adopts a “closed state” in which its C-terminal tail folds over and covers the transamidation active site located within the catalytic core domain (24). When tTG adopts this more compact conformation, substrates are unable to gain access to the transamidation active site, thus preventing tTG from functioning as an acyltransferase and catalyzing the cross-linking of proteins (3). This overall structure is strikingly different from that determined for tTG bound to a substrate-mimetic gluten peptide, which is much more open and extended, such that the C-terminal tail has moved away from the catalytic core domain and the active center is fully accessible to substrates as well as transamidation inhibitory molecules (26). Given the ability of tTG to exhibit such striking differences in structural conformations, one might then imagine how tTG, when adopting the open-state structure for extended periods of time, not only fails to provide the signaling functions essential for cell survival normally provided by tTG in a closed state, but in some way actually interferes with those functions, with the net result then being cell death instead of cell survival.
We tested this idea by closely examining the two structural states of tTG so as to identify key interaction sites that when disrupted would be expected to convert a closed-state form of tTG to an open state (24, 26). Two candidate pairs of hydrogen bonds were identified and predicted to form between residues located within the most C-terminal β-barrel domain and the catalytic core domain of tTG. Indeed, we found that disrupting either of these hydrogen bonds through site-directed mutagenesis, generated tTG mutants that were capable of constitutively assuming an open-state conformation, as indicted by their inability to bind GTP, their enhanced sensitivity to trypsin proteolysis, and as an outcome of SAXS analyses. We further showed that these same open-state properties were exhibited by tTG-Short as well as by GTP binding-defective point mutants. We then demonstrated that the ectopic expression of any mutant form or isoform of tTG (i.e. tTG-Short) that is capable of constitutively adopting the open-state conformation in cells results in a strong apoptotic response that was not dependent on transamidation (protein cross-linking activity). These findings when taken together led us to conclude that the ability of tTG to assume the open-state conformation for extended periods of time is highly toxic to cells (Fig. 8).
FIGURE 8.
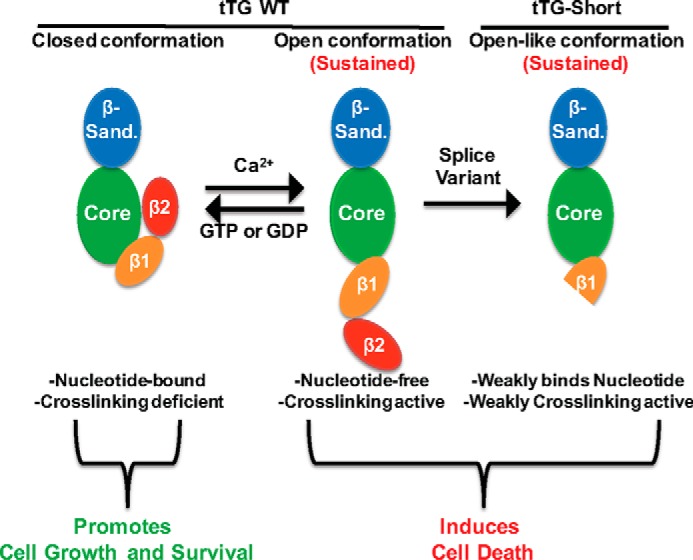
Diagram highlighting the different conformational states of tTG and the effects they have on cells. tTG WT primarily adopts a closed conformation in cells due to the relatively high levels of GTP or GDP and low levels of Ca2+ (left panel). Under these conditions tTG has been shown to promote cell growth and survival. However, in response to cell stresses, intracellular Ca2+ levels can be increased, resulting in less GTP/GDP binding to tTG and allowing for it to adopt an open, catalytically active conformation (middle panel). If tTG persists in the open conformation for extended lengths of time, it induces cell death. Interestingly, tTG-Short is a splice variant of tTG that lacks the portion of the C terminus determined to be important for keeping it in the closed conformation. Consistent with this finding, tTG-Short expression in cells induces apoptosis (right panel).
We are now left with the question of why open-state forms of tTG have such deleterious effects for cell survival. The results of recent studies examining the actions of tTG as they pertain to EGFR signaling during cancer progression might begin to offer a possible explanation. By building on various lines of evidence suggesting that tTG was a downstream signaling partner of the EGFR in cancer cells (9, 10, 44–46), we discovered that a key role for tTG when overexpressed in brain cancer cells was to extend the signaling lifetimes of EGFRs (10). Specifically, we found that tTG was able to protect EGFRs against the negative regulatory actions of c-Cbl, an E3 ubiquitin ligase that has been shown to play important roles in promoting EGFR down-regulation and degradation in cells (47, 48). Brain cancer cells showing high expression of tTG also showed a corresponding reduction in EGFR ubiquitination and enhanced EGFR-signaling lifetimes. The ability of tTG to block the actions of c-Cbl was not dependent on its transamidation activity but appeared to require its GTP binding capability, as GTP binding-defective point mutants of tTG were ineffective in conferring this protection (10). We then discovered that tTG was also capable of acting as a signaling scaffold for at least two other EGFR signaling partners, namely the c-Src tyrosine kinase and PI 3-kinase (9, 28). Moreover, by functioning in this capacity, tTG helped to optimize the c-Src-dependent phosphorylation of the 85-kDa-regulation subunit (p85) of PI 3-kinase (49, 50) as well as ensure the maximal activation of its kinase activity (28). Here again, the GTP binding capability of tTG seemed to be necessary, as GTP binding-defective tTG point mutants were ineffective in promoting p85 phosphorylation and PI 3-kinase activation. Importantly, these point mutants were able to form a complex with PI 3-kinase and thereby could act as dominant-negative inhibitors of PI 3-kinase activation.
Thus, a picture is starting to emerge where tTG plays important roles as a protein scaffold functioning in EGFR signaling as well as possibly in the actions of other growth factor receptors that signal through c-Src and PI 3-kinase (51, 52). When in its GTP-bound state, tTG would be capable of assembling signaling partners and regulators in a manner that sustains EGFR-signaling lifetimes and enhances PI 3-kinase activation (10, 28). However, forms of tTG that are incapable of binding GTP and thus adopt an open conformational state apparently cannot fulfill the entirety of these functions while still being able to associate with some of the signaling partners such as PI 3-kinase (28), thereby preventing the necessary signals that lead to its optimal activation. Given the roles played by PI 3-kinase in cell survival, especially within the context of cancer cell growth and malignant transformation (51), the presence of excessive amounts of open-state forms of tTG could have significant deleterious effects on overall cell survival, eventually contributing to cell death.
It is possible that the ability of tTG to transition between its closed and open states provides a mechanism by which to regulate certain aspects of EGFR-signaling, particularly EGFR lifetimes in cells and the finite duration of maximal PI 3-kinase activation (10, 28). However, disruptions of the normal transitioning between these states, as might occur with the overexpression of wild type tTG in cancer cells, such that a greater than normal amount of tTG is present in a closed conformational state could greatly enhance EGFR-signaling and cell survival. On the other hand, the overexpression of tTG forms that would assume the open state, such as tTG-Short, could likely have the opposite outcome and be severely detrimental to survival. It is also likely that cellular localization will be an important factor, as we know that when tTG is intracellular, its closed-state is the preferred structural conformation for positive EGFR signaling (9, 10, 36–38), whereas when tTG is secreted from cells as part of the cargo of extracellular vesicles and thus not exposed to sufficient concentrations of GTP, it can adopt an open-state conformation and catalyze protein cross-linking events that play highly beneficial roles for tumor cells within their microenvironment (53). Nevertheless, our expectation would be small molecules that selectively induce tTG to adopt an open-state within cancer cells could have therapeutic value. Therefore, it will be interesting to see whether classes of compounds that disrupt the interactions that we have described here and have shown to be important for adopting the open-state conformation can in fact be identified and tested for their clinical benefits.
Author Contributions
M. A. A. and R. A. C. designed the study, prepared the figures, and wrote the manuscript. J. Z., G. S., and Y. M. designed and generated vectors for expression of tTG isoforms and mutants in bacteria and mammalian cells, performed SAXS analysis, and performed GTP binding and enzymatic transamidation activity assays as well as carried out all cell-based experiments.
Acknowledgments
We thank Dr. Richard Gillilan and MacCHESS (National Institutes of Health Grant P41 GM103485) for SAXS data collection and help with the analyses and Cindy Westmiller for expert secretarial assistance.
This work was supported by National Institutes of Health Grant R01 GM040654 and by National Cancer Institute Grant CA201402. The authors declare that they have no conflicts of interest with the contents of this article. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- tTG
- tissue transglutaminase
- tTG-Short
- tissue transglutaminase-short isoform
- BPA
- biotinylated pentylamine
- SAXS
- small angle x-ray scattering
- RA
- retinoic acid
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- FL
- full-length
- CS
- calf serum
- EGFR
- epidermal growth factor receptor
- PI 3-kinase
- phosphatidylinositol 3-kinase
- Z-DON
- 6-diazo-5-oxo-norleucine tetrapeptide.
References
- 1. Folk J. E. (1980) Transglutaminases. Annu. Rev. Biochem. 49, 517–531 [DOI] [PubMed] [Google Scholar]
- 2. Nakaoka H., Perez D. M., Baek K. J., Das T., Husain A., Misono K., Im M. J., and Graham R. M. (1994) Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science 264, 1593–1596 [DOI] [PubMed] [Google Scholar]
- 3. Gundemir S., Colak G., Tucholski J., and Johnson G. V. (2012) Transglutaminase 2: a molecular Swiss army knife. Biochim. Biophys. Acta 1823, 406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aeschlimann D., Wetterwald A., Fleisch H., and Paulsson M. (1993) Expression of tissue transglutaminase in skeletal tissues correlates with events of terminal differentiation of chondrocytes. J. Cell Biol. 120, 1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Z., Telci D., and Griffin M. (2011) Importance of syndecan-4 and syndecan-2 in osteoblast cell adhesion and survival mediated by a tissue transglutaminase-fibronectin complex. Exp. Cell Res. 317, 367–381 [DOI] [PubMed] [Google Scholar]
- 6. Telci D., and Griffin M. (2006) Tissue transglutaminase (TG2): a wound response enzyme. Front Biosci. 11, 867–882 [DOI] [PubMed] [Google Scholar]
- 7. Verderio E. A., Johnson T., and Griffin M. (2004) Tissue transglutaminase in normal and abnormal wound healing: review article. Amino Acids 26, 387–404 [DOI] [PubMed] [Google Scholar]
- 8. Mehta K., and Lopez-Berestein G. (1986) Expression of tissue transglutaminase in cultured monocytic leukemia (THP-1) cells during differentiation. Cancer Res. 46, 1388–1394 [PubMed] [Google Scholar]
- 9. Li B., Antonyak M. A., Druso J. E., Cheng L., Nikitin A. Y., and Cerione R. A. (2010) EGF potentiated oncogenesis requires a tissue transglutaminase-dependent signaling pathway leading to Src activation. Proc. Natl. Acad. Sci. U.S.A. 107, 1408–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J., Antonyak M. A., Singh G., and Cerione R. A. (2013) A mechanism for the upregulation of EGF receptor levels in glioblastoma. Cell Rep. 3, 2008–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan L., Siegel M., Choi K., Khosla C., Miller C. R., Jackson E. N., Piwnica-Worms D., and Rich K. M. (2007) Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene 26, 2563–2573 [DOI] [PubMed] [Google Scholar]
- 12. Singer C. F., Hudelist G., Walter I., Rueckliniger E., Czerwenka K., Kubista E., Huber A. V. (2006) Tissue array-based expression of transglutaminase-2 in human breast and ovarian cancer. Clin. Exp. Metastasis 23, 33–39 [DOI] [PubMed] [Google Scholar]
- 13. Kim D. S., Park S. S., Nam B. H., Kim I. H., and Kim S. Y. (2006) Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-κB inactivation. Cancer Res. 66, 10936–10943 [DOI] [PubMed] [Google Scholar]
- 14. Kim S. Y., Grant P., Lee J. H., Pant H. C., and Steinert P. M. (1999) Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer's disease. J. Biol. Chem. 274, 30715–30721 [DOI] [PubMed] [Google Scholar]
- 15. Karpuj M. V., Garren H., Slunt H., Price D. L., Gusella J., Becher M. W., and Steinman L. (1999) Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington's disease brain nuclei. Proc. Natl. Acad. Sci. U.S.A. 96, 7388–7393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lesort M., Chun W., Johnson G. V., and Ferrante R. J. (1999) Tissue transglutaminase is increased in Huntington's disease brain. J. Neurochem. 73, 2018–2027 [PubMed] [Google Scholar]
- 17. Jeitner T. M., Muma N. A., Battaile K. P., and Cooper A. J. (2009) Transglutaminase activation in neurodegenerative diseases. Future Neurol. 4, 449–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katt W. P., Antonyak M. A., and Cerione R. A. (2015) Simultaneously targeting tissue transglutaminase and kidney type glutaminase sensitizes cancer cells to acid toxicity and offers new opportunities for therapeutic intervention. Mol. Pharm. 12, 46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caccamo D., Currò M., and Ientile R. (2010) Potential of transglutaminase 2 as a therapeutic target. Expert Opin. Ther. Targets 14, 989–1003 [DOI] [PubMed] [Google Scholar]
- 20. Siegel M., and Khosla C. (2007) Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol. Ther. 115, 232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Festoff B. W., SantaCruz K., Arnold P. M., Sebastian C. T., Davies P. J., and Citron B. A. (2002) Injury-induced “switch” from GTP-regulated to novel GTP-independent isoform of tissue transglutaminase in the rat spinal cord. J. Neurochem. 81, 708–718 [DOI] [PubMed] [Google Scholar]
- 22. Datta S., Antonyak M. A., and Cerione R. A. (2007) GTP binding-defective forms of tissue transglutaminase trigger cell death. Biochemistry 46, 14819–14829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Datta S., Antonyak M. A., and Cerione R. A. (2006) Importance of Ca2+-dependent transamidation activity in the protection afforded by tissue transglutaminase against doxorubicin-induced apoptosis. Biochemistry 45, 13163–13174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu S., Cerione R. A., and Clardy J. (2002) Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. U.S.A. 99, 2743–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bernardi P., and Rasola A. (2007) Calcium and cell death: the mitochondrial connection. Subcell. Biochem. 45, 481–506 [DOI] [PubMed] [Google Scholar]
- 26. Pinkas D. M., Strop P., Brunger A. T., and Khosla C. (2007) Transglutaminase 2 undergoes a large conformational change upon activation. PLos Biol. 5, e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eckert R. L., Fisher M. L., Grun D., Adhikary G., Xu W., and Kerr C. (2015) Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol. Carcinog. 54, 947–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boroughs L. K., Antonyak M. A., and Cerione R. A. (2014) A novel mechanism by which tissue transglutaminase activates signaling events that promote cell survival. J. Biol. Chem. 289, 10115–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gundemir S., Colak G., Feola J., Blouin R., and Johnson G. V. (2013) Transglutaminase 2 facilitates or ameliorates HIF signaling and ischemic cell death depending on its conformation and localization. Biochim. Biophys. Acta 1833, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee K. N., Arnold S. A., Birckbichler P. J., Patterson M. K. Jr., Fraij B. M., Takeuchi Y., and Carter H. A. (1993) Site-directed mutagenesis of human tissue transglutaminase: Cys-277 is essential for transglutaminase activity but not for GTPase activity. Biochim. Biophys. Acta 1202, 1–6 [DOI] [PubMed] [Google Scholar]
- 31. Citron B. A., Suo Z., SantaCruz K., Davies P. J., Qin F., and Festoff B. W. (2002) Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem. Int. 40, 69–78 [DOI] [PubMed] [Google Scholar]
- 32. Citron B. A., SantaCruz K. S., Davies P. J., and Festoff B. W. (2001) Intron-exon swapping of transglutaminase mRNA and neuronal Tau aggregation in Alzheimer's disease. J. Biol. Chem. 276, 3295–3301 [DOI] [PubMed] [Google Scholar]
- 33. Lai T. S., and Greenberg C. S. (2013) TGM2 and implications for human disease: role of alternative splicing. Front. Biosci. 18, 504–519 [DOI] [PubMed] [Google Scholar]
- 34. Antonyak M. A., Jansen J. M., Miller A. M., Ly T. K., Endo M., and Cerione R. A. (2006) Two isoforms of tissue transglutaminase mediate opposing cellular fates. Proc. Natl. Acad. Sci. U.S.A. 103, 18609–18614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nielsen S. S., Toft K. N., Snakenborg D., Jeppesen M. G., Jacobsen J. K., Vestergaard B., Kutter J. P., and Arleth L. (2009) BioXTAS RAW, a software program for high-throughput automated small-angle x-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 42, 959–964 [Google Scholar]
- 36. Svergun D. I. (1991) Mathematical methods in small-angle scattering data analysis. J. Appl. Crystallogr. 24, 485–492 [Google Scholar]
- 37. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 38. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 39. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kozin M. B., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 42. Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dias S. M., Wilson K. F., Rojas K. S., Ambrosio A. L., and Cerione R. A. (2009) The molecular basis for the regulation of the cap-binding complex by the importins. Nat. Struct. Mol. Biol. 16, 930–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Antonyak M. A., Miller A. M., Jansen J. M., Boehm J. E., Balkman C. E., Wakshlag J. J., Page R. L., and Cerione R. A. (2004) Augmentation of tissue transglutaminase expression and activation by epidermal growth factor inhibits doxorubicin-induced apoptosis in human breast cancer cells. J. Biol. Chem. 279, 41461–41467 [DOI] [PubMed] [Google Scholar]
- 45. Antonyak M. A., Li B., Regan A. D., Feng Q., Dusaban S. S., and Cerione R. A. (2009) Tissue transglutaminase is an essential participant in the epidermal growth factor-stimulated signaling pathway leading to cancer cell migration and invasion. J. Biol. Chem. 284, 17914–17925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Z., Xu X., Bai L., Chen W., and Lin Y. (2011) Epidermal growth factor receptor-mediated tissue transglutaminase overexpression couples acquired tumor necrosis factor-related apoptosis-inducing ligand resistance and migration through c-FLIP and MMP-9 proteins in lung cancer cells. J. Biol. Chem. 286, 21164–21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levkowitz G., Waterman H., Zamir E., Kam Z., Oved S., Langdon W. Y., Beguinot L., Geiger B., and Yarden Y. (1998) c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 12, 3663–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Waterman H., Levkowitz G., Alroy I., and Yarden Y. (1999) The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor. J. Biol. Chem. 274, 22151–22154 [DOI] [PubMed] [Google Scholar]
- 49. Auger K. R., Carpenter C. L., Shoelson S. E., Piwnica-Worms H., and Cantley L. C. (1992) Polyoma virus middle T antigen-pp60c-src complex associates with purified phosphatidylinositol 3-kinase in vitro. J. Biol. Chem. 267, 5408–5415 [PubMed] [Google Scholar]
- 50. Liu X., Marengere L. E., Koch C. A., and Pawson T. (1993) The v-Src SH3 domain binds phosphatidylinositol 3′-kinase. Mol. Cell. Biol. 13, 5225–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Polivka J. Jr., and Janku F. (2014) Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 142, 164–175 [DOI] [PubMed] [Google Scholar]
- 52. Chen Q., Zhou Z., Shan L., Zeng H., Hua Y., and Cai Z. (2015) The importance of Src signaling in sarcoma. Oncol Lett. 10, 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Antonyak M. A., Li B., Boroughs L. K., Johnson J. L., Druso J. E., Bryant K. L., Holowka D. A., and Cerione R. A. (2011) Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]