

Abstract
Age-associated decline in organ function governs life span. We determined the effect of aging on lung function and cellular/molecular changes of 8- to 32-month old mice. Proteomic analysis of lung matrix indicated significant compositional changes with advanced age consistent with a profibrotic environment that leads to a significant increase in dynamic compliance and airway resistance. The excess of matrix proteins deposition was associated modestly with the activation of myofibroblasts and transforming growth factor-beta signaling pathway. More importantly, detection of senescent cells in the lungs increased with age and these cells contributed toward the excess extracellular matrix deposition observed in our aged mouse model and in elderly human samples. Mechanistic target of rapamycin (mTOR)/AKT activity was enhanced in aged mouse lungs compared with those from younger mice associated with the increased expression of the histone variant protein, MH2A, a marker for aging and potentially for senescence. Introduction in the mouse diet of rapamycin, significantly blocked the mTOR activity and limited the activation of myofibroblasts but did not result in a reduction in lung collagen deposition unless it was associated with prevention of cellular senescence. Together these data indicate that cellular senescence significantly contributes to the extracellular matrix changes associated with aging in a mTOR 1-dependent mechanism.
Key Words: Aging, Senescence, Remodeling, Rapamycin, mTOR pathway
Despite ample documentation of age-related changes in physiologic parameters (1), mechanisms governing the process of aging of the lungs have not been well studied. With an increase in the elderly population, more people are being diagnosed with aged-related pulmonary diseases such as chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis (2), and yet the basis/impact of the molecular changes associated with age on the onset and pathology of these diseases is not well established. The accumulation of extracellular matrix (ECM) or physiological remodeling is characteristic of aging organs and tied to loss of function. Advanced age has been associated with increased activity of transforming growth factor-beta 1 (TGF-β1), the main cytokine that drives the activation of myofibroblasts and expression of ECM proteins. However the activation of this signaling pathway in aged tissues is modest in comparison to what it is reported for fibrotic disorders.
Cellular senescence is a biologic phenotype whereby cells experiencing genotoxic stress become nonreplicative and yet remain metabolically active. Senescent cells secrete numerous cytokines, proteases, and growth factors. This secretory profile is known as the senescence-associated secretory phenotype (SASP) (3). In healthy individuals, protease secretion is believed to facilitate the enlargement of airway spaces, similar to tobacco-related emphysema (4). In chronic obstructive pulmonary disease patients, cell senescence from cigarette smoke-induced oxidative stress may be central to the proinflammatory state of the lungs and exacerbate proinflammatory episodes. Similarly, in the bleomycin mouse model of lung fibrosis cellular senescence promotes fibrosis (5); along such lines reduced epithelial cell senescence was associated with reduced collagen deposition (6).
Several signaling pathways have been implicated in aging; however, a limited number of interventions have been described with potential for enhancing life span (7). Among these, the mechanistic target of rapamycin (mTOR) pathway has emerged as highly interesting since mice treated with an enteric formulation of rapamycin (called eRapa), a putative immunomodulatory drug and inhibitor of mTOR, are characterized by increased life span (8). The latest publication by the Intervention Testing Program showed that chronic rapamycin treatment results in an increase in maximum life span that is sex- and dose-dependent, and which appears to be metabolically distinct from dietary restriction (9). Studies of models of lung disease in young mice show that rapamycin decreases TGF-α–induced pulmonary fibrosis in young mice through inhibition of mTOR, as well as airway remodeling during chronic airway obstruction (10,11). Despite promising evidence that rapamycin ameliorates numerous age-associated diseases and increases life span, the effect of rapamycin on aged lungs is largely unknown.
The aim of this study was to determine whether senescent cells play a key role in the physiologic remodeling of the aging lungs. To test this hypothesis, we assessed the expression of ECM proteins such as collagen and tenascin-c (TNC-c) and the physiologic consequences on lung functions. We also showed that the number of senescent cells increased over time and more so in the small vasculature wall than the parenchyma. Part of the SASP, these senescent cells also secreted ECM proteins. Our findings indicate that the increased presence of senescent cells contributes to an aged-dependent tissue response manifesting with qualitative and quantitative alteration of the ECM that are in part regulated by mTOR-dependent signaling.
Methods
Mouse Housing Conditions and Samples Collection
Male C57BL/6J mice from the National Institute of Aging colony were housed at the University of Texas Health Science Center at San Antonio in the Lab Animal Housing Facility until sample collection. For the rapamycin diet, male C57BL/6 mice (The Jackson Laboratories, Bar Harbor, ME) were fed chow containing 14- or 42-ppm rapamycin (details in Supplementary Methods) (12). The different animal groups are summarized in Supplementary Table 1. All experiments were done in accordance to University of Texas Health Science Center at San Antonio policies and procedures.
Decellularization of Lung Matrices and Proteomics Analysis
Decellularization of murine lungs and analysis were achieved as previously described (13).
Characterization of Collagen Deposition
Collagen deposition was detected by picrosirius red stain on paraffin-embedded lung sections (5). For each group, the quantification was performed on the entire upper right lobe (n = 5). Sircol assay (#S1000; Accurate Chemical and Scientific Corp., Westbury, NY) was performed three times on approximately 10mg of lung tissue according to manufacturer’s instruction (five animals per age group). Pyridinoline level: The level of urinary pyridinoline/desoxypyridinoline was assayed using the MicroVue PYD EIA kit (#8010; Quidel Corporation, San Diego, CA) and MicroVue Creatinine Assay kit (cat. No. 8009; Quidel Corporation). The data are reported as the ratio of nmol pyridinoline and desoxypyridinoline to mmol creatinine. Detection of osteopontin and TNC-c by immunofluorescence: Paraffin-embedded tissue sections were stained following standard procedures (Supplementary Table 2).
Lung Mechanics and Tissue Collection
Following anesthesia, mice were placed on the FlexiVent (Scireq, Montreal, Quebec, Canada) as previously described (6).
Detection of α-Smooth Muscle Actin and TGF-β Signaling
Paraffin-embedded tissue sections were stained following standard procedure using VectaStain Elite ABC kit (#PK-6102; Vector Laboratories). The expression level was detected by Western blot (WB) using previously published methods (5).
Detection of Cellular Senescence
Both detection of protein level expression (WB) and immunohistochemistry were performed as previously described (5) (Supplementary Table 2).
Human Lung Tissue Samples
Human lungs unsuitable for organ transplantation but free of pathology was obtained from Gift of Life Michigan as previously reported (13). Individual samples were chosen based on patient age only. Four and eight samples were included in the 20- to 30-year-old and in the 50- to 65-year-old group, respectively.
Statistical Analysis
All statistical analyses of the data were carried out using GraphPad Prism software (GraphPad, La Jolla, CA).
Results
Changes in ECM Proteins Are Associated With Aged Lungs
To evaluate changes in the composition of young and aged mouse lung ECM, known to be a highly dynamic complex that varies in composition depending on physiological circumstances, lung sections from 8- and 24-month-old mice were decellularized and lysates subjected to liquid chromatography/tandem mass spectrometry. Our gross analysis indicated that the main upregulated groups of proteins in 24-month-old compared with the 8-month old mice were collagen and collagen-related proteins, including basement membrane proteins (laminin family, collagen IV family). Interestingly, proteins such as caveolin-1 and -2 and latent TGF-β binding protein involved in the inhibition of TGF-β pathway are among the most downregulated proteins, as well as surfactant protein (surfactant-associated protein B) and elastin-related proteins (lysyl oxidase like-1). The latter are also known to be downregulated in chronic obstructive pulmonary disease patients (14). We also observed the downregulation of tight junction proteins (zona occludens-1), which are critical for the maintenance of the endothelium/epithelium barrier (15) (Supplementary Table 3). These findings indicate that the changes occurring in aged lungs were both qualitative and quantitative. The environment in the aged lungs appeared remodeled (increased of ECM proteins such as collagen, decorin, and lysyl oxidase like for example).
Increased collagen levels were confirmed by three independent analyses. Levels of cross-linked collagen detected by staining with picrosirius red stain was significantly increased in the lungs as the mice aged (Figure 1A). This primarily occurred in the perivascular and peribronchial regions. The newly synthesized collagen chains detected in the weak acid-soluble form by Sircol also increased (Supplementary Figure 1A). The level of pyridinoline cross-link, a marker of collagen degradation in the urine, was also increased with age (Supplementary Figure 1B). This indicator of physiological remodeling was preceded in mice greater than 12 months old by increased expression of extracellular structural proteins such as osteopontin or TNC-c, which usually serve as an anchor for the activated myofibroblasts and the neo-synthesized collagen fibers (Figure 1B).
Figure 1.
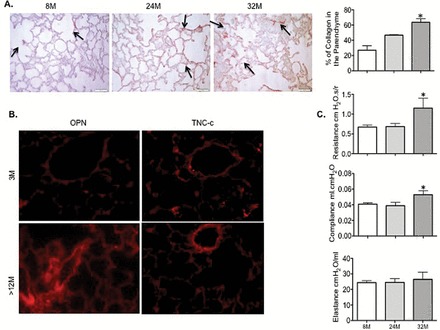
Aged lungs are associated with a profibrotic environment and changes in lung mechanics in wild-type C57BL/6J (WT). (A) PSR staining and quantitation of collagen deposition in 8-, 24-, and 32-mo old lung tissues. Arrows indicate the collagen deposition. Magnification ×200. The bar graph represents the quantification of the collagen deposition using ImagePro software. (B) Representative photomicrographs of IHC staining for OPN and TNC-c indicative of an increase in extracellular matrix proteins expression in the parenchyma and vessel walls in mice older than 12-mo compared with 3-mo old mice. Magnification ×400. (C) Endotracheal intubation was performed on deeply anesthetized mice. Ventilation was set at 150 breaths/min (normal breathing rate) with a positive end-expiratory pressure of 2 cmH2O. Airway resistance and dynamic compliance were significantly increased overtime in the lungs of the animals from 8 to 32 mo of age; dynamic elastance did not vary over time (n = 8–10 animals per age group). Data presented are mean ± SEM. One-way analysis of variance Tukey post-test, *p < .05. IHC = immunohistochemistry; OPN = osteopontin; PSR = picrosirius red; TNC-c = tenascin-c.
To determine the effect of these changes on lung mechanics, lung compliance, elastance, and airway resistance in mice were then evaluated using a Flexivent instrument at baseline with no other stimulation. By 32 months of age, male mice displayed a significant increase in airway resistance and dynamic compliance compared with 8-month-old animals (p = .034 and p = .0266, respectively) (Figure 1C). Elastance or the quality of recoil of mouse lungs remained unaltered in all age groups (Figure 1C). These data suggested a stiffening of lung tissue, including the airways, in agreement with the increased ECM deposition that we observed, and previously published studies (16). The potential mechanisms leading to these changes is the activation of TGF-β signaling due to the reduced expression of its inhibitors and the loss of epithelium barrier known to result in profibrotic responses.
Activation of TGF-β Signaling and Fibroblasts
The cells most likely responsible for the secretion of this augmented expression of ECM proteins are the activated fibroblasts or myofibroblasts upon TGF-β activation. We detected the levels of α-smooth muscle actin (α-SMA), a marker of fibroblast activation by immunohistochemical analysis and WB (Supplementary Figure 2A and B). They were modestly increased in 24- and 32-month-old mice as compared with 8-month-old controls. The staining of α-SMA was observed mainly around the alveoli and in the peribronchial and perivascular areas.
We assessed the activation of TGF-β by measuring the level of expression of TGF-β receptor I and phosphorylated Smad3 protein. A statistical increased level of expression of both proteins was measured overtime and reached significance in the lung homogenate of 24- to 32-month-old animals (Supplementary Figure 2C).
These data suggested that a modest activation of myofibroblasts due to the activation of TGF-β signaling could partially explain the increased ECM deposition associated with aging.
Cellular Senescence Associated With Aging Mouse Lungs Suggests a Role in ECM Changes
Because senescent cells release cytokines and chemokines that could stimulate resident fibroblasts to become the myofibroblasts (17), exemplified by the expression of α-SMA in the lungs, we explored whether an accumulation of senescence cells occurred in aged lungs and which cellular senescence pathway was most likely activated. To test for this possiblitiy, we examined the level of expression of proteins implicated in the p53/p21 response system, the p16/pRb pathway, and AKT/mTOR effectors. We also analyzed the expression of γH2AX, a marker of DNA damage due to its coregulation with the activation of the p53/p21 pathway. We detected the expression of mH2AC (histone variant macro H2A) as a marker for the formation the senescence-associated heterochromatin foci. The formation of the senescence-associated heterochromatin foci is mediated by the pRb/p16 pathway (18). Compared with younger animals, we observed a significant increase in p53 in the 24-month-old mice (p < .05) and a significant increase in the p21 expression levels in the 32-month-old lungs (p < .01). Levels of γH2AX were significantly increased in all age groups about twofold (p < .01) in the 24-month-old lungs and 1.5-fold (p < .05) in the 32-month-old samples (Figure 2A). The level of expression of pRb, p16, and MH2A was significantly increased in the mature mouse group; both pRb and MH2A levels were up to strongly elevated in the older group and undetectable in their 8-month-old counterparts (Figure 2B). Immunohistochemical staining of the senescence marker, MH2A also showed increased bronchiolar and more of the alveolar staining in the 24- and 32-month-old mice, as compared with the less-stained 8-month-old mouse lung sections (Figure 2C). Using p16, an upstream effector leading to the activation of MH2A and also a marker of senescence, we determined that the increase in senescent cells could be detected in 1-year-old animals (Figure 3). Interestingly, the localization of the p16-positive cells was mainly in the wall of the vasculature (Figure 3). Likewise, p21-positive cells increased with time and mainly in the wall of the vasculature (data not shown). We further investigated whether these senescent cells could also be directly implicated in the ECM changes. Indeed, osteopontin and TNC-c expression was colocalized within p16-positive cells. The secretion of osteopontin was mainly associated with senescent cells within the vasculature, whereas TNC-c was mainly secreted by senescent cells in the parenchyma (Figure 3). Thus, our data are consistent with senescence cells being active via both the p53/p21 and the pRb/p16 pathways, and these cells are actively contributing to ECM deposition.
Figure 2.
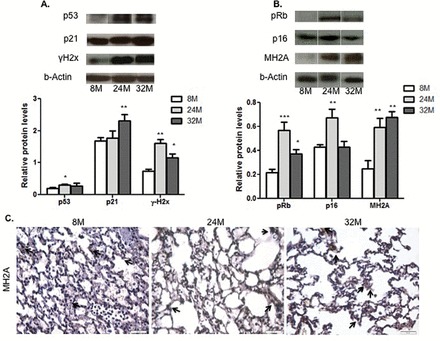
Cellular senescence increased in mouse lungs with age. (A) DNA damage response signaling pathway. Densitometric quantifications of p53, p21, and γH2x WB analysis carried out with whole lung homogenates of 8-, 24-, and 32-mo old male C57B/6J mice. Relative levels were calculated relative to actin level as the loading control. (B) Oxidative stress signaling pathway. Densitometric quantification of pRb, p16, and MH2A WB analysis. Relative levels were calculated relative to actin level as the loading control analysis carried out with whole lung homogenates of 8-, 24-, and 32-mo old male C57B/6J mice. (C) Representative IHC staining of MH2A in 8-, 24-, and 32-month old male mouse lungs. n = 5; statistical significance: p value *<.05; **<.01; ***<.005. IHC = immunohistochemistry; WB = Western blot.
Figure 3.
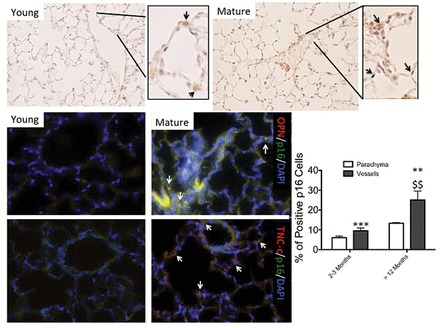
Extracellular matrix proteins are secreted by senescent cells. Representative microphotographs of the detection of p16-positive cells in young mice (2–3 mo old) and mature (older than 12 mo) by IHC. Arrows are indicative of positive-stained cells. Bar graph representation of the percentage of positive cells counting using ImageJ software in the parenchyma and vessel walls. Original magnification ×400. n = 5 animals and at least five sections per animal. OPN and TNC-c protein expression was assessed by immunofluorescence staining (red) in relation to the senescence status of the cells evaluated by immunofluorescence staining of p16 (green). Arrows are indicative of positive-stained cells. Statistical significance: p value **<.01; ***<.005 between young and mature animals; $$<.01 between parenchyma and vessel walls. n = 3. IHC= immunohistochemistry; OPN = osteopontin; TNC-c = tenascin-c.
Detection of Increased Senescent Cells Secreting ECM Proteins in Human Aged Lungs
To confirm that the aging process in murine lungs is similar in human lungs, we tested in human lungs of young (20–30 years old) and mature (50–65 years old) adults whether remodeling was evidenced and associated with increased number of p16-positive cells that were expressing ECM protein. Increased collagen deposition was observed in lungs of a 50- to 60-year-old persons compared with a 20- to 30-year-old persons using picrosirius red staining (Supplementary Figure 4A). Likewise, increased expression of MH2A was observed in human aged lungs. The localization of MH2A-positive cells was also predominantly in the vasculature (Supplementary Figure 4A). Overall we observed an increased number of p16-positive cells in the lungs from older persons. Interestingly, these cells also associated with TNC-c staining (Supplementary Figure 4B), clearly suggestive of a causative role of senescent cells in the changes of ECM observed in aged lungs. In summary, the aged-related changes in human lungs as shown in our murine model, involved increased senescence cells mainly in the vasculature but also in parenchyma and secretion of ECM proteins partially from the senescent cells.
Rapamycin Limits Senescence and Age-Related Collagen Accumulation
Oral rapamycin treatment has been shown to improve health and life span in mice (19); however, its effect on lung aging has yet to be addressed. We first determined whether the mTORC1 pathway was activated in aged lungs. Indeed in 24-month-old lungs, AKT phosphorylation was significantly increased (up to twofold; p < .05), whereas phosphorylated mTORC1 was elevated gradually at 24 months, and led to a significant fivefold increase (p < .05) in 32-month-old mouse lungs (Figure 4A and B). The downstream effector kinase, p70-S6 kinase, showed a strong trend of activation, although not significant, in the aged mice (Figure 4C).
Figure 4.
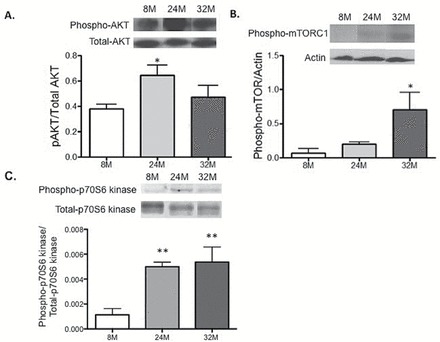
AKT/mTOR activation in aged mouse lungs. Western blot analyses of AKT/mTOR pathway activation (A) Akt, (B) mTOR, (C) mTOR downstream effector p70S6 kinase and densitometric quantitation carried out in whole lung homogenates of lungs isolated from 8-, 24-, and 32-mo old male mice. One-way ANOVA Tukey post-test: *p value <.05 and **p value<.01. n = 3. ANOVA = analysis of variance; mTOR = mechanistic target of rapamycin.
Subsequently, we fed mice with eRapa diets for 4 weeks (short-term; ST rapa) and 20 months (long-term; LT rapa) either at a 14- or 42-ppm rapamycin concentration. A significant decrease (p < .001) in the levels of P-p70-S6 kinase was observed in all treatment cohorts (Figure 5A) indicating that the mice did receive rapamycin and that the mTORC1 pathway has been effectively inhibited. Interestingly, we only observed a decrease in the number of p16-positive cells in the vasculature not in the parenchyma in the lungs of the mice that were on the 42 ppm ST diet. The length of the 14 ppm diet (ST or LT) did not affect the number of senescence cells as shown by the level of expression of p16 (immunohistochemistry) and MH2A detected by WB and immunohistochemistry detection of MH2A showing a high number of MH2A-positive cells in the bronchial and alveolar linings (Supplementary Figure 5A).
Figure 5.
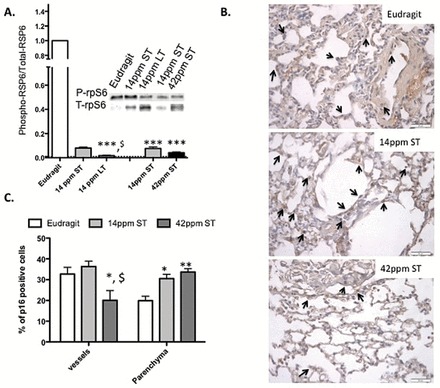
Oral rapamycin treatment reduces mTOR activation and cellular senescence in mouse lungs. (A) WB analyses and quantitation of phospho-p70S6 kinase/total p70S6 kinase in whole lung homogenates of mice from control (Eudragit), rapamycin fed for short-term (ST rapa) and long-term (LT rapa) for a duration of 3wk and 4 mo, respectively, whereas 14 and 42 ppm denotes the concentration of rapamycin fed to mice for the same amount to time (4 mo). (B) Positive p16 cells in the parenchyma and vessel walls as indicated by the arrows. (C) The quantification of the positive p16 cells in the parenchyma and the vessel walls was determined using ImageJ software. N = 5 animals per treatment group/2–3 slides per animal. Statistical significance: p value *<.05; **<.01; ***<.001 demonstrate statistical significance of the rapamycin treatment compared with Eudragit diet within a tissue compartment, and p value $<.05 demonstrates statistical significance between two tissue compartment within the same treatment group. mTOR = mechanistic target of rapamycin; WB = Western blot.
Collagen deposition was significantly increased in both the 14 ppm ST rapa and 14 ppm LT rapa-treated lung sections (Figure 6A and B). The staining pattern was similar to the aged mouse tissue sections at 24 months, with increased collagen deposition around the bronchial layers and vasculature, with little alveolar lining staining. In contrast, the mice fed with 42 ppm diet had significantly less collagen in their lungs (about 45% and 28% collagen in Eudragit diet and 42 ppm ST samples, respectively, p < .001) (Figure 6A and B). In contrast, the levels of α-SMA were reduced in all diet groups as shown by both WB and immunohistochemistry (Figure 6C and Supplementary Figure 5B).
Figure 6.
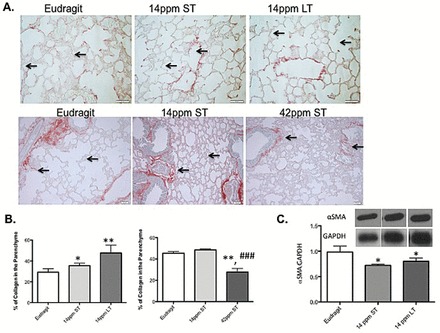
Rapamycin inhibits profibrotic environment in the lungs in a dose-dependent manner. (A and B) PSR staining and quantitation of collagen deposition in the lung tissue sections from control mice (Eudragit), rapamycin fed for short-term (St rapa) and long-term (Lt rapa) for a duration of 3wk and 4 mo, respectively, whereas 14 and 42 ppm denotes the concentration of rapamycin fed to mice for the same amount to time (4 mo). (C) Representative WB of α-SMA and their densitometric quantitations carried out using the whole lung homogenates of mice from control, rapamycin fed for short-term (14 ppm ST) and long-term (14 ppm LT). p value *<.05; **<.01 between control and treatment group and p value ###<.001 between treatment group. α-SMA = α-smooth muscle actin; PSR = picrosirius red; WB = Western blot.
Discussion
Our study demonstrates that as humans and mice advance in age, the lungs undergo physiological remodeling characterized by increased levels of ECM proteins and an increase in the number of senescent cells. This increase in ECM proteins is most likely due in part to modest activation of TGF-β signaling and activation of resident fibroblasts. Not previously recognized, the increased number of senescent cells in aged lungs appeared to be a prime contributor to these changes. Inclusion of rapamycin in the diet prevented not only cellular senescence, most specifically in the vasculature, but also limited collagen deposition. Our data suggest that the increased senescent cells play a significant and unrecognized role in remodeling.
It is important to notice that as the mice aged, the amplitude of the biological responses varied more. This phenomena has been previously reported in healthy aging and it is known that chronological age is not necessary relevant on how aging occurs (20). In addition, even though the regulation of the signaling pathways leading to senescence are better understood, they are intertwined and additional studies are needed to elucidate their regulation. Some of our data can illustrate these general observations reported by many investigators (20,21).
Fundamental changes with age in lung mechanics are well documented (22). In elderly subjects, structural changes impair total respiratory system compliance. Despite our appreciation of the age-dependent decline in respiratory function in patients, the underlying cellular and molecular basis is poorly understood. Defining a good animal model represents a first step in discerning these age-dependent changes in respiratory functions. Indeed, our observations indicate changes in compliance in our animals that mimic decreased compliance reported in the elderly. Changes in parenchyma such as the dilation of air spaces or senile emphysema correlates in the mouse model with changes in the expression of proteins associated with basement membrane. We previously demonstrated the change in the TGF-β–dependent pathway causing a more profibrotic microenvironment (16). Our current data suggested that the underlying cellular and molecular pathways activated in the mouse model were similar in the human samples such as the increased senescent cells via the p16-dependent pathways and the expression of ECM proteins by these p16-positive cells. Overall, studying aging processes in mouse model appears relevant to humans.
To establish whether changes in aged lungs are relevant to the development of age-related diseases, we analyzed cellular senescence. Evidence indicates that oxidative stress, chromosomal instability due to DNA damage, and chronic inflammation promote aging and all of which associate with cellular senescence (23). Interestingly, senescent epithelial cells have been considered relevant in the development of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis, two of the most important age-associated lung disorders (24,25). Most likely, the senescent cells contribute to the remodeling in aged lungs by their SASP. This SASP profile has been implicated in several beneficial processes (eg, embryonic development (26,27)) or detrimental (eg, promotion of profibrotic responses to bleomycin-induced pulmonary fibrosis (5)). Our data suggest that as cells become senescent, they secrete increasing amounts of ECM proteins. Localization of these senescent cells (vasculature or parenchyma) determines the type of ECM proteins secreted. This is important to understand the triggers of senescence and how some environmental factors (tobacco, chemicals, etc…) could activate specific senescence-related signaling pathways.
Our findings identify a novel role for senescent cells: the ability to promote remodeling by the secretion of ECM proteins. The upstream signals that induce ECM proteins secretion are as yet unknown; our data indicated that mTORC1 might be contributing. Previous studies have shown increased health span in mice receiving rapamycin, a classical mTORC1 inhibitor (8). mTORC1 activation/deactivation is a rate-limiting step in the cell cycle. To our knowledge, ours observation are the first to show that in mammals mTORC1 activity increases with age, and that rapamycin oral therapy inhibits or lowers its activity, which further validates the crucial link of mTORC1 activation with the cellular aging in the lung (8). Several studies have linked activated mTOR to cellular senescence in various organs during aging, and rapamycin appears able to rescue cell revival capacity and expand the age limit by inhibiting mTORC1 activation (8). The use of rapamycin in chronic treatment could be controversial. The lung toxicity is known as one of the major complication associated with rapamycin therapy in patients but beneficial in mouse models (28). However, the underlying mechanism is not clear and may not be associated with the inactivation of the mTOR pathway (29). In contrast, chronic administration of enteric rapamycin in mice improves health span and life span in multiple studies (8,9). No toxicity on any organ has been noted even at doses as high as 6 mg/kg body weight (except for testicular degeneration, which is reversible, and cataracts). The effect on longevity is dose-related and the maximum dose that benefits longevity has not been reached yet. This difference between human and mouse is worth noting from the translational potential of the use of rapamycin. However, it does not diminish the importance of our findings presenting that inhibition of the mTOR pathway prevents cellular senescence and remodeling of the lungs with age. It is noteworthy that although rapamycin-fed mouse lungs had reduced levels of α-SMA, a marker of myofibroblast activation, we only observed reduced collagen deposition when the rapamycin-fed mice also presented reduced cellular senescence. Our data clearly indicate that reduced senescence by blocking mTORC1, beneficial for reducing the profibrotic responses in a bleomycin mouse model (5), limits the profibrotic environment of aged organs. The blockage of mTOR by rapamycin resulted in the decrease of senescent cells mostly in the vasculature. Our work does not directly propose a rationale for this observation. It is interesting to report that mTOR inhibitors are pharmacological approaches to treat pulmonary vascular remodeling (30).
In conclusion, senescent cells in the lungs appear to be a significant contributing factor in physiological and pathological (age-associated fibrosis) remodeling in the lungs. Further investigations are needed to reconcile the apparent contradictory role of senescence most likely by determining the different triggers of senescence and their contributions to SASP. Understanding the novel role of senescent cells in these age-related changes offers the opportunity to design interventions to lower the risk for the development of pulmonary aged-related diseases and their complications.
Supplementary Material
Supplementary material can be found at: http://biomedgerontology.oxfordjournals.org/
Funding
The Janey Briscoe Center of Excellence in Cardiovascular Research (C.J. Le S.), NIHAG036613 (C.J.O.), Glenn Award (C.J.O.), RC2AG036613 (Z.D.S.), and U01 HL111016 (E.S.W.).
Supplementary Material
Acknowledgment
We would like to thank for their technical assistance: Ms. Ashley M. Cornett and Ms. Sherry Dodds.
References
- 1. Wahba WM. Influence of aging on lung function–clinical significance of changes from age twenty. Anesth Analg. 1983;62:764–776. PMID: 6346955. [PubMed] [Google Scholar]
- 2. Faner R, Rojas M, Macnee W, Agustí A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:306–313. doi:10.1164/rccm.201202-0282PP [DOI] [PubMed] [Google Scholar]
- 3. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi:10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174:886–893. PMID: 16888288. [DOI] [PubMed] [Google Scholar]
- 5. Shivshankar P, Brampton C, Miyasato S, Kasper M, Thannickal VJ, Le Saux CJ. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am J Respir Cell Mol Biol. 2012;47:28–36. doi:10.1165/rcmb.2011-0349OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jourdan Le Saux C, Davy P, Brampton C, et al. A novel telomerase activator suppresses lung damage in a murine model of idiopathic pulmonary fibrosis. PLoS One. 2013;8(3):e58423. doi:10.1371/journal.pone.0058423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newgard CB, Sharpless NE. Coming of age: molecular drivers of aging and therapeutic opportunities. J Clin Invest. 2013;123:946–950. doi:10.1172/JCI68833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilkinson JE, Burmeister L, Brooks SV, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–682. doi:10.1111/j.1474-9726.2012.00832.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miller RA, Harrison DE, Astle CM, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13:468–477. doi:10.1111/acel.12194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Korfhagen TR, Le Cras TD, Davidson CR, et al. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2009;41:562–572. doi:10.1165/rcmb.2008-0377OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kramer EL, Hardie WD, Mushaben EM, et al. Rapamycin decreases airway remodeling and hyperreactivity in a transgenic model of noninflammatory lung disease. J Appl Physiol. 2011;111:1760–1767. doi:10.1152/japplphysiol.00737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi:10.1038/nature08221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Booth AJ, Hadley R, Cornett AM, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186:866–876. doi:10.1164/rccm.201204-0754OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W, Zhou J, Chen L, Luo Z, Zhao Y. Lysyl oxidase, a critical intra- and extra-cellular target in the lung for cigarette smoke pathogenesis. Int J Environ Res Public Health. 2011;8:161–184. doi:10.3390/ijerph8010161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mazzon E, Cuzzocrea S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir Res. 2007;8:75. PMID: 17971210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sueblinvong V, Neujahr DC, Mills ST, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. 2012;344:41–51. doi:10.1097/MAJ.0b013e318234c132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Velarde MC, Demaria M, Campisi J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip Top Gerontol. 2013;38:17–27. doi:10.1159/000343572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang R, Poustovoitov MV, Ye X, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–30. PMID: 15621527. [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y, Bokov A, Gelfond J, et al. Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci. 2014;69:119–130. doi:10.1093/gerona/glt056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lowsky DJ, Olshansky SJ, Bhattacharya J, Goldman DP. Heterogeneity in healthy aging. J Gerontol A Biol Sci Med Sci. 2014;69:640–649. doi:10.1093/gerona/glt162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amdam GV. Social context, stress, and plasticity of aging. Aging Cell. 2011;10:18–27. doi:10.1111/j.1474-9726.2010.00647.x [DOI] [PubMed] [Google Scholar]
- 22. Sharma G, Goodwin J. Effect of aging on respiratory system physiology and immunology. Clin Interv Aging. 2006;1:253–260. PMID: 18046878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. doi:10.1016/j.molcel.2009.09.021 [DOI] [PubMed] [Google Scholar]
- 24. Aoshiba K, Nagai A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:596–601. doi:10.1513/pats.200904-017RM [DOI] [PubMed] [Google Scholar]
- 25. Minagawa S, Araya J, Numata T, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–L401. doi:10.1152/ajplung.00097.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muñoz-Espín D, Cañamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–1118. doi:10.1016/j.cell.2013 [DOI] [PubMed] [Google Scholar]
- 27. Storer M, Mas A, Robert-Moreno A, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. doi:10.1016/j.cell.2013.10.041 [DOI] [PubMed] [Google Scholar]
- 28. Olyaei AJ, de Mattos AM, Bennett WM. Nephrotoxicity of immunosuppressive drugs: new insight and preventive strategies. Curr Opin Crit Care. 2001;7:384–389. doi:10.1159/000321857 [DOI] [PubMed] [Google Scholar]
- 29. Pham PT, Pham PC, Danovitch GM, et al. Sirolimus-associated pulmonary toxicity. Transplantation. 2004;77:1215–1220. PMID: 15114088. [DOI] [PubMed] [Google Scholar]
- 30. Goncharova EA. mTOR and vascular remodeling in lung diseases: current challenges and therapeutic prospects. FASEB J. 2013;27:1796–1807. doi:10.1096/fj.12-222224 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.