

Abstract
During G1-phase of the cell cycle, normal cells respond first to growth factors that indicate that it is appropriate to divide and then later in G1 to the presence of nutrients that indicate sufficient raw material to generate two daughter cells. Dividing cells rely on the “conditionally essential” amino acid glutamine (Q) as an anaplerotic carbon source for TCA cycle intermediates and as a nitrogen source for nucleotide biosynthesis. We previously reported that while non-transformed cells arrest in the latter portion of G1 upon Q deprivation, mutant KRas-driven cancer cells bypass the G1 checkpoint, and instead, arrest in S-phase. In this study, we report that the arrest of KRas-driven cancer cells in S-phase upon Q deprivation is due to the lack of deoxynucleotides needed for DNA synthesis. The lack of deoxynucleotides causes replicative stress leading to activation of the ataxia telangiectasia and Rad3-related protein (ATR)-mediated DNA damage pathway, which arrests cells in S-phase. The key metabolite generated from Q utilization was aspartate, which is generated from a transaminase reaction whereby Q-derived glutamate is converted to α-ketoglutarate with the concomitant conversion of oxaloacetate to aspartate. Aspartate is a critical metabolite for both purine and pyrimidine nucleotide biosynthesis. This study identifies the molecular basis for the S-phase arrest caused by Q deprivation in KRas-driven cancer cells that arrest in S-phase in response to Q deprivation. Given that arresting cells in S-phase sensitizes cells to apoptotic insult, this study suggests novel therapeutic approaches to KRas-driven cancers.
Keywords: aspartate (aspartic acid), cell cycle, glutamine, GTPase Kras (KRAS), nucleotide, ATR, KRas, S-Phase arrest
Introduction
One of the emerging hallmarks of cancer is metabolic transformation (1). Cancer cells are highly dependent on aerobic glycolysis, also known as the Warburg effect (2). To meet the higher nutrient requirements of proliferation cancer cells rely on glutamine (Q)2 as a carbon and nitrogen source to support cell growth and promote survival (3). Q, which is considered as “conditionally essential” amino acid, is the most significant amino acid for providing carbon for TCA cycle intermediates as well as nitrogen for nucleotide and non-essential amino acid synthesis (4). Q donates its amide nitrogen directly into the purine and pyrimidine ring structures during nucleotide biosynthesis and is converted into glutamate (5). Glutamate further undergoes a transamination reaction with oxaloacetate catalyzed by glutamate oxaloacetate transaminase (GOT) yielding α-ketoglutarate (α-KG), which can enter into the TCA cycle, and aspartate, which is a key intermediate for both purine and pyrimidine nucleotide biosynthesis.
The most common driver mutations found in cancer cells are in genes encoding proteins that regulate progression through the G1 phase of the cell cycle (6). The Myc oncoprotein enhances the transcription of genes that are involved in Q metabolism and makes cancer cells addicted to glutamine (7, 8). Cells have checkpoints late in G1 that check for sufficient nutrients prior to committing to replicating the genome and dividing into two cells (6, 9). In addition to nutrient sensing checkpoints, there are also checkpoints that monitor genomic integrity and DNA damage. When the DNA is damaged, the cell cycle is halted at late G1, S-phase and prior to cytokinesis (10). The S-phase checkpoints are mediated by two protein kinases: ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) (11). The outcome of activating these pathways is either cell cycle arrest or apoptosis. We have recently distinguished a series of late G1 nutrient sensing checkpoints that are distinct from the mid-G1 growth factor-dependent restriction point (9). One of the nutrient-sensing checkpoints is for Q (9). Significantly, KRas-driven cancer cells deprived of Q override the G1 checkpoint and instead arrest in S-phase (9, 12). This finding has important therapeutic implications for KRas-driven cancers because cells in S-phase can be selectively killed by chemotherapeutic agents such as capecitabine and rapamycin (9, 12, 13).
In this study, we have investigated the mechanism for the S-phase arrest observed in KRas-driven cancer calls in response to Q deprivation. We report here that the S-phase arrest in response to Q deprivation is due to a lack of aspartate generated by the transamination reaction between glutamate and oxaloacetate. The lack of aspartate impairs nucleotide biosynthesis and the shortage of nucleotides generates single-stranded DNA that activates the ATR DNA damage pathway and arrests cells in S-phase.
Experimental Procedures
Cells and Cell Culture Conditions
The human cancer cell lines MDA-MB-231, MCF-7, Calu-1, and BJ-hTERT cells were obtained from the American Tissue Type Culture Collection (ATCC). All the cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma D6429) supplemented with 10% fetal bovine serum (Sigma F4135).
Materials
Reagents were obtained from the following sources: Antibodies against, phospho-Chk1 (Ser-345) (2348), phospho-Chk-2 (Thr-68) (2197), Chk-1 (2360), Chk-2 (2662), and actin (8457) were obtained from Cell Signaling; Antibody against PPAT (HPA036092) was obtained from Atlas Antibodies; anti-mouse and anti-rabbit HRP-conjugated secondary antibodies were obtained from Promega. For the Gln deprivation; Dulbecco's modified Eagle's medium without Q (D5546), dialyzed fetal bovine serum (F0392), l-glutamine (G7513), aminooxyacetate (AOA) (C13408), DMKG (349631) and aspartate (β-MD) (A8291) were obtained from Sigma-Aldrich. MK-8776 (sc364611) was obtained from Santa-Cruz Biotechnology. Negative control scrambled siRNA (d-001206-13-05), siRNAs targeted against Chk-1(M-003255-04-0005), and siRNAs targeted against phosphoribosyl pyrophosphate amidotransferase (PPAT) (M-006003-01–0005) was obtained from Dharmacon. dNSs (ES-008-D) were purchased from EMD Millipore. Ultima Gold scintillation fluid (6013681) and [3H]thymidine deoxyribose (TdR) (20 Ci/mmol, 1 mCi/ml) (NET-027E) were obtained from PerkinElmer.
Transient Transfections
Cells were plated in 6-well plates in medium containing 10% FBS. The next day, transfections with siRNAs (100 nm) in Lipofectamine RNAiMAX were performed. After 6 h, reagents were replaced with fresh 10% FBS, and cells were allowed to incubate for indicated times as explained in the experiments.
Flow Cytometric Analysis
Cell cycle distribution was determined by flow cytometry as described previously (9). Briefly, cells were fixed in 70% ethanol, stained using propidium iodide, and passed through 70-μm meshes to remove cell aggregates. Fluorescence intensity corresponding to DNA content in different phase of cell cycle was measured by flow cytometry (FACSCalibur; Becton Dickinson), and analyzed using WinCycle software (Phoenix Flow Systems). Each experiment was performed in duplicate and two-way ANOVA tests were performed in all statistical analyses. p values for the S-phase population in MDA-MB-231, MCF-7, and BJ-hTERT and Calu-1 cells, across the samples are expressed relative to control Q. Raw data for flow cytometry experiments are provided as supplemental figures.
Western Blot Analysis
Proteins were extracted from cultured cells in M-PER (Thermo Scientific 78501). Equal amounts of proteins were subjected to SDS-PAGE on polyacrylamide separating gels. Electrophoresed proteins were then transferred to nitrocellulose membrane. After transfer, membranes were blocked in an isotonic solution containing 5% nonfat dry milk in phosphate-buffered saline. Membranes were then incubated with primary antibodies as described in the text. The dilutions were used per vendors instructions. Depending on the origin of the primary antibody, either anti-mouse or anti-rabbit HRP-conjugated IgG was used for detection using ECL system (Thermo Scientific 34080).
Thymidine Incorporation Assay
Cells were labeled with 1μCi/ml [3H]thymine deoxyribose (TdR). At indicated times, cells were washed twice with 1 ml phosphate-buffered saline, and then precipitated twice with 1 ml of 10% trichloroacetic acid. The precipitates were solubilized in 0.5 ml of 0.5% SDS/0.5 M NaOH solution, and the extent of TdR incorporation was quantified using 75 μl of sample and 3 ml of scintillation fluid. Each experiment was performed in duplicate, and one-way ANOVA tests were performed in all statistical analyses.
Results
Deoxynucleosides Reverse the S-phase Arrest Caused by Q Deprivation in KRas-driven Cancer Cells
Since Q provides nitrogen for purine and pyrimidine nucleotide biosynthesis (14, 15), Q deprivation could disrupt the pool of available nucleotides in cells by interfering with de novo purine and pyrimidine biosynthesis. To test this hypothesis, we subjected KRas-driven MDA-MB-231 breast cancer cells, non-KRas-driven MCF-7 breast cancer cells, and non-cancerous BJ-hTERT fibroblasts to Q deprivation for 48 h. As observed previously (9, 12, 13), the MDA-MB-231 cells arrested in S-phase, whereas the MCF7 and BJ-hTERT cells arrested in G1-phase upon Q deprivation (Fig. 1A). After 48 h of Q deprivation, a mixture of deoxynucleosides was added exogenously, and cell cycle progression was monitored by flow cytometric analysis. As shown in Fig, 1A, we were able to reverse the S-phase cell cycle arrest caused by Q deprivation with the addition of nucleosides to the MDA-MB-231 cells. The deoxynucleoside mixture had no effect on the G1-arrested MCF7 and BJ-hTERT cells (Fig. 1A). These data indicate that the S-phase arrest of the KRas-driven MDA-MB-231 cells induced by Q deprivation was due to the depletion of deoxynucleotides needed for DNA synthesis.
FIGURE 1.

Reversal of S phase arrest in K-Ras mutant cells upon exogenous addition of nucleosides. A, MDA-MB-231, MCF-7, and BJ-hTERT cells were plated at 30% confluence in 10-cm plates in DMEM containing 10% fetal bovine serum (FBS). After 24 h, cells were shifted to DMEM containing or lacking Q for 48 h. Both +Q and -Q medium contained 10% dialyzed fetal bovine serum. At this point, cells were transferred into medium lacking Q with or without nucleosides. The lysates were collected at different time intervals and analyzed for cell cycle distribution by measuring DNA content/cell using flow cytometric analysis. The error bars represent S.D. for experiments repeated for two times. p values for the S-phase population in MDA-MB-231, MCF-7, and BJ-hTERT cells, across the samples are expressed relative to control Q. B, cells were plated and treated as in A. Cells were harvested at indicated time points, stained using crystal violet and quantified by light microscopy. Error bars represent S.D. for experiments repeated for two times. C, cells were plated in 12-well plates with 30% confluence and shifted to the medium as explained in A. After 48 h of Q deprivation, cells were shifted to medium lacking Q with or without deoxynucleosides (dNSs) for 24 h. [3H]thymine deoxyribose (TdR) (20 Ci/mmol, 1 mCi/ml) was added for the last 2 h of treatment. The samples were then subjected to scintillation counting to measure labeled DNA. Values were normalized to the cpm for +Q, which were given values of 100%. Error bars represent S.D. for experiments repeated two times. ns, not significant is p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
We next examined whether the deoxynucleosides promoted cell proliferation in the MDA-MB-231 cells deprived of Q. As shown in Fig. 1B, addition of deoxynucleosides promoted cell proliferation in MDA-MB-231, but not in MCF7 or BJ-hTERT cells, which were arrested in G1 upon glutamine starvation. This finding demonstrates that providing deoxynucleosides not only allows progression through S-phase, but allows cells to undergo mitosis as well. The lack of an effect on the MCF7 and BJ-hTERT cells suggests that the Q requirement for nucleotide biosynthesis is restricted to S-phase arrest only.
To further establish that the deoxynucleosides were restoring DNA synthesis we examined the impact of deoxynucleosides on DNA synthesis as indicated by the incorporation of [3H]thymine deoxyribose (TdR). KRas-driven MDA-MB-231 breast and Calu-1 lung cancer cells along with the non-KRas-driven MCF7 breast cancer cells and the BJ-hTERT cells were subjected to Q deprivation for 48 h and then treated with the deoxynucleoside mix for 24 h as indicated. [3H]TdR was added for the last 2 h at which time, the cells were harvested, and the level of [3H]TdR incorporation was determined. As shown in Fig. 1C, Q deprivation resulted in the suppression of 80% of the [3H]TdR incorporation in all cell lines. When deoxynucleosides were provided, [3H]TdR incorporation was restored in the S-phase arrested MDA-MB-231 and Calu-1 cells, but not in the G1-arrested MCF-7 and BJ-hTERT cells. These data demonstrate that DNA synthesis was restored by the presence of deoxynucleosides in Q-deprived KRas-driven cancer cells.
Aspartate Reverses the S-phase Arrest in KRas-driven Cancer Cells
We previously reported that we could mimic Q-deprivation with the transaminase inhibitor AOA (12), which blocks the conversion of Q-derived glutamate to α-KG in KRas-driven cancer cells (16). Importantly, the conversion of glutamate to α-KG is coupled with the conversion of oxaloacetate to aspartate by the enzyme GOT. Thus, inhibition of GOT with AOA suppresses the production of both α-KG and aspartate. KRas-driven MDA-MB-231 and Calu-1 cells were treated with AOA for 48 h and as reported previously (12, 13), the cells arrested in the S- and G2-phases of the cell cycle (Fig. 2B). As was observed with Q deprivation in Fig. 1a, the S-phase arrest induced by AOA could be reversed with deoxynucleosides (Fig. 2A). Thus, the S-phase arrest in KRas-driven cancer cells induced by inhibiting Q utilization with AOA can be reversed by providing deoxynucleosides. Of significance, aspartate is a critical metabolite in the synthesis of both purines and pyrimidines. We therefore examined whether the AOA-induced S-phase arrest could be reversed by providing cell permeable analogs of α-KG and/or aspartate, the products of GOT. As shown in Fig. 2B, aspartate especially reversed AOA-induced suppression of [3H]TdR in MDA-MB-231 and Calu-1 cells, whereas α-KG had very little effect. In contrast, both aspartate and α-KG could reverse the suppression of [3H]TdR incorporation in MCF7 cells. Similar results were obtained using flow cytometry where aspartate could reverse the S-phase arrest seen in the KRas-driven cancer cells, whereas both α-KG and aspartate could only partially reverse the G1 arrest seen in MCF7 cells (Fig. 2C). Thus, it appears that suppression of aspartate production by the transaminase reaction catalyzed by GOT is a key factor for inducing S-phase arrest in response to Q deprivation. Since aspartate is a metabolite in purine and pyrimidine biosynthesis, deprivation of aspartate could reduce the pool of nucleotides needed for DNA synthesis and cause S-phase arrest.
FIGURE 2.

S-G2 phase arrest caused by inhibition of utilization of glutamine can be reversed by addition of nucleosides and aspartate exogenously. A, MDA-MB-231 and Calu-1 were plated at 30% confluence in 10-cm plates in Complete medium (CM) containing 10% FBS. After 24 h, cells were shifted to medium containing or lacking 0.5 mm AOA for 48 h. At this point, cells were transferred into media containing AOA with or without dNSs. Lysates were collected after 48 h and analyzed for cell cycle distribution by measuring DNA content/cell using flow cytometric analysis. Error bars represent S.D. for experiments repeated two times. p values for the S-phase and G2-phase population in MDA-MB-231 and Calu-1 cells respectively, across the samples are expressed relative to control Q. B, MDA-MB-231, Calu-1, and MCF-7 cells were plated and treated with AOA for 48 h as in A. Along with AOA, cells were treated with cell permeable analogues of α-KG (DMKG; 4 mm) and aspartate (β-MD, 10 mm). [3H]thymidine deoxyribose (TdR) (20 Ci/mmol, 1 mCi/ml) was added for the last 24 h of the treatment. Cells were then subjected to scintillation counting to measure labeled DNA. Error bars represent S.D. for experiments repeated two times. C, MDA-MB-231, Calu-1, and MCF-7 cells were plated and treated with AOA for 48 h as described in A. Along with AOA, cells were treated with the cell permeable analogues of α-KG and aspartate. Cells were harvested after 48 h and analyzed for cell cycle distribution by measuring DNA content/cell using flow cytometric analysis. Error bars represent S.D. for experiments repeated two times. p values for the S-phase in MDA-MB-231; MCF-7 and G2-phase population in Calu-1 cells, across the samples are expressed relative to control Q (ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.).
Blocking Nucleotide Biosynthesis Causes an S-Phase Arrest
The data in Figs. 1 and 2 suggest that the S-phase arrest observed in the absence of Q is due the lack of Q-derived precursors for purine and pyrimidine biosynthesis. We therefore investigated whether suppressing de novo purine and pyrimidine biosynthetic pathways would, like Q deprivation, also lead to S-phase arrest in KRas-driven cancer cells. A rate-limiting step in the de novo biosynthetic pathway for purine nucleotides is conversion of 5-phosphoribosyl-α-pyrophosphate and Q into glutamate and β-5-phosphoribosylamine, which is catalyzed by phosphoribosyl pyrophosphate amidotransferase (PPAT). Thus, knockdown of PPAT should block the utilization of Q for purine nucleotide biosynthesis and mimic Q deprivation. We therefore used siRNA targeted against PPAT to suppress its expression in the KRas-driven cancer cell lines MDA-MB-231 and Calu-1. As shown in Fig. 3A, complete knockdown of PPAT was observed after 72 h treatment with the siRNA. At this point MDA-MB-231, Calu-1, and MCF7 cells showed increased S-phase cell populations (Fig. 3B). These data are consistent with the hypothesis that Q deprivation disrupts nucleotide biosynthesis and leads to S-phase arrest.
FIGURE 3.

S-phase arrest upon blocking de novo nucleotide biosynthesis pathway. A, MDA-MB-231 and Calu-1 cells were plated at 60% confluence in 6-well plates in CM. After 24 h, cells were transfected with either scrambled (Scr) or PPAT siRNA. Western blot was performed on lysates collected at the indicated time points to check the levels of PPAT. The data shown are representative of experiments repeated two times. B, cells were prepared as in A and then shifted to fresh medium for 96 h. The cells were collected, and flow cytometric analysis was performed for cell cycle distribution by measuring DNA content/cell. The error bars represent S.D. for experiments repeated two times. p values for the S-phase population in MDA-MB-231 and Calu-1 cells, across the samples are expressed relative to control Q (ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.).
Glutamine Deprivation Activates the ATR-mediated DNA Damage Pathway
During DNA replication, cells are on high alert for DNA abnormalities such as strand breaks or base modifications that interfere with replicating the genome. Throughout this process, if anything is wrong, checkpoints mediated by DNA damage are activated, and cell cycle progression stops (17, 18). We postulated that since Q-deprived KRas mutant cells lack sufficient nucleotides to replicate the genome, stretches of single-stranded DNA would be generated from unreplicated DNA caused by the lack of deoxynucleotides. Single-stranded DNA stimulates the ATR pathway whereas double strand breaks caused by ionizing radiation stimulate the ATM pathway. In response to unreplicated single-stranded DNA, ATR phosphorylates checkpoint kinase 1 (Chk1); and in response double strand breaks, ATM phosphorylates Chk2 (19). We therefore examined the impact of Q deprivation on the phosphorylation of Chk1 and Chk2 in cells harboring KRas mutations and those without KRas mutations. As shown in Fig. 4A, both Q deprivation and AOA stimulated a robust increase in Chk1, but not Chk2 phosphorylation in the KRas-driven MDA-MB-231 and Calu-1 cells. Neither Q deprivation nor AOA was able to induce Chk1 phosphorylation in the MCF7 or BJ-hTERT cells that are arrested in G1 by the treatments (Fig. 4A). These data indicate that in K-Ras mutant cells, glutamine starvation leads to replication stress due to insufficient precursors for deoxynucleotide synthesis, which in turn causes stretches of single-stranded DNA that activates the ATR-mediated DNA damage pathway and causes S-phase arrest. The stimulation of Chk1 phosphorylation by Q deprivation could be overcome with the addition of deoxynucleosides (Fig. 4B). These data implicate the ATR-mediated DNA damage pathway in the S-phase arrest that occurs in KRas-driven cancer cells deprived of Q.
FIGURE 4.

Activation of ATR-mediated DNA damage pathway upon deprivation or inhibition of utilization of the glutamine. A, MDA-MB-231, Calu-1, MCF-7, and BJ-hTERT cells were plated at 30% confluence in CM. After 24 h, cells were transferred to medium with Q, without Q and medium containing Q and AOA both for 48 h. Cells were then collected, and Western blots were performed for phospho-Chk1 (Ser-345), phospho-Chk2 (Thr-68), Chk1, Chk2, and actin. Cisplatin (10 μm) and hydroxurea (5 mm) treated cells were used as positive controls to stimulate the phosphorylation of Chk1 and Chk2, respectively. The data shown are representative of experiments repeated two times. B, MDA-MB-231 and Calu-1 cells were plated at 30% confluence in 10-cm plates in CM. After 24 h, cells were shifted to DMEM containing Q or DMEM without Q for 48 h. At this point, cells were transferred into medium lacking Q with or without nucleosides. The lysates were collected after 24 h and subjected to Western blots analysis for phospho-Chk1 and Chk-1. The data shown are representative of experiments repeated two times. C, MDA-MB-231 and Calu-1 cells were plated at 30% confluence in 10-cm plate in CM. Next day; cells were shifted to medium containing no Q alongside DMSO or MK-8776 (10 μm) for 48 h. Cells were then collected, and cell cycle distribution was determined by flow cytometry. The error bars represent S.D. for experiments repeated two times. Western blot was also performed to check the phosphorylation of Chk-1 and total Chk-1. The data shown are representative of experiments repeated two times. p values for the S-phase population in MDA-MB-231 and Calu-1 cells, across the samples are expressed relative to control Q. D, MDA-MB-231 and Calu-1 cells were plated at 60% confluence in 6-well plates in CM. After 24 h, cells were transfected with either scrambled or Chk-1 siRNA. 48 h later, cells were transferred to Q-deprived medium another 48 h. Cells were then collected, and cell cycle distribution was determined as above. The error bars represent S.D. for experiments repeated for two times. Western blot was also performed to check the levels of Chk-1. The data shown are representative of experiments repeated two times. p values for the S-phase population in MDA-MB-231 and Calu-1 cells, across the samples are expressed relative to control Q (ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.).
To further establish that the S-phase arrest observed in KRas-driven cancer cells upon Q deprivation was dependent of the ATR-Chk1 stress response pathway, we investigated whether suppression of Chk1 phosphorylation of Chk1 expression could reverse the S-phase arrest seen with Q deprivation. We first examined the effect of the ATR kinase inhibitor MK-8776 (20), and as shown in Fig. 4C, this compound suppressed the phosphorylation of Chk1 induced by Q deprivation and reduced the S-phase population of both MDA-MB-231 and Calu-1 cells. The treatment with this compound for 48 h did not result in significant cell death (data not shown). We also examined the impact of suppressing Chk1 expression with siRNA. As shown in Fig. 4D, suppressed Chk1 expression also reduced the population of cells with S-phase DNA content in both the MDA-MB-231 and Calu-1 cells when deprived of Q. These data further demonstrate that the S-phase arrest in KRas-driven cancer cells observed with Q deprivation is dependent on the ATR-Chk1 stress pathway.
Discussion
We previously reported that KRas-driven cancers bypass a distinct Q-dependent late G1 checkpoint and arrest in S-phase in response to Q deprivation (9). Significantly, the cells arrested in S-phase are selectively killed by rapamycin (13) and other cytotoxic agents (12). Thus, the observation that KRas-driven cancer cells arrest in S-phase in response to Q deprivation has important clinical implications, in that KRas itself has been considered undruggable (21). In this report, we have identified the mechanism for S-phase arrest observed with Q deprivation. The lack of Q led to reduced levels of precursors needed for nucleotide biosynthesis, which induced replicative stress leading to an ATR and Chk1-mediated S-phase arrest.
The ATR/Chk1 stress response pathway responds to stretches of single-stranded DNA caused by insufficient deoxynucleotides. Q, a conditionally essential amino acid, serves a critical anaplerotic agent that feeds the TCA cycle when citrate is shuttled out of the mitochondria for generating cytosolic acetyl-CoA for fatty acid synthesis in dividing cells. Q can be deamidated to glutamate and then deaminated to generate the TCA cycle intermediate α-KG, which is just downstream from citrate in the TCA cycle (Fig. 5). However, in KRas-driven cancer cells, the conversion of glutamate to α-KG is accompanied by the conversion of oxaloacetate to aspartate in the transamination reaction catalyzed by GOT2 (12, 16). Significantly, the S-phase arrest induced by inhibition of GOT2 could be overcome largely with aspartate. α-KG had little effect on the S-phase arrest induced by suppression of GOT2. Aspartate exits the mitochondria via the malate-aspartate shuttle that is usually used for shuttling electrons into the mitochondria, but is also important for generating cytosolic aspartate that can contribute to redox balance and amino acid and nucleotide biosynthesis (Fig. 5) (22). It is also of interest that the deamidation reaction catalyzed by PPAT generates cytosolic glutamate from Q. This is important because it generates cytosolic glutamate that can be used to facilitate mitochondrial export of aspartate via the malate-aspartate shuttle where the exit of aspartate is coupled to the uptake of glutamate. Aspartate is a critical metabolite for purine and pyrimidine biosynthesis. Several recent reports have similarly identified aspartate derived from several metabolic pathways as being critical in dividing cells for nucleotide biosynthesis including fatty acid metabolism (23), the urea cycle (24), and TCA cycle-derived citrate (25, 26). Consistent with this role for aspartate, the S-phase arrest of KRas-driven cancer cells caused by inhibition of GOT could also be reversed with deoxynucleosides and mimicked with suppression of nucleotide biosynthesis. Collectively, the data provided here reveal that the S-phase arrest observed in KRas-driven cancer cells deprived of Q is due to replicative stress created by the lack of aspartate needed for deoxynucleotide biosynthesis. Thus, strategies for targeting aspartate synthesis pathways may be an effective means to sensitize KRas-driven cancer cells to drugs that are specific for S-phase-arrested cells.
FIGURE 5.
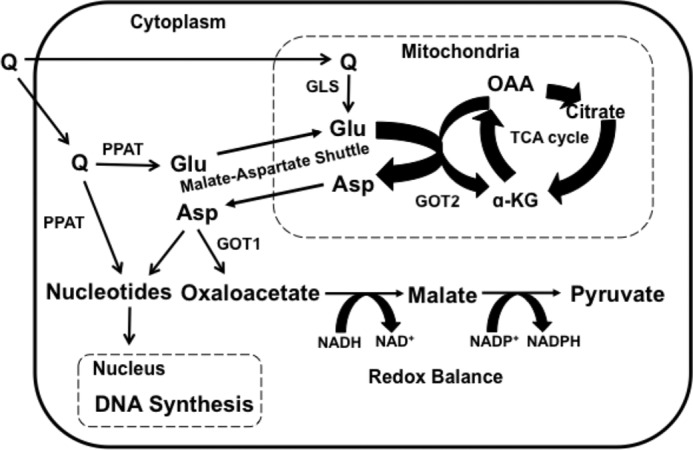
Anaplerotic utilization of Q in KRas-driven cancer cells. Q is converted to glutamate (Glu) by glutaminase (GLS) in the mitochondria; however Q is also converted to Glu by PPAT during both purine and pyrimidine synthesis. The generation of Glu by PPAT represents a cytosolic source of Glu that can facilitate the export of aspartate (ASP) from the mitochondria via the malate-Asp shuttle. A mitochondrial transaminase reaction catalyzed by GOT2 converts Glu to α-KG, and oxaloacetate (OAA) to Asp. Asp exits the mitochondria via the malate-Asp shuttle where Asp can be converted to OAA by GOT1. OAA can then be reduced by malate dehydrogenase to malate using NADH. Malate can be oxidized to pyruvate with concomitant production of NADPH and maintain redox balance. Conversely, Asp can be used for the synthesis of nucleotides needed DNA and RNA synthesis.
Author Contributions
Conceptualization, D. P., D. F., D. L.; Methodology, D. P., and D. F.; Investigation, D. P., D. M., E. B., V. M., S. K.; Writing of manuscript, D. P., D. F., D. L.
Supplementary Material
This work was supported by Grants R01-CA046677, R01-CA179542 from the NCI, National Institutes of Health. Research Centers in Minority Institutions Award RR-03039 from the National Center for Research Resources of the National Institutes of Health, which supports infrastructure and instrumentation in the Biological Sciences Dept. at Hunter College, is also acknowledged. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility ofthe authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental figures.
- Q
- glutamine
- α-KG
- α-ketoglutarate
- AOA
- aminooxyacetate
- ATM
- ataxia telangiectasia-mutated
- ATR
- ataxia telangiectasia and Rad3-related
- CM
- complete medium
- dNSs
- deoxynucleosides
- GOT
- glutamate oxaloacetate transaminase
- PPAT
- phosphoribosyl pyrophosphate amidotransferase
- TdR
- thymine deoxyribose.
References
- 1. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DeBerardinis R. J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., and Thompson C. B. (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wise D. R., and Thompson C. B. (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 35, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang J., Fan J., Venneti S., Cross J. R., Takagi T., Bhinder B., Djaballah H., Kanai M., Cheng E. H., Judkins A. R., Pawel B., Baggs J., Cherry S., Rabinowitz J. D., and Thompson C. B. (2014) Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell 56, 205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foster D. A., Yellen P., Xu L., and Saqcena M. (2010) Regulation of G1 Cell Cycle Progression: Distinguishing the Restriction Point from a Nutrient-Sensing Cell Growth Checkpoint(s). Genes Cancer 1, 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wise D. R., DeBerardinis R. J., Mancuso A., Sayed N., Zhang X. Y., Pfeiffer H. K., Nissim I., Daikhin E., Yudkoff M., McMahon S. B., and Thompson C. B. (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 105, 18782–18787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuneva M., Zamboni N., Oefner P., Sachidanandam R., and Lazebnik Y. (2007) Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 178, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saqcena M., Menon D., Patel D., Mukhopadhyay S., Chow V., and Foster D. A. (2013) Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS ONE 8, e74157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goodarzi A. A., Block W. D., and Lees-Miller S. P. (2003) The role of ATM and ATR in DNA damage-induced cell cycle control. Prog Cell Cycle Res 5, 393–411 [PubMed] [Google Scholar]
- 11. Gaillard H., García-Muse T., and Aguilera A. (2015) Replication stress and cancer. Nat. Rev. Cancer 15, 276–289 [DOI] [PubMed] [Google Scholar]
- 12. Saqcena M., Mukhopadhyay S., Hosny C., Alhamed A., Chatterjee A., and Foster D. A. (2015) Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K-Ras mutant cancer cells to cytotoxic drugs. Oncogene 34, 2672–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saqcena M., Patel D., Menon D., Mukhopadhyay S., and Foster D. A. (2015) Apoptotic effects of high-dose rapamycin occur in S-phase of the cell cycle. Cell Cycle 14, 2285–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wasa M., Bode B. P., Abcouwer S. F., Collins C. L., Tanabe K. K., and Souba W. W. (1996) Glutamine as a regulator of DNA and protein biosynthesis in human solid tumor cell lines. Ann. Surg. 224, 189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boza J. J., Moennoz D., Bournot C. E., Blum S., Zbinden I., Finot P. A., and Ballevre O. (2000) Role of glutamine on the de novo purine nucleotide synthesis in Caco-2 cells. Eur. J. Nutrition 39, 38–46 [DOI] [PubMed] [Google Scholar]
- 16. Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., Perera R. M., Ferrone C. R., Mullarky E., Shyh-Chang N., Kang Y., Fleming J. B., Bardeesy N., Asara J. M., Haigis M. C., DePinho R. A., Cantley L. C., and Kimmelman A. C. (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abraham R. T. (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15, 2177–2196 [DOI] [PubMed] [Google Scholar]
- 18. Brown E. J., and Baltimore D. (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 17, 615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reinhardt H. C., and Yaffe M. B. (2009) Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dai Y., Chen S., Kmieciak M., Zhou L., Lin H., Pei X. Y., and Grant S. (2013) The novel Chk1 inhibitor MK-8776 sensitizes human leukemia cells to HDAC inhibitors by targeting the intra-S checkpoint and DNA replication and repair. Mol. Cancer Ther. 12, 878–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., and Der C. J. (2014) Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 13, 828–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Vranken J. G., and Rutter J. (2015) You Down With ETC? Yeah, You Know D! Cell 162, 471–473 [DOI] [PubMed] [Google Scholar]
- 23. Schoors S., Bruning U., Missiaen R., Queiroz K. C., Borgers G., Elia I., Zecchin A., Cantelmo A. R., Christen S., Goveia J., Heggermont W., Goddé L., Vinckier S., Van Veldhoven P. P., Eelen G., Schoonjans L., Gerhardt H., Dewerchin M., Baes M., De Bock K., Ghesquière B., Lunt S. Y., Fendt S. M., and Carmeliet P. (2015) Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rabinovich S., Adler L., Yizhak K., Sarver A., Silberman A., Agron S., Stettner N., Sun Q., Brandis A., Helbling D., Korman S., Itzkovitz S., Dimmock D., Ulitsky I., Nagamani S. C., Ruppin E., and Erez A. (2015) Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527, 379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sullivan L. B., Gui D. Y., Hosios A. M., Bush L. N., Freinkman E., and Vander Heiden M. G. (2015) Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Birsoy K., Wang T., Chen W. W., Freinkman E., Abu-Remaileh M., and Sabatini D. M. (2015) An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.