

Abstract
SMYD3 is a SET domain-containing N-lysine methyltransferase associated with multiple cancers. Its reported substrates include histones (H3K4 and H4K5), vascular endothelial growth factor receptor 1 (VEGFR1 Lys831) and MAP3 kinase kinase (MAP3K2 Lys260). To reveal the structural basis for substrate preference and the catalytic mechanism of SMYD3, we have solved its co-crystal structures with VEGFR1 and MAP3K2 peptides. Our structural and biochemical analyses show that MAP3K2 serves as a robust substrate of SMYD3 because of the presence of a phenylalanine residue at the −2 position. A shallow hydrophobic pocket on SMYD3 accommodates the binding of the phenylalanine and promotes efficient catalytic activities of SMYD3. By contrast, SMYD3 displayed a weak activity toward a VEGFR1 peptide, and the location of the acceptor lysine in the folded kinase domain of VEGFR1 requires drastic conformational rearrangements for juxtaposition of the acceptor lysine with the enzymatic active site. Our results clearly revealed structural determinants for the substrate preference of SMYD3 and provided mechanistic insights into lysine methylation of MAP3K2. The knowledge should be useful for the development of SMYD3 inhibitors in the fight against MAP3K2 and Ras-driven cancer.
Keywords: cancer, chromatin, crystal structure, protein methylation, substrate specificity, SET domain
Introduction
Su(var), E(z), Trithorax (SET)6 domain-containing proteins catalyze the transfer of the methyl group from S-adenosyl-l-methionine (SAM) onto the Nϵ group of a lysine in the substrate protein. The best known and the most common substrates of the SET domain family of enzymes are histones. The tight site and methylation state specificities of SET domain proteins reflect the important roles of histone lysine methylation in epigenetic control of eukaryotic gene expression and regulation of higher order chromatin structure (1–5). Some SET domain proteins also methylate non-histone substrates, although only a small number of them are known to date. One example is the methylation of p53 by SET7/SET9 (6), which was first identified as an H3K4 monomethylase (7). There are more than 90 SET domain proteins in the human proteome, many of which remain poorly characterized, and more non-histone substrates may turn up through further studies. Recently, a group of SET and myeloid-Nervy-DEAF-1 (MYND) domain-containing proteins (SMYD) have been characterized as versatile lysine methyltransferases (8–10). SMYD1 and SMYD3 were reported to methylate histone H3K4, and SMYD3 also methylates H4K5. SMYD2 had been shown to catalyze the dimethylation at H3K36 and repress the transcription of reporter genes. SMYD proteins also have non-histone substrates. It has been reported that p53 and estrogen receptor α are good substrates of SMYD2, and VEGFR1 and MAP3K2 are substrates of SMYD3 (11–14).
SMYD3 was found to be significantly overexpressed in multiple types of cancer, including colorectal, liver, and breast cancer (15–18). It was proposed that the reported H3K4 trimethylase activity, enhanced by the help of heat shock protein HSP90, was responsible for the oncogenic property of SMYD3 (9). SMYD3 has also been shown to methylate histone H4K5 and regulate cancer cell phenotypes (19). VEGFR1, which is a single-pass membrane protein with an extracellular ligand-binding domain and a cytoplasmic tyrosine kinase domain (20–23), has also been reported to be a substrate of SMYD3 (12). SMYD3 catalyzes the methylation on Lys831 of VEGFR1 in vitro and in HEK293 cells. It is reported that this methylation enhanced autophosphorylation and kinase activity of VEGFR1, which is important for its role in cancer cell progression. A most recent study identified MAP3K2 as a cytoplasmic substrate of SMYD3. The study shows that in tumors or in LAC and PDAC cells, nearly all SMYD3 are located in the cytoplasm (13, 24). In vivo studies show that SMYD3 catalyzes the methylation of MAP3K2 at Lys260, activates MAP kinase signaling module, and promotes Ras-driven tumorigenesis.
In this study, we set out to investigate the substrate preference of SMYD3 through structural and biochemical approaches. We showed by in vitro methyltransferase activity assays that histones and VEGFR1 are poor substrates of SMYD3 compared with MAP3K2. By solving the high resolution crystal structures of SMYD3 in complex with VEGFR1 and MAP3K2 peptides, we determined the structural basis for the substrate specificity of SMYD3, and our findings reinforce the notion that MAP3K2 is a physiological substrate of SMYD3. These results also provide novel insights for the design of SMYD3 inhibitors, which may be beneficial for the treatment of MAP3K2 and Ras-driven cancer.
Experimental Procedures
Protein Preparations
cDNA encoding full-length human SMYD3 was amplified by PCR and cloned into an engineered pET-28a-SMT3 vector between the EcoRI and XhoI restriction sites. The plasmid expresses a His6-SUMO N-terminally tagged fusion protein, His-SUMO-SMYD3. The fusion protein was expressed in the Escherichia coli BL21(DE3) codon Plus RIL strain by inducing with 0.2 mm isopropyl β-d-thiogalactopyranoside at 16 °C for 20 h when the cell density reached A600 ∼0.9. The cells were then harvested by centrifugation, resuspended in a lysis buffer (20 mm Tris, pH 8.0, 300 mm NaCl, 10 mm imidazole), and lysed by sonication. Cell debris was removed by centrifugation, and the supernatant was loaded onto a nickel-nitrilotriacetic acid column (Novagen) pre-equilibrated with lysis buffer. After washing with 10 column volumes of washing buffer (20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 20 mm imidazole), the target protein was eluted with the elution buffer (20 mm Tris-HCl, 300 mm NaCl, 300 mm imidazole) and treated with SUMO protease at 4 °C for 2 h to cleave the His-SUMO tag. Then the samples were diluted with buffer A (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 5% glycerol, 1‰ β-mercaptoethanol) and loaded onto a HiTrapTM SP HP column (GE Healthcare). The untagged protein was eluted with a 100 to 1,000 mm NaCl gradient and was pooled, concentrated, and further purified through a HiLoadTM 16/60 SuperdexTM 75 column (GE Healthcare) pre-equilibrated with 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 5% glycerol, 1‰ β-mercaptoethanol. High purity fractions were pooled and concentrated for crystallization.
Site-directed mutagenesis of SMYD3 was performed using the TaKaRa MutanBEST kit and verified by DNA sequencing. Expression and purification procedures for mutant proteins were the same as that for the wild-type protein.
Peptides
Peptides with 26 amino acids in length were used in the methyltransferase activity assay. The following is a list of the peptides used: MAP3K2 (a.a. 249–274, DYDNPIFEKFGK260GGTYPRRYHVSYHH); human VEGFR1 (a.a. 820–845, WEFARERLKLGK831SLGRGAFGKVVQAS); human histone H3 (a.a. 1–26, ARTK4QTARKSTGGKAPRKQLATKAAR); and histone H4 (a.a. 1–26, SGRGK5GGKGLGKGGAKRHRKVLRDNI). The following peptides were used for co-crystallization: MAP3K2 (a.a. 256–265, EKFGK260GGTYP) and VEGFR1 (a.a. 828–834, KLGK831(me2)SLG). All chemically synthesized peptides were purchased from SciLight Biotechnology (Beijing, China).
Crystallization
The purified SMYD3 protein was concentrated to ∼8–9 mg/ml, mixed with 1 mm SAH and 5–6 mm MAP3K2 peptide, and incubated on ice for 1 h before being used for crystal screening. Crystallization was performed using the hanging drop vapor diffusion method, with an equal volume of the protein solution and the reservoir solution. Crystals were obtained in a solution containing 3.4 m sodium acetate (pH 7.5) at 16 °C. In case of for the SMYD3-VEGFR1 complex, SMYD3 was crystallized first in a solution containing 2.8 m sodium acetate (pH 7.0) at 20 °C, and the VEGFR1 peptide was soaked in by the addition of 10 mm peptide.
Data Collection and Structure Determination
By adding 25 mm peptide to the crystallization drop, the SMYD3-MAP3K2 crystals were soaked in a higher concentration of MAP3K2 peptide for ∼2 h before x-ray diffraction data collection. The crystals were flash-frozen in liquid nitrogen using a cryoprotectant prepared from the reservoir solution supplemented with 15% (v/v) glycerol. Diffraction data were collected at Beamline BL17U of the Shanghai Synchrotron Radiation Facility using an ADSC Q315r detector. The data were processed using HKL2000 software (25). Single-wavelength anomalous dispersion phasing using anomalous signals from three endogenous zinc atoms was carried out to solve the apo structure of SMYD3. The complex structures containing substrate peptide of MAP3K2 or VEGFR1 were solved by molecular replacement with the PHASER software (26), using the apo-SMYD3 structure as the search model. COOT (27) and PHENIX (28) were used for model rebuilding and refinement. Detailed statistics for data collection and refinement are shown in Table 1. Structure figures were prepared using PyMOL.
TABLE 1.
Data collection and refinement
| SMYD3-MAP3K2 | SMYD3-VEGFR1 | |
|---|---|---|
| Data collection | ||
| Wave length (Å) | 0.9791 | 0.9788 |
| Space group | P212121 | P212121 |
| Unit cell (Å) | a = 53.60, b = 104.18, c = 117.22 | a = 53.38, b = 104.66, c = 117.60 |
| Resolution (Å) | 50.00–2.70 (2.80–2.70) | 30.00–2.40 (2.49–2.40) |
| Rmerge | 0.111 (0.732) | 0.119 (0.529) |
| I/σI | 15.8 (2.6) | 13.0 (4.3) |
| Completeness (%) | 99.9 (100.0) | 99.9 (100.0) |
| Total/unique reflections | 113,209/18,908 | 179,578/26,005 |
| Refinement statistics | ||
| Rwork/Rfree | 0.178/0.225 | 0.184/0.223 |
| Root mean square deviation bonds (Å) | 0.004 | 0.008 |
| Root mean square deviation angles (°) | 0.798 | 1.153 |
| No. atom | ||
| Protein | 3455 | 3426 |
| Peptide | 67 | 50 |
| Ligand/ion | 37 | 45 |
| water | 117 | 96 |
| B factor (Å2) | ||
| Protein | 36.1 | 64.9 |
| Peptide | 55.0 | 74.1 |
| Ligand/ion | 33.9 | 75.5 |
| Water | 36.2 | 65.4 |
| Ramachandran plots | ||
| Favored | 98.4% | 97.9% |
| Allowed | 1.6% | 2.1% |
| Outlier | 0.0% | 0.0% |
In Vitro Methyltransferase Assay
A 20-μl reaction mixture containing 1 μm 3H-labeled S-adenosyl-l-methionine (PerkinElmer Life Science), 1.5 μg of recombinant SMYD3, and 1 μg of peptide in methyltransferase assay buffer (20 mm HEPES, pH 8.0, 10 mm KCl, 20 mm MgCl2, 10 mm DTT, 5% (v/v) glycerol) was incubated for 1 h at 30 °C. The reaction products were separated by 13% SDS-PAGE, transferred onto PVDF membranes, and then subjected to autoradiography. For quantification, the membranes were stained by Coomassie Blue G250, followed by liquid scintillation counting for each peptide band.
Computational Docking of VEGFR1 Kinase Domain to the SMYD3 Structure
Comparison between the structures of SMYD1 (PDB code 3N71) and SMYD3 shows SMYD1 may have a more open conformation in the substrate-binding cleft. By homology modeling, an open conformation of SMYD3 was modeled using the structure of SMYD1 as a template. DynDom program (29) was used to simulate conformational rotate of the C-terminal domain of SMYD3. An extremely large widening of the substrate-binding cleft of SMYD3 is required if the structure of VEGFR1 kinase domain is docked in using ZDOCK program (30) in Discovery Studio (Accelrys).
Results
Substrate Preference of SMYD3
Previous studies suggested that SMYD3 may have several nuclear and cellular substrates, including histones H3 and H4, VEGFR1, and MAP3K2. We first set out to examine the substrate preference of SMYD3 in vitro. 26-amino acid peptides encompassing the reported lysine methylation sites were used in the in vitro methyltransferase assay (MTA), including histones H3 (a.a. 1–26, Lys4 site) and H4 (a.a. 1–26, K5 site), VEGFR1 (a.a. 820–845, Lys831 site), and MAP3K2 (a.a. 249–274, Lys260 site). MTA results show that the catalytic activity of SMYD3 on histones H3K4 and H4K5 are too weak to be detected. In contrast, under the same condition, robust methylase activities of SMYD3 were detected with the MAP3K2 peptide as the substrate, although the activity for the VEGFR1 peptide was considerably lower compared with that for MAP3K2. The catalytic activity of SMYD3 on MAP3K2 peptide is ∼14-fold higher than that on VEGFR1 peptide (Fig. 1). Furthermore, SMYD3 catalyzes methylation equally well on the unmethylated, monomethylated, and dimethylated MAP3K2 peptides (Fig. 1). These results demonstrate that SMYD3 is a trimethylase catalyzing mono-, di-, and trimethylation on lysine residues, but histones and VEGFR1 are poor substrates of SMYD3 compared with MAP3K2.
FIGURE 1.
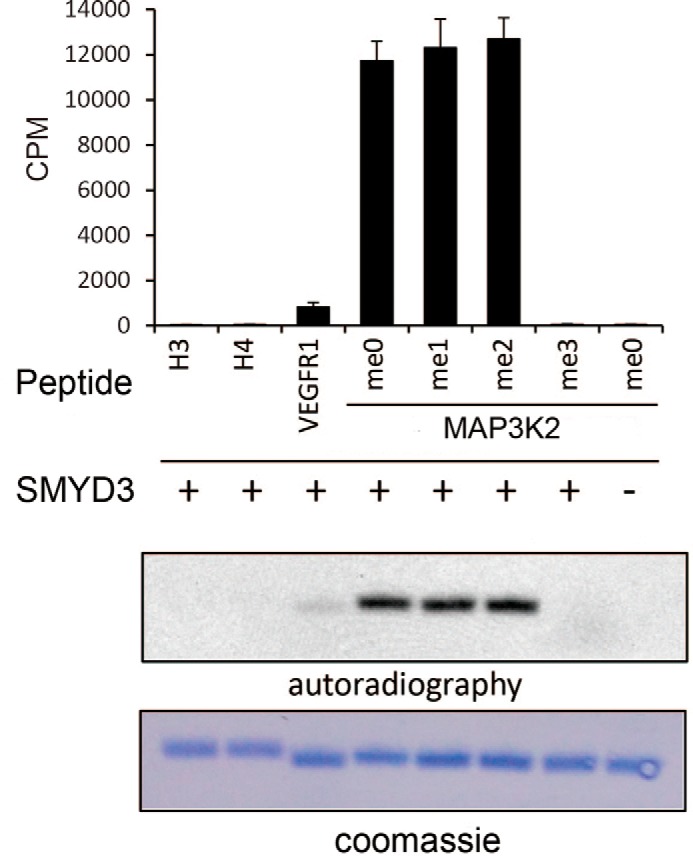
In vitro methyltransferase assays show the substrate preference of SMYD3. 1 μg of substrate peptides were used in each reaction, including histones H3 (a.a. 1–26) and H4 (a.a. 1–26), VEGFR1 (a.a. 820–845), and MAP3K2 (a.a. 249–274) carrying four different states of methylated Lys260 residue. 1.5 μg of recombinant SMYD3 was added in each reaction except the negative control result represented at the far right column. Each parallel reaction was performed three times. CPM values counted by liquid scintillation are show in a bar diagram (top panel). Autoradiography of the 3H-fluorography using [3H]AdoMet as the methyl donor is shown in the middle panel, and Coomassie Brilliant Blue staining of the peptides is shown in the bottom panel.
Structures of SMYD3 in Complex with MAP3K2 and VEGFR1 Peptides
We solved the crystal structure of full-length SMYD3 in complex with substrate peptides of MAP3K2 (a.a. 256–265, EKFGK(me0)GGTYP) and VEGFR1 (a.a. 828–834, KLGK(me2)SLG) in the presence and absence of S-adenosylhomocysteine (SAH), a reaction product of the methyl group donor SAM, at 2.7 and 2.4 Å resolution, respectively. The overall structure of the ternary complex of SMYD3 in complex with MAP3K2 and SAH is very similar to the previously reported apo form of SMYD3 (PDB code 3QWP) and to the binary complexes of SMYD3 with co-factors SAM (PDB code 3MEK) or SAH (PDB code 3OXL) (31) and with the small molecule inhibitor sinefungin (PDB codes 3PDN and 3RU0) (32, 33) (supplemental Fig. S1). In brief, SMYD3 is composed of three domains, an N-terminal catalytic domain (SET and post-SET), a MYND domain inserted into the SET domain, and a C-terminal domain (Fig. 2A). A topological diagram of the arrangement of secondary structure elements of SMYD3 is shown in supplemental Fig. S2. Similar to that found in a previous study (31), three zinc ions are bound to one SMYD3 molecule; two of them are in the MYND domain, and the third one is in the post-SET region. The zinc ions are coordinated either by four cysteine residues or by three cysteine residues and one histidine residue. Co-factor SAH is bound at the SAM-binding pocket of SMYD3, which is composed of three loop regions, including the β1-β2 loop and the η1-η2 loop of the SET domain and the α6-α7 loop of the post-SET region (supplemental Fig. S1B). The surrounding environment of the SAH molecule is highly similar to that of SAM (in 3MEK) and SAM analog Sinefungin (in 3PDN) (supplemental Fig. S1B).
FIGURE 2.

Overall structure of SMYD3 in complex with substrate peptides. A, top panel, a schematic drawing of SMYD3 domain organization. Bottom panel, a ribbon diagram showing the overall structure of SMYD3 in complex with the MAP3K2 peptide and SAH. The SET, MYND, post-SET, and C-terminal domains of SMYD3 are shown in green, purple, yellow, and pink, respectively. SAH and the MAP3K2 peptide are shown in a stick model (carbon, magenta, and cyan, respectively; nitrogen, blue; oxygen, red). Zinc ions are shown as gray spheres. The same coloring scheme is used in all figures unless explicitly specified. B, electrostatic potential and substrate-binding cleft on the surface of SMYD3. MAP3K2 and VEGFR1 peptides, which are shown in a stick model with carbon in cyan and orange, respectively, bind to the same amphiphilic substrate-binding pocket embraced by the SET, post-SET, and C-terminal domains of SMYD3. C, a close view of MAP3K2 and SAH binding by SMYD3. Residues involved in intermolecular interactions are shown as a stick model. The ϵ-amino group of Lys260 and the sulfur atom of SAH are highlighted with dots. Black dashed lines indicate intermolecular hydrogen bonds. D, superposition of MAP3K2 and VEGFR1 peptides in their complex structures with SMYD3. Residues of SMYD3 interacting with MAP3K2 and VEGFR1 are shown as a stick model (carbon, green, and gray, respectively).
The MAP3K2 and VEGFR1 peptides bind to the same amphiphilic pocket of SMYD3, which is a deep cavity embraced by the SET domain, the post-SET region, and the C-terminal domain (Fig. 2, A and B). All of the residues of MAP3K2 and VEGFR1 peptides have respectable electron density allowing unambiguous assignment of all residues except the first Glu256 of MAP3K2 (supplemental Fig. S3). Substrate peptides principally interact with the SET domain residues of SMYD3 by intermolecular hydrogen bonds and hydrophobic interactions. Detailed interactions between MAP3K2 peptide, SAH molecule, and SMYD3 are shown in Fig. 2C. Lys260 is stabilized through hydrophobic contacts with the phenyl rings of Phe183, Tyr239, and Tyr257. Distance between the Nϵ group of Lys260 and sulfur atom of SAH is ∼3.6 Å, which is suitable for the transfer of the methyl group when a SAM molecule, with the donor methyl group attached to the sulfur atom, is in place of SAH. Phe258 of MAP3K2 is situated in a shallow hydrophobic pocket formed by Val178, Ile179, and Val195 of SMYD3 (Figs. 2B and 3B). Intermolecular hydrogen bonds between MAP3K2 and SMYD3 include following: Nϵ group of Lys257 of MAP3K2 (M-Lys257) interacts with both the hydroxyl group of Thr263 and the side chain carboxylate of Glu192 of SMYD3 (S-Thr263 and S-Glu192); the main chain amide group of M-Gly261 and the main chain carboxyl group of S-Tyr239; the hydroxyl group of M-Thr263 and the side chain carboxyl group of S-Glu192; and the amide group of M-Tyr264 and the side chain carboxyl group of S-Asp241. The SAH molecule is stabilized both by a π-π stacking and by hydrogen bonds as previously described (31, 32) (Fig. 2C).
FIGURE 3.

Crucial roles of the −2 position residue of the substrates. A, amino acid sequence alignment of the reported substrates of SMYD3. The methyl-acceptor lysine residues are colored in yellow and annotated as position 0. Residues at the −2 and +3 and +4 positions are highlighted in red boxes. B, an enlarged view of the −2 residue-binding pocket of SMYD3. −2 residues of the substrates bind to a shallow hydrophobic pocket formed by two valine and one isoleucine residues, and two hydrophilic serine residues are located at the perimeter of the pocket. C, in vitro MTA results of SMYD3 activities on mutated MAP3K2 peptides. D, in vitro MTA results examine the methyltransferase activity of SMYD3 mutants.
In the SMYD3-VEGFR1 binary structure, the peptide binds to the same pocket as that bound by the MAP3K2 peptide (Fig. 2, B and D). Seven residues of VEGFR1 are visible in the structure. The first four residues of VEGFR1 (Lys828–Lys831) can be aligned with corresponding MAP3K2 residues (Lys257–Lys260), whereas the last three residues point to different directions and are less involved in interaction with SMYD3 (Fig. 2D). VEGFR1 interacts with SMYD3 in a manner similar to the MAP3K2 peptide. The dimethylated Lys831 residue of VEGFR1 is surrounded by hydrophobic residues of SMYD3 including Phe183, Tyr239, and Tyr257 as well. Intermolecular hydrogen bonds are nearly identical in the two complex structures, except that in the structure of the VEGFR1 complex, the side chain conformations of Glu192 and Asp241 of SMYD3 are altered by interaction with the side chains of Lys828 and Ser832 of VEGFR1, respectively.
Structural Basis for Substrate Preference of SMYD3
Structural comparison between the MAP3K2 and VEGFR1 bound structures shows that the first four residues superimpose well, whereas the conformations of the remaining residues are more divergent (Fig. 2D). This observation suggests a common substrate-binding mechanism of SMYD3 involving the first four residues of the substrate peptides. Sequence alignment of the reported substrates of SMYD3, including MAP3K2, VEGFR1, and histones H3 and H4 shows that major differences lie at the −2 position and the +3 and +4 position with respect to the acceptor lysine of the substrates (Fig. 3A). The residues at the −2 position are hydrophobic in MAP3K2 (Phe258) and VEGFR1 (Leu829). By contrast, an arginine residue is found at the corresponding position of histones H3 and H4. As shown in the complex structures, Phe258 of MAP3K2 and Leu829 of VEGFR1 are inserted into a shallow hydrophobic pocket of SMYD3 formed by Val178, Ile179, and Val195 (Figs. 2B and 3B). It is energetically unfavorable to place a positively charged arginine residue into the hydrophobic pocket. Furthermore, it is difficult to accommodate the long side chain of an arginine residue in the shallow pocket. Residues at the +3 and +4 positions are similar in VEGFR1 and H3K4 (Gly834-Arg835 and Ala7-Arg8, respectively), but they differ greatly from those in MAP3K2 and H4K5 (Thr263-Tyr264 and Lys8-Gly9, respectively). Thr263 and Tyr264 of MAP3K2 interact with SMYD3 mainly through their main chain amide groups, and Gly834 of VEGFR1 does not interact with SMYD3 directly.
Next, we tested the roles of residues at the −2, +3, and +4 positions of the substrate by in vitro MTA using mutated MAP3K2 peptides, including −2 position mutants F258L (changing to a leucine residue as in VEGFR1) and F258R (mutating to an arginine residue as in histones H3 and H4), and a double mutant, T263G/Y264R (mutate to glycine and arginine residues as in VEGFR1), carrying changes at +3 and +4 positions. MTA results showed that the enzymatic activity of SMYD3 is greatly reduced when the F258L or F258R mutant peptides of MAP3K2 were used as substrates (Fig. 3C). The catalytic activity of SMYD3 toward the F258L peptide decreased ∼6-fold compared with that with the wild-type peptide, whereas no methylation activity was detected when a F258R peptide was used as a substrate. In contrast, SMYD3 methylates the +3 and +4 position mutant peptide T263G/Y264R at a level comparable with that with the wild-type MAP3K2 peptide. These results indicate that the phenylalanine residue at the −2 position of MAP3K2 is crucial for determining the substrate preference of SMYD3.
As previously noted, Phe258 of MAP3K2 and Leu829 of VEGFR1 are situated in a shallow hydrophobic pocket of SMYD3 formed by residues with hydrophobic side chains including Val178, Ile179, and Val195. However, two serine residues, Ser101 and Ser182, are located at the perimeter of this pocket as well (Fig. 3B). The side chain hydroxyl groups of the serine residues are hydrophilic and may not be optimal for the hydrophobic environment surrounding the phenyl ring of Phe258. To test our prediction that the hydrophobic environment of the shallow SMYD3 pocket is important for substrate binding, we changed the composition of this pocket in two opposite ways. First, we changed the existing hydrophobic residues to smaller, less hydrophobic residues and tested their enzymatic activities by the in vitro MTA assay using the MAP3K2 peptide as a substrate. The results show that the V195A mutant lost approximately half of its activity compared with the wild-type SMYD3, whereas the activity of the V195T mutant decreased ∼5-fold (Fig. 3D). Similarly, an I179A mutation reduced the SMYD3 activity to a level comparable with that of the V195T mutant. Second, we changed the hydrophilic serines to more hydrophobic alanines. Our prediction implies that the serine to alanine mutants should be beneficial to the methylase activity of SMYD3. Reassuringly, we find that both the S101A and S182A mutants of SMYD3 showed ∼2-fold higher activities than the wild-type enzyme (Fig. 3D). Therefore, we conclude that the implicated shallow hydrophobic pocket is a crucial determinant of substrate specificity of SMYD3.
Discussion
Confusing claims of various substrates for SMYD3 have been made, including histone H3 at lysine 4, histone H4 at lysine 5, VEGFR1 at lysine 831, and MAP3K2 at lysine 260 (9, 12, 13, 19). Could they be all correct, only one correct, or some correct? The answer to this question is important for the interpretation of experimental data, but arriving at a definitive answer may be complicated because in vivo situations, such as interacting with other cellular factors, could alter the activity of SMYD3. Our approach to this question is to examine the intrinsic properties of SMYD3 using a combined structural and biochemical analysis, hoping to identify key elements in SMYD3 that would tell its substrate preference, and this information may be used in conjunction with in vivo analysis to aid reasonable interpretation and prediction of the biological functions of SMYD3. Judging by our structural and biochemical results, we conclude that lysine 260 of MAP3K2 is the most likely substrate of SMYD3, because of the presence of a shallow hydrophobic substrate-binding pocket hosting the binding of the −2 residue of the substrate peptide.
Previous structural and biochemical studies have examined the important roles of residues in the SAM-binding pocket and the lysine-binding channel for the catalytic activities of SMYD3 using histone mixtures extracted from calf thymus as substrate. These residues include Phe259, Asn132, and Tyr124 in the SAM-binding pocket and Tyr239 and Phe183 in the lysine-binding channel (31). These results agree well with our structural information derived from the ternary structure of SMYD3 in complex with MAP3K2 and SAH (Fig. 2C). In this study, we identified MAP3K2 as the preferred substrate of SMYD3 compared with VEGFR1 and histones H3 and H4. Our analysis shows that the identity of the amino acid at the −2 position of the substrate peptides is key for being an optimal substrate of SMYD3. The shallow hydrophobic pocket of SMYD3 that hosts the binding of the −2 position phenylalanine residue of MAP3K2 is critical for substrate binding and enzymatic activity. Optimal substrate-enzyme interaction through this shallow hydrophobic pocket largely accounts for the preference of SMYD3 for the reported substrates.
Obviously, natural features of the substrate other than the property of the −2 position residue, such as the folding of substrate protein, may pose additional hurdles for the catalytic activity of SMYD3. For example, Lys831 is located in the kinase domain of VEGFR1 (23), residing in the middle of an outer β-strand of the N-terminal β-sheet domain (Fig. 4). Assuming that the kinase domain of VEGFR1 is not unfolded under a physiological setting, it is not possible to position Lys831 next to the substrate-binding channel of SMYD3 without a large rearrangement of SMYD3 domains. Docking of a folded kinase domain of VEGFR1 with SMYD3 shows the requirement of a dramatic and unnatural widening (at least 30° is required) of the substrate-binding cleft formed by C-terminal domain and post-SET domain of SMYD3 (Fig. 4). In addition, the docked Lys831 residue of VEGFR1 is ∼90° away from the dimethylated Lys831 residue of the co-crystallized VEGFR1 peptide, and the lysine is far away from the sulfur atom of the SAH molecule, which is next to the enzyme active site. In the case with histone H3, SMYD3 has a low methyltransferase activity on recombinant histone H3 to begin with and shows no detectable activities toward recombinant histone octamer, nucleosome, or oligonucleosome substrates (34) (supplemental Fig. S4A). For MAP3K2, Lys260 is located in a long loop region between the N-terminal PB1 domain and C-terminal kinase domain, which leaves adequate flexibility for SMYD3 binding (supplemental Fig. S4B).
FIGURE 4.
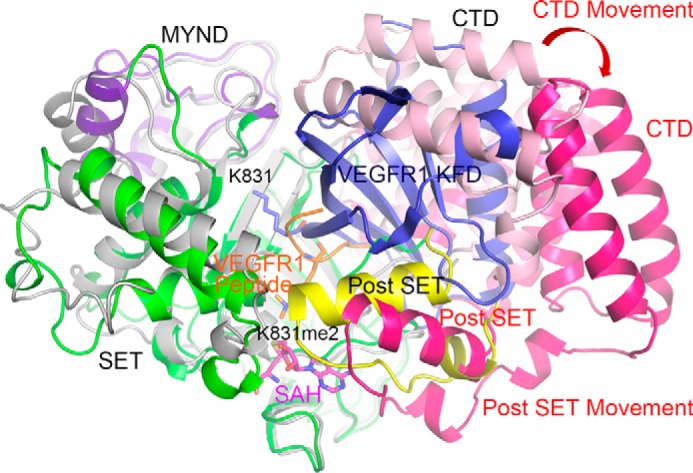
A computational model of the kinase fold domain of VEGFR1 docked into the substrate-binding cleft of a stimulated SMYD3 structure. The crystal structure of SMYD3 is shown as in Fig. 2A, and the simulated N- and C-terminal domains of SMYD3 are shown in light gray and hot pink, respectively. VEGFR1 peptide is shown in orange, and the dimethylated Lys831 residue is highlighted in a stick model. The kinase fold domain of VEGFR1 is shown in blue, and the Lys831 residue is highlighted in a stick model, which is far away from the catalytic center. Movement of the C-terminal domain and the post-SET domain after simulation is denoted by a red arrow.
Taken together, our structural and biochemical analysis provided mechanistic understanding of the substrate preference of SMYD3, and the mechanistic insights should shed light on its biological functions. The structural information should also be useful for the development of SMYD3 inhibitors, which may serve as therapeutic agents against MAP3K2 and Ras-driven cancer.
Author Contributions
W. F., N. L., Q. Q., B. Z., R.-M. X., and N. Y. designed research; W. F., N. L., Q. Q., and M. W. performed research; W. F., N. L., Q. Q., M. W., J. M., B. Z., R.-M. X., and N. Y. analyzed data; and N. Y., R.-M. X., and B. Z. wrote the paper.
Supplementary Material
Acknowledgments
We thank the staff at Shanghai Synchrotron Radiation Facility for help in diffraction data collection, Dr. Hongjie Zhang for technical support of radioactivity detection, and Drs. Zhi Chen and Zhuo Mao for participation at an early stage of the work.
This work was supported by Chinese Ministry of Science and Technology Grants 2015CB856200 and 2012CB910702, Natural Science Foundation of China Grants 31521002, 31430018, and 31370734, Chinese Academy of Sciences Strategic Priority Research Program Grant XDB08010100, and the National Key New Drug Creation and Manufacturing Program of China Grant 2014ZX09507002. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Experimental Procedures and supplemental Figs. S1–S4.
The atomic coordinates and structure factors (codes 5EX0 and 5EX3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- SET
- Su(var), E(z), Trithorax
- SAM
- S-adenosyl-l-methionine
- MYND
- myeloid-Nervy-DEAF-1
- SMYD
- SET and MYND domain-containing proteins
- a.a.
- amino acid(s)
- PDB
- Protein Data Bank
- MTA
- methyltransferase assay
- SAH
- S-adenosyl-l-homocysteine.
References
- 1. Grewal S. I., and Elgin S. C. (2002) Heterochromatin: new possibilities for the inheritance of structure. Curr. Opin. Genet. Dev. 12, 178–187 [DOI] [PubMed] [Google Scholar]
- 2. Lachner M., and Jenuwein T. (2002) The many faces of histone lysine methylation. Curr. Opin. Cell Biol. 14, 286–298 [DOI] [PubMed] [Google Scholar]
- 3. Strahl B. D., and Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 4. Wu J., and Grunstein M. (2000) 25 years after the nucleosome model: chromatin modifications. Trends Biochem. Sci. 25, 619–623 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y., and Reinberg D. (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360 [DOI] [PubMed] [Google Scholar]
- 6. Ivanov G. S., Ivanova T., Kurash J., Ivanov A., Chuikov S., Gizatullin F., Herrera-Medina E. M., Rauscher F. 3rd, Reinberg D., and Barlev N. A. (2007) Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell Biol. 27, 6756–6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nishioka K., Chuikov S., Sarma K., Erdjument-Bromage H., Allis C. D., Tempst P., and Reinberg D. (2002) Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Gene Dev. 16, 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown M. A., Sims R. J. 3rd, Gottlieb P. D., and Tucker P. W. (2006) Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamamoto R., Furukawa Y., Morita M., Iimura Y., Silva F. P., Li M., Yagyu R., and Nakamura Y. (2004) SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 6, 731–740 [DOI] [PubMed] [Google Scholar]
- 10. Tan X., Rotllant J., Li H., De Deyne P., and Du S. J. (2006) SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl. Acad. Sci. U.S.A. 103, 2713–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang J., Perez-Burgos L., Placek B. J., Sengupta R., Richter M., Dorsey J. A., Kubicek S., Opravil S., Jenuwein T., and Berger S. L. (2006) Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632 [DOI] [PubMed] [Google Scholar]
- 12. Kunizaki M., Hamamoto R., Silva F. P., Yamaguchi K., Nagayasu T., Shibuya M., Nakamura Y., and Furukawa Y. (2007) The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 67, 10759–10765 [DOI] [PubMed] [Google Scholar]
- 13. Mazur P. K., Reynoird N., Khatri P., Jansen P. W., Wilkinson A. W., Liu S., Barbash O., Van Aller G. S., Huddleston M., Dhanak D., Tummino P. J., Kruger R. G., Garcia B. A., Butte A. J., Vermeulen M., Sage J., and Gozani O. (2014) SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X., Tanaka K., Yan J., Li J., Peng D., Jiang Y., Yang Z., Barton M. C., Wen H., and Shi X. (2013) Regulation of estrogen receptor alpha by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl. Acad. Sci. U.S.A. 110, 17284–17289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamamoto R., Silva F. P., Tsuge M., Nishidate T., Katagiri T., Nakamura Y., and Furukawa Y. (2006) Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 97, 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zou J. N., Wang S. Z., Yang J. S., Luo X. G., Xie J. H., and Xi T. (2009) Knockdown of SMYD3 by RNA interference down-regulates c-Met expression and inhibits cells migration and invasion induced by HGF. Cancer Lett. 280, 78–85 [DOI] [PubMed] [Google Scholar]
- 17. Tsuge M., Hamamoto R., Silva F. P., Ohnishi Y., Chayama K., Kamatani N., Furukawa Y., and Nakamura Y. (2005) A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5′ flanking region of SMYD3 is a risk factor for human cancers. Nat. Genet. 37, 1104–1107 [DOI] [PubMed] [Google Scholar]
- 18. Silva F. P., Hamamoto R., Kunizaki M., Tsuge M., Nakamura Y., and Furukawa Y. (2008) Enhanced methyltransferase activity of SMYD3 by the cleavage of its N-terminal region in human cancer cells. Oncogene 27, 2686–2692 [DOI] [PubMed] [Google Scholar]
- 19. Van Aller G. S., Reynoird N., Barbash O., Huddleston M., Liu S., Zmoos A. F., McDevitt P., Sinnamon R., Le B., Mas G., Annan R., Sage J., Garcia B. A., Tummino P. J., Gozani O., and Kruger R. G. (2012) Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 7, 340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dales J. P., Garcia S., Bonnier P., Duffaud F., Carpentier S., Djemli A., Ramuz O., Andrac L., Lavaut M., Allasia C., and Charpin C. (2003) [Prognostic significance of VEGF receptors, VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) in breast carcinoma]. Ann. Pathol. 23, 297–305 [PubMed] [Google Scholar]
- 21. Wu Y., Hooper A. T., Zhong Z., Witte L., Bohlen P., Rafii S., and Hicklin D. J. (2006) The vascular endothelial growth factor receptor (VEGFR-1) supports growth and survival of human breast carcinoma. Int. J. Cancer 119, 1519–1529 [DOI] [PubMed] [Google Scholar]
- 22. Yang A. D., Camp E. R., Fan F., Shen L., Gray M. J., Liu W., Somcio R., Bauer T. W., Wu Y., Hicklin D. J., and Ellis L. M. (2006) Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 66, 46–51 [DOI] [PubMed] [Google Scholar]
- 23. Starovasnik M. A., Christinger H. W., Wiesmann C., Champe M. A., de Vos A. M., and Skelton N. J. (1999) Solution structure of the VEGF-binding domain of Flt-1: comparison of its free and bound states. J. Mol. Biol. 293, 531–544 [DOI] [PubMed] [Google Scholar]
- 24. Colón-Bolea P., and Crespo P. (2014) Lysine methylation in cancer: SMYD3-MAP3K2 teaches us new lessons in the Ras-ERK pathway. BioEssays 36, 1162–1169 [DOI] [PubMed] [Google Scholar]
- 25. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 26. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee R. A., Razaz M., and Hayward S. (2003) The DynDom database of protein domain motions. Bioinformatics 19, 1290–1291 [DOI] [PubMed] [Google Scholar]
- 30. Chen R., Li L., and Weng Z. (2003) ZDOCK: An initial-stage protein-docking algorithm. Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 31. Xu S., Wu J., Sun B., Zhong C., and Ding J. (2011) Structural and biochemical studies of human lysine methyltransferase Smyd3 reveal the important functional roles of its post-SET and TPR domains and the regulation of its activity by DNA binding. Nucleic Acids Res. 39, 4438–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sirinupong N., Brunzelle J., Doko E., and Yang Z. (2011) Structural insights into the autoinhibition and posttranslational activation of histone methyltransferase SmyD3. J. Mol. Biol. 406, 149–159 [DOI] [PubMed] [Google Scholar]
- 33. Foreman K. W., Brown M., Park F., Emtage S., Harriss J., Das C., Zhu L., Crew A., Arnold L., Shaaban S., and Tucker P. (2011) Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS One 6, e22290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luger K., Rechsteiner T. J., and Richmond T. J. (1999) Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.