

Abstract
Leukocyte transendothelial migration (TEM) requires two major events: local dissociation of adherens junctions manifested as gaps in vascular endothelial (VE)-cadherin staining at the site of TEM and targeted trafficking of the lateral border recycling compartment (LBRC) to the site of TEM. However, the association between LBRC recycling and VE-cadherin gaps remains unknown. We found that when targeting of the LBRC is selectively inhibited using established methods, such as a function blocking anti–platelet endothelial cell adhesion molecule 1 antibody, depolymerizing microtubules, or microinjection of an antibody that inhibits kinesin, VE-cadherin gaps do not form around the blocked leukocyte. This is the first time that the LBRC has been implicated in this process. We obtained similar results for neutrophils and monocytes and in studies using live cell imaging microscopy conducted under fluid shear conditions. Depolymerizing microtubules did not affect the ability of leukocytes to induce tyrosine phosphorylation of VE-cadherin. A VE-cadherin double mutant (Y658F, Y731F) expressed in endothelial cells acted as a dominant negative and inhibited VE-cadherin gap formation and TEM, yet targeting of the LBRC still occurred. These data suggest that targeting of the LBRC to the site of TEM precedes VE-cadherin clearance. Recruitment of the LBRC may play a role in clearing VE-cadherin from the site of TEM.
Leukocytes attach to vascular endothelial cells at the site of inflammation via a series of adhesive steps that involve tethering, rolling, adhesion, and locomotion.1, 2, 3, 4 Although a lot is known about these earlier steps leading to diapedesis or transendothelial migration (TEM), the molecular mechanisms regulating TEM have not been fully elucidated. Here, we examined the relationship between two major events required for diapedesis: dissociation of adherens junctions at, and lateral border recycling compartment (LBRC) trafficking to, the site of TEM. These two processes have been previously demonstrated to be required for efficient leukocyte TEM, but the association between these two events has not been reported.5, 6, 7, 8, 9, 10
Vascular endothelial (VE)-cadherin (cadherin-5, CD144) is a type 1 transmembrane protein that is exclusively expressed in endothelial cells. It is concentrated at adherens junctions, where it forms calcium-dependent homophilic interactions and participates in barrier function and TEM.11, 12, 13 Not surprisingly, VE-cadherin is a tightly regulated protein. Its surface expression is stabilized by its association with the cytoplasmic protein p120 catenin, and it can link to the actin cytoskeleton via its interaction with β-catenin and plakoglobin, members of the armadillo gene family.7, 14, 15, 16 Adhesion of leukocytes to the endothelial cell activates downstream signaling pathways that induce VE-cadherin to be cleared from the site of transmigration to produce what appears as a gap in VE-cadherin staining along the junction.5, 6 However, VE-cadherin may not be internalized but rather pushed aside along the plane of the junction and diffused back to refill the junction once transmigration is complete.6, 7 The mechanism(s) by which this occurs is not clear.
Platelet endothelial cell adhesion molecule (PECAM, CD31) is a-130 kDa type 1 transmembrane glycoprotein expressed by platelets, leukocytes, and endothelial cells. In endothelial cells, PECAM is almost exclusively concentrated at cell-cell borders.17, 18 Homophilic interaction between PECAM on the leukocyte and endothelial cells induces downstream signaling pathways in each of these cell types that are required for TEM.17, 18, 19, 20, 21 Indeed, disrupting homophilic interactions between these two cell types by the addition of blocking antibodies against extracellular domain 1 of PECAM or by addition of soluble recombinant PECAM-Fc protein inhibits TEM in vitro and inflammation in vivo22, 23, 24, 25 in all mouse strains except C57Bl/6.25, 26, 27, 28
The lateral border recycling compartment (LBRC) is a unique reticulum of interconnecting vesicle-like structures along the lateral borders of endothelial cells at cell-cell contacts.8, 9 The LBRC traffics to the site of TEM to surround the transmigrating leukocyte. Approximately one-third of total PECAM is in the LBRC, as are other molecules important for TEM, such as JAM-A, poliovirus receptor, and CD99.10, 29, 30 Even though VE-cadherin is present at cell-cell junctions, it is excluded from the LBRC.10, 30, 31
Homophilic interactions between PECAM on the leukocyte and at the endothelial cell border initiate signals that recruit the LBRC.8 Trafficking of the LBRC requires an intact microtubule cytoskeleton and kinesin, a plus end molecular motor. Anything that prevents targeted trafficking of the LBRC inhibits TEM.8, 9, 10 Thus, two major events occur during diapedesis. The LBRC is recruited to the site of TEM, and VE-cadherin moves out of the way. Despite the requirement for these two events for efficient paracellular transmigration, the association between them has not been characterized.
We used a pharmacologic and genetic approach to delineate the spatial and temporal association between the LBRC and VE-cadherin gap formation. We have two experimental systems in which to study the association of these phenomena. In one we block movement of the LBRC to the site of TEM; in the other we block the ability of VE-cadherin to leave the site of leukocyte adhesion. The analysis of both systems will be compatible with only one answer to this question. We found that the LBRC is recruited at an early step during TEM and can occur in the absence of formation of VE-cadherin gaps. The LBRC may have a dual function, not only to provide a gateway for the leukocyte across the endothelial cell barrier but also to help clear VE-cadherin specifically from sites of TEM.
Materials and Methods
Reagents
Monoclonal antibodies against domain 4 of human VE-cadherin (clone hec1) or the N-terminal domain of human PECAM (clone hec7) were prepared as previously described.17, 32 P1.1 mouse monoclonal IgG ascites was a kind gift from Dr. Peter Newman (Blood Center of Wisconsin, Milwaukee, WI). P1.1 is a nonblocking antibody against PECAM domain 5. Fab fragments were prepared from purified IgG as previously described.8 Polyclonal antibody HD against Drosophila kinesin-1 was a kind gift from Dr. Geri Kreitzer (Weill Cornell Medical Center, New York, NY).9 Goat polyclonal anti-mouse F(ab')2 IgG was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). The goat polyclonal anti-mouse F(ab')2 IgG Alexa568 and the antiphosphorylated Y731 VE-cadherin antibody were purchased from Invitrogen (Carlsbad, CA). IB4 mouse monoclonal IgG recognizing anti-human CD18 was used to label leukocytes and was prepared as previously described.33 Monoclonal antitubulin antibody was purchased from Sigma-Aldrich (St. Louis, MO). Demecolcine (DCN) was obtained from Calbiochem (Billerica, MA). 4G10 Platinum (an antiphosphotyrosine mouse monoclonal cocktail IgG2b) was purchased from Millipore (Billerica, MA).
Cell Culture
Human umbilical endothelial cells (HUVECs) were isolated and cultured as previously described17, 34 on plastic or hydrated collagen gels and were used at passage 2. 293A cells were cultured in Dulbecco's modified Eagle medium (Gibco/Life Technologies, Carlsbad, CA) that contained 10% fetal bovine serum (FBS) and supplemented with 10 mmol/L modified Eagle medium nonessential amino acids, l-glutamine, and 100 U/mL of penicillin and streptomycin. Human polymorphonuclear (PMN) cells (>95% pure) were isolated from whole blood obtained from healthy volunteers using a Histopaque 1119 (Sigma-Aldrich): Ficoll-Paque gradient (GE Healthcare, Atlanta, GA). Contaminating red blood cells were lyzed at 4°C for 10 minutes with ice-cold ammonium chloride (STEMCELL Technologies, Vancouver, BC, Canada) by diluting 1 mL of ammonium chloride stock (0.8%) with 4 mL of 1× Dulbecco's phosphate-buffered saline (PBS) (Mediatech, Inc., Corning, Manassas, VA). Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation on Ficoll-Paque Plus (GE Healthcare) as previously described.35 All experimental protocols were approved by the Northwestern University Institutional Review Board.
Quantitative End Point TEM Assay
Quantitative end point TEM assay was performed and quantified as previously described.19 Briefly, HUVECs were cultured on hydrated collagen gels on a 96-well plate until confluent. In some experiments with monocytes and all experiments with PMN cells, endothelial cells were first activated by co-culture in 10 ng/mL of tumor necrosis factor (TNF)-α for 4 hours. To assess the role of the VE-cadherin wild-type (WT) and mutant green fluorescent protein (GFP) adenoviral constructs, HUVECs were washed several times with M199 without human serum and transduced with virus expressing GFP-tagged VE-cadherin WT or double mutant (DMT) overnight in the presence of 50 μL of M199 and 2% FBS. Media was removed and replaced with complete M199 medium that contained 20% human serum. Monolayers were used for TEM assays 24, 48, or 72 hours after transduction as indicated. Greater than 95% transduction rates were obtained in each of our studies.
Targeted Recycling Assay
Targeted recycling experiments were performed as previously described.8, 9 In brief, HUVEC monolayers grown on hydrated collagen on 96-well plates were incubated with P1.1 Fab fragment, a non–function-blocking monoclonal antibody against PECAM, at 37°C. Movement of PECAM serves as a surrogate marker for the LBRC. Cells were washed several times with ice-cold M199 medium that contained 0.1% human serum albumin to remove any unbound antibody. An excess of goat anti-mouse F(ab')2 fragment was premixed in incubation medium to mask all exposed P1.1 Fab. In studies in which pharmacologic agents were included [eg, dimethyl sulfoxide (DMSO) or 1 μmol/L of DCN], drugs were included in the premix. After 1 hour of incubation on ice, unbound antibody and drugs were washed away. In experiments where PBMCs were used, a cell suspension of 2 × 106 cells/mL was mixed with 20 μg/mL Alexa 568–conjugated goat anti-mouse Fab2 fragment and 100 μL of this was added per well on ice. For studies that involved PMN cells, the cell suspension was adjusted to 5.0 × 105 cells/mL.
Leukocytes were allowed to settle on the HUVEC monolayer on ice, followed by a short (9-minute) incubation at 37°C to catch the leukocytes in the process of TEM. The 96-well plates were washed several times on ice with ice-cold M199 medium that contained 0.1% human serum albumin to remove any unbound leukocyte and secondary antibodies. Cells were fixed for 10 minutes at room temperature and processed for immunofluorescence as described in this section.
VE-Cadherin GFP Constructs
A plasmid that expressed full-length GFP-tagged VE-cadherin cDNA (pCad5(s) cDNA1neo) was used as a template to generate VE-cadherin WT adenoviral vector. The adenoviral vector, pAD-CMV-V5 Destination Vector, was from Gateway Technology, Invitrogen. Mutations at positions tyrosine 658 and tyrosine 731 in VE-cadherin cDNA were generated using the QuikChange II XL Site-Directed Mutagenesis Kit from Agilent Technologies (Santa Clara, CA) after the procedure of the manufacturer. Plasmids were used to transform XL10-Gold ultracompetent cells and were plated in selective media. Plasmids from positive clones were isolated using QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA) and sequenced on an ABI 3730 DNA automated sequencer (Applied Biosystems, Core Sequencing Facility, Northwestern University, Chicago, IL) to confirm the presence of the appropriate point mutations. Plasmid DNA was purified using the GenEluteHP Plasmid MidiPrep Kit (Sigma-Aldrich). The 293A cells were transfected with adenoviral vector according to the manufacturer's protocol (Gateway Technology, Invitrogen). Approximately 7 days after transfection, the adenovirus-containing 293A cells were harvested and lyzed to prepare a crude viral stock. The resultant viral stock was amplified.
Gel Electrophoresis and Immunoblotting
To detect phosphotyrosine proteins, endothelial cells were rinsed with ice-cold PBS and lyzed in ice-cold cell lysis buffer: 10 mmol/L Tris, pH 7.4, 100 mmol/L NaCl, 1 mmol/L EGTA, 1 mmol/L EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, and 10% glycerol. Cell lysis buffer was supplemented with 1 tablet of protease inhibitor and PhosPhoStop (Roche, Basel, Switzerland) per 10 mL of lysis buffer before use. Lysates were diluted in a 1:1 ratio with 2× Laemmli Sample Prep Buffer (213 mmol/L Tris, pH 6.8, 34% glycerol, 3.41% SDS). For immunoprecipitation studies, lysates were incubated with the indicated antibodies for 3 hours at 4°C followed by 1 hour with protein G beads. Samples were heated at 95°C for 5 minutes, and proteins were separated on 8% to 10% SDS-PAGE after standard procedures and subsequently processed for Western blot analysis.
Microinjection
HUVECs were grown on 35-mm glass bottom dishes (MatTek Corporation, Ashland, MA) coated with fibronectin until confluent and were treated with 10 ng/mL of TNF-α for 4 hours. Microinjection was performed using an Olympus Disk Scanning Unit confocal microscope, under 20× phase, along with an Eppendorf FemtoJet Microinjector and micromanipulator. Cells were microinjected with polyclonal antikinesin IgG (HD) antibody or an isotype-matched control IgG antibody that were diluted in 1× PBS to a concentration of 5 mg/mL. To identify injected cells, 1 mg/mL of dextran–Alexa 488 was included with each of the antibodies. Approximately 100 contiguous endothelial cells were microinjected per field per condition. After microinjection, endothelial cells were allowed to recover for 2 hours in the presence of 10 ng/mL of TNF-α at 37°C. Freshly isolated PBMCs from healthy volunteers were resuspended to 4 × 106 cells/mL in ice-cold M199 medium that contained 0.1% human serum albumin. One hundred microliters of the leukocyte cell suspension was added to the HUVEC monolayers and allowed to settle for 15 minutes. Plates were placed at 37°C for 10 minutes to allow leukocytes to transmigrate across the endothelial layer. Cells were fixed with 2% paraformaldehyde in Dulbecco's PBS (Cellgro) with Ca++Mg++ and prepared for immunofluorescence microscopy as described below.
Immunofluorescence Microscopy
Single- and double-label immunofluorescence was performed as detailed previously.9 Collagen gels in 96-well plates were transferred onto 35-mm glass bottom dishes and viewed on a PerkinElmer UltraVIEW VoX Confocal Spinning Disk Microscope (Duluth, GA), fitted with appropriate filters for visualization of GFP, as well as Alexa 568 (Invitrogen), Dylight 488, and Dylight 650 conjugated probes (Thermo Fisher Scientific, Waltham, MA). Images were acquired with a UPlanApo 40× oil immersion objective using Volocity software version 6.3 (PerkinElmer). Recorded images were analyzed offline with ImageJ version 1.45s (NIH, Bethesda, MD; http://imagej.nih.gov/ij) and Volocity software version 6.3 (PerkinElmer).
Live Cell Imaging Microscopy
Endothelial cells were grown to confluence on 35-mm glass bottom dishes (MatTek Corporation) or Ibidi chambers (Ibidi USA Inc., Madison, WI) coated with fibronectin. Cells were treated with 10 ng/mL of TNF-α for 4 hours before an experiment and treated with 0.1% DMSO or 1 μmol/L of DCN (Calbiochem) for 10 minutes at 37°C. Cells were rinsed several times after drug treatment before leukocytes were added. Cells were imaged in a heated closed chamber. Images were captured using a Leica 40× HCX PL APO 40X/0.85 lens, a Leica inverted microscope, a Hamamatsu ORCA-ER digital camera, and Simple PCI 6 software version 6.6.0.14 (W. Nuhsbaum Inc, McHenry, IL).
For experiments that involved DCN treatment, endothelial cells were fixed and processed for immunofluorescence at the end of each recording to confirm that microtubules in the DCN-treated group remained depolymerized.
Statiscal Analysis
All experiments with quantification are expressed as means ± SEM. Each condition was conducted in triplicate and represent data from three separate experiments and >100 leukocyte/endothelial cell interactions. Differences between two groups were analyzed with the t-test. P ≤ 0.05 was considered statistically significant. Statistical analysis was performed using GraphPad Prism software version 5 (GraphPad, San Diego, CA).
Results
A Function Blocking Antibody against PECAM Inhibits Leukocyte-Induced VE-Cadherin Gaps
A major event controlling diapedesis is the loosening of adherens junctions at the site of transmigration.5, 14, 36, 37, 38, 39 This produces what appears as a gap in VE-cadherin staining.6 Because VE-cadherin gaps are required for efficient TEM, lower TEM is associated with reduced gaps. However, the mechanism(s) by which this occurs is not clear. There is evidence that intercellular adhesion molecule 1 (ICAM-1) clustering can increase phosphorylation of VE-cadherin tyrosine residues and regulate VE-cadherin gap formation.40 Locomotion on the endothelial surface involves adhesive interactions between β2-integrins on the leukocyte and ICAM-1 on the endothelial cell.41, 42 This has been found to cluster ICAM-1 on endothelial cells. However, VE-cadherin gap formation may require additional signals that are downstream of ICAM-1 signaling.7, 40 For example, in a study conducted by Allport et al,5 a function-blocking antibody directed against the extracellular domain of PECAM inhibited VE-cadherin gap formation and TEM. In addition, we previously found that inhibiting homophilic interactions between PECAM in leukocytes and endothelial cells inhibited recruitment of the LBRC and TEM.8 On the basis of these data, we wanted to characterize the association between LBRC recruitment and VE-cadherin gap formation in endothelial cells during TEM. Figure 1 is a schematic of the experimental approach that would allow us to determine the association between these two critical events: LBRC recruitment and VE-cadherin gap formation.
Figure 1.

Outline of logic for determining the association of vascular endothelial (VE)-cadherin gap formation and targeted recycling (TR) of the lateral border recycling compartment (LBRC). Transmigration assays are performed under conditions in which one or the other of the processes is inhibited. A: Two complementary approaches are taken to block trafficking (TR) of the LBRC to the site of TEM. B: TEM experiments are performed under conditions where formation of VE-cadherin gaps is inhibited. The successful completion of both arms of this study is compatible with only one of the four conclusions.
We first determined whether we would obtain similar findings with freshly isolated monocytes instead of the monocyte cell line that was used in the Allport study. We compared the extent of TEM and adherens junction disruption between control and anti-PECAM–treated endothelial cells by performing three-dimensional confocal imaging of monocytes fixed in the act of TEM. Under the monocyte/endothelial cell ratios used in these experiments, there is no change in junctional permeability or transendothelial electrical resistance during TEM,43 so we used the appearance of a gap in VE-cadherin staining5, 6 or VE-cadherin gap as a surrogate marker for junction disruption. As expected, under control conditions, a VE-cadherin gap formed (there was an absence of VE-cadherin staining) at cell-cell junctions where monocytes were in the act of transmigrating (Figure 2A). In the presence of hec7, a function-blocking anti-PECAM monoclonal antibody, monocytes positioned over a cell-cell junction did not induce a VE-cadherin gap (VE-cadherin staining was intact at this site) and failed to transmigrate (orthogonal XZ view). There was no difference in adhesion or locomotion of monocytes to cell-cell junctions between the hec7-treated group compared with control19 (data not shown). Quantification of these results indicated that under control conditions after 1 hour approximately 80% of the monocytes initiated TEM and had a VE-cadherin gap associated with them (Figure 2B). In contrast, anti-PECAM treatment significantly inhibited TEM and VE-cadherin gaps (Figure 2B). Only 20% of monocytes transmigrated and had VE-cadherin gaps associated with them. TEM was essentially always associated with a VE-cadherin gap. Because ICAM-1 function was intact in these cells, these results suggest that ICAM-1 signaling is not sufficient to induce VE-cadherin gap formation and requires additional signal(s). One consequence of PECAM clustering is recruitment of the LBRC to the site of TEM.8, 9, 10
Figure 2.

A function-blocking antibody against platelet endothelial cell adhesion molecule 1 (PECAM) inhibits vascular endothelial (VE)-cadherin gaps. A: In control (CTL) there is a VE-cadherin gap (green arrows) where a monocyte transmigrates (merge and XZ view orthogonal). In the presence of anti-PECAM antibody, VE-cadherin staining is not cleared at the site where a monocyte is on an adherens junction (green arrows, merge) and there is no transmigration (XZ view orthogonal). The dotted lines in the merged view represent the plane of the orthogonal image, and the dotted lines in the XZ view indicate the plane of the endothelial monolayer. B: Quantification shows that hec7 treatment inhibits VE-cadherin gaps and transendothelial migration (TEM) (1 hour) compared with CTL. Data are expressed as the means ± SEM percentage of monocytes at the junction that underwent TEM or had VE-cadherin gaps. Each condition was conducted in triplicate and represents data from three separate experiments and over 100 leukocyte/endothelial cell interactions. The horizontal dotted line in the merged view represents the plane of the orthogonal image, and the dotted line in the XZ view indicates the plane of the endothelial monolayer. ∗∗P < 0.01, t-test. Scale bars = 10 μm. XZ, orthogonal view displays the XZ plane of a three-dimensional image at a given point.
VE-cadherin Gaps Do Not Form when Trafficking of the LBRC Is Inhibited
To test the association of the LBRC in VE-cadherin gap formation, we first used a pharmacologic approach to inhibit the movement of the LBRC and examined the ability of VE-cadherin gaps to form. We have previously found that in the brief absence of polymerized microtubules, targeting of the LBRC does not occur.9 Leukocytes can still adhere tightly to the apical surface of the endothelial borders and migrate to cell-cell junctions but do not initiate transmigration.9 Under these experimental conditions, homophilic interactions between PECAM in endothelial cells and leukocytes are left intact because no function-blocking antibodies are included.
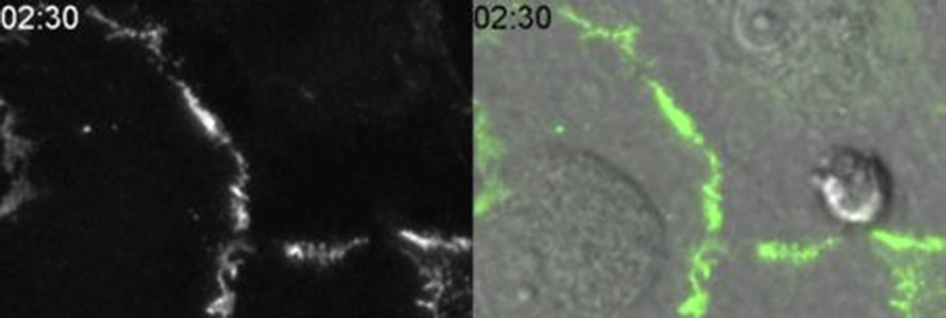
We exposed HUVEC monolayers grown on hydrated collagen gels to DCN under conditions that we have previously found to selectively induce microtubule depolymerization without perturbing the structure or function of the endothelial cell junction.9 Under control conditions, the LBRC surrounded the transmigrating monocyte (Figure 3A). We quantified enrichment of the LBRC at sites of monocyte-endothelial cell interaction and found that there was a mean threefold enrichment along the cell border at this site. As expected, there was a corresponding VE-cadherin gap at this site. An orthogonal XZ view reveals that the monocyte was in the act of TEM (Figure 3A). In contrast, in endothelial cells treated with DCN, the monocyte was arrested at the apical surface of the endothelial monolayer on an adherens junction unable to transmigrate. There was no corresponding enrichment of immunofluorescence around the monocyte, indicating that trafficking of the LBRC was inhibited by DCN. A VE-cadherin gap did not form (Figure 3A). Importantly, pretreatment of the endothelial cell monolayer with DCN did not impair monocyte adhesion or locomotion to cell-cell junctions (data not shown). We included eluate controls in our studies, as shown in the study by Mamdouh et al,9 to rule out the possibility that residual DCN in the media was inhibiting monocytes directly (data not shown). Quantification of the targeted recycling experiment revealed that LBRC enrichment correlated tightly with VE-cadherin gap formation (Figure 3B). In control samples, 80% of the monocytes at the junctions were in the process of transmigration and associated with a rim of recycling LBRC and a gap in VE-cadherin staining. In DCN-treated cells, LBRC targeted recycling was inhibited. Less than 10% of monocytes were associated with recycled LBRC and a VE-cadherin gap. We did not observe a full block because it is likely that the dissociation of microtubules was not complete at the low concentration of DCN that were used in the assay. When targeting of the LBRC was inhibited, VE-cadherin gaps did not form around the 90% of monocytes that were arrested at cell-cell contact sites. Endothelial cells were fixed immediately after the experiment to confirm that microtubules remained depolymerized during the experiment (Figure 3C). As previously reported, the number of monocytes at the junctions was similar in both the control and DCN-treated group (data not shown).9 Moreover, the overall intensity of VE-cadherin at the junctions was unaffected by DCN treatment (means ± SEM intensity = 1709 ± 188 for controls versus 1705 ± 159 for DCN treated). The targeted recycling assay focuses on monocytes caught in the act of TEM; it does not measure total TEM of the population. In parallel, we confirmed that under the conditions used in the targeted recycling assay, TEM of monocytes was inhibited by DCN treatment (Figure 3D). We observed a similar level of inhibition in longer TEM assays (Figure 3E) in which 80% of control cells transmigrated. These results suggest that VE-cadherin gap formation may be downstream of LBRC recruitment.
Figure 3.

VE-cadherin gaps do not form when targeting of the lateral border recycling compartment (LBRC) is inhibited. Targeted recycling assay was performed as in Materials and Methods. A: Under control (CTL) conditions, recycled LBRC are enriched around the transmigrating monocyte (red arrows). There is a corresponding VE-cadherin gap at this site (green arrows). The merged image indicates a monocyte is transmigrating at this site (blue color; XZ view orthogonal). In contrast, in the demecolcine (DCN)–treated group, recycled LBRC is not enriched around the monocyte (red arrows), and a VE-cadherin gap does not form (green arrows). The merged image indicates that a monocyte is present at this site (blue color) that was unable to transmigrate (XZ view orthogonal). The dotted lines in the merge view represent the plane of the orthogonal image, and the dotted lines in the XZ view indicate the plane of the endothelial monolayer. B: Quantification of monocytes in CTL and DCN-treated cells that were at cell-cell junctions that had enriched LBRC staining around the monocyte and were associated with a VE-cadherin gap. C: Human umbilical endothelial cell (HUVEC) monolayers treated as above were processed for immunofluorescence microscopy and labeled with antibodies against antitubulin to reveal that microtubules were still largely depolymerized the end of the assay. D and E: HUVEC monolayers were treated with Dulbecco's modified Eagle medium or DCN and transendothelial migration (TEM) assay was conducted for 8.5 minutes (the time of the targeted recycling assay, D) or 1 hour at 37°C (E). The percentage of TEM in CTL and after DCN treatment was quantitated. Data in B, D, and E are expressed as means ± SEM and represent >100 endothelial/monocyte interactions from at least three independent experiments; each condition repeated in triplicates. ∗∗P < 0.01, ∗∗∗P < 0.001, t-test. Scale bars: 10 μm (A and C). XZ, orthogonal view displays the XZ plane of a three-dimensional image at a given point.
We repeated the experiments using PMN cells and obtained similar results. Under control conditions, the LBRC was recruited to the transmigrating PMN shown as an enhanced immunofluorescence ring (Figure 4A). A corresponding VE-cadherin gap formed at this site. When DCN inhibited targeting of the LBRC, there was no increase in immunofluorescence around the PMN cells (Figure 4A), and no gap in VE-cadherin staining formed. Figure 4B quantifies these results and indicates that VE-cadherin gaps did not form when targeting of the LBRC was inhibited. Under these conditions, TEM is inhibited by DCN (Figure 4C).
Figure 4.

Polymorphonuclear (PMN) cell transmigration and vascular endothelial (VE)-cadherin gap formation require lateral border recycling compartment (LBRC) trafficking. Targeted recycling assay was performed on cytokine-activated human umbilical endothelial cell (HUVEC) monolayers using PMN cells. A: Under control (CTL) conditions, LBRCs are enriched around a PMN cell (red arrows). There is a corresponding VE-cadherin gap at this site (green arrows). The merged image indicates a transmigrating PMN cell is present at this site (blue color; XZ view orthogonal). In contrast, in endothelial cells that were pretreated with 1 μmol/L of demecolcine (DCN) (right panels), PMN cells do not transmigrate (orthogonal), there is no enriched LBRC immunofluorescence at this site (red arrows), and there is no VE-cadherin gap (green arrows). The horizontal dotted lines in the merged view represent the plane of the orthogonal image, and the dotted lines in the XZ view indicate the plane of the endothelial monolayer. B: Quantification of the targeted recycling assay illustrates that under control conditions, targeted recycling occurs and is associated with VE-cadherin gaps, whereas in HUVECs treated with DCN, targeted recycling is markedly inhibited and is associated with absence of VE-cadherin gaps. C: HUVECs were treated with pharmacologic agents as above. PMN cells were allowed to transmigrate for 20 minutes at 37°C. Data in B and C are expressed as means ± SEM and represent data from three independent experiments from >100 PMN/endothelial interactions; each condition repeated in triplicates. ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001, t-test. Scale bars = 10 μm. XZ, orthogonal view displays the XZ plane of a three-dimensional image at a given point.
The conditions used to depolymerize microtubules have no effect on junctional permeability or VE-cadherin distribution.9 However, pharmacologic agents can have off target effects, and disrupting the microtubule network could have blocked VE-cadherin gap formation independently of LBRC. Therefore, we took a complementary approach to inhibit targeting of the LBRC during TEM while keeping the microtubule network intact. Previously, we found that we could inhibit targeting of the LBRC by microinjecting endothelial cells with a function blocking antibody (HD) that was raised against a highly conserved motor domain of Drosophila kinesin heavy chain.9
We used the HD antibody in a TEM assay to assess VE-cadherin gap formation in endothelial cells in which kinesin motor activity was inhibited (Figure 5A). VE-cadherin staining was intact at cell-cell junctions under the conditions used in this assay. However, because dextran-488 was mixed in with the IgG or the HD antibody to identify the injected endothelial cells, the gain in the green channel had to be turned down to avoid saturating the channel in the images presented. VE-cadherin gaps and recycling LBRC were measured as in previous figures. In cells injected with control antibody, a monocyte that was in the process of TEM recruited the LBRC, and a VE-cadherin gap formed at this site (Figure 5A). However, in the presence of the HD antibody, most monocytes were arrested at cell borders unable to transmigrate. There was no LBRC recruitment to this site and no clearance of VE-cadherin from this site (Figure 5A). HD antibody microinjection did not inhibit the ability of monocytes to adhere to the endothelial monolayer or locomote to the adherens junctions (data not shown). For microinjection, endothelial monolayers are grown on coverslip bottom dishes. The rate and extent of TEM are lower under these conditions compared with cells grown on collagen gels in the previous figures. Nevertheless, quantification revealed that HD treatment significantly inhibited recruitment of the LBRC and inhibited VE-cadherin gap formation compared with control IgG (Figure 5B). In the short time for TEM in the targeted recycling assay, approximately half of the adherent control monocytes had transmigrated, associated with targeted recycling and VE-cadherin gaps. In three independent experiments, each with triplicate samples and a total of ≥100 monocytes counted at the junctions, we did not observe LBRC recruitment around the monocyte, VE-cadherin gap formation, or TEM of monocytes in HD-treated samples. Together these findings are consistent with our earlier results and suggest that VE-cadherin gap formation may be downstream of LBRC recruitment and implicates the LBRC in de novo VE-cadherin gap formation associated with transmigrating leukocytes.
Figure 5.

Microinjection of antikinesin antibody inhibits transendothelial migration (TEM), lateral border recycling compartment (LBRC) trafficking, and vascular endothelial (VE)-cadherin gaps. Cytokine-activated endothelial cells were microinjected with control IgG antibody or with HD antibody, directed against the kinesin motor domain. To identify injected cells, 1 mg/mL of dextran-Alexa 488 was included with each of the antibodies. A: Under control IgG conditions, a monocyte at a cell-cell junction (merge) that is in the process of transmigration (XZ view orthogonal) has a corresponding VE-cadherin gap (green arrows). In the HD microinjected cells, VE-cadherin staining is intact at a site where a monocyte is on a cell-cell junction (merge). This monocyte fails to transmigrate (XZ view orthogonal), and a VE-cadherin gap does not form at this site (green arrows). Red arrows denote recycled LBRC. The horizontal dotted lines in the merge view represent the plane of the orthogonal image, and the dotted lines in the XZ view indicate the plane of the endothelial monolayer. B: Quantification of the microinjection studies. C: In a TEM assay run for the time of the targeted recycling assay (9 minutes), HD inhibits TEM of monocytes compared to control IgG. Data are expressed as means ± SEM and represent three independent experiments; each condition repeated in triplicate. ∗∗P < 0.01, t-test. Scale bars = 10 μm. XZ, orthogonal view displays the XZ plane of a three-dimensional image at a given point.
We occasionally observed that at cell borders over which PMN cells were arrested in the targeted recycling assay, VE-cadherin staining appeared less intense than elsewhere around the cell or had small (<1-μm) interruptions in VE-cadherin staining (Figure 4A). Adherens junctions on endothelial cells are dynamic. Previous studies have found that PMN cells tend to seek out sites for TEM where VE-cadherin expression is low.6 Thus, it was possible that the differences in VE-cadherin expression that we observed were present before the PMN arrived rather than due to PMN cell–endothelial cell interactions. However, it is impossible to determine this using a one-time point assay. Therefore, we observed VE-cadherin gap formation associated with TEM events in real time using immunofluorescence microscopy. This also allowed us to verify that treatment of endothelial cells with DCN did not inhibit the ability of PMN cells to migrate to the cell borders. We used a monoclonal antibody against VE-cadherin that does not inhibit VE-cadherin function38 or TEM19, 44 to label the adherens junctions in endothelial cells (Figure 6A and Supplemental Video S1). Under control conditions, PMN cells migrated along cell borders on endothelial cells and underwent paracellular transmigration in approximately 35% of the interactions observed during 20 minutes. As with the microinjection experiments, TEM is considerably less robust when endothelial cells are plated on coverslip dishes (Figure 6B) than on collagen gels (Figure 2, Figure 3, and 4). Nonetheless, even under these conditions, PMN cells transmigrated and formed de novo VE-cadherin gaps (Figure 6A and Supplemental Video S1), apparently ignoring preexisting VE-cadherin gaps (Figure 6A). In contrast, PMN TEM was greatly inhibited after DCN treatment; only 5% of cells transmigrated despite the ability of PMN cells to adhere and locomote along adherens junctions (Figure 6B). Notably, even if preexisting VE-cadherin gaps were present in the monolayer, the PMN cells did not migrate across these areas under conditions in which the LBRC could not undergo targeted recycling (Figure 6A and Supplemental Video S2), even when observed for prolonged periods. We repeated these studies in endothelial cells that expressed VE-cadherin GFP and obtained similar results (data not shown). At the end of each recording, endothelial cells were fixed and stained for tubulin to confirm that microtubules remained depolymerized during the duration of the recording (Figure 6C). Under control conditions, the microtubules in endothelial cells extended to the periphery at sites of cell-cell contact (Figure 6, C and D). Treatment of endothelial cells with DCN clearly depolymerized microtubules. The images in Figure 6, C and D, were taken at the end of the experiment to reveal that microtubules were still depolymerized. At this time, the junctions might not have been as intact as at the start of the recording. Nevertheless, these studies indicate that PMN TEM requires intact microtubules. PMN cells fail to transmigrate even when adherens junctions were disrupted or preformed gaps were present if targeting of the LBRC is inhibited.
Figure 6.

Time-lapse immunofluorescence microscopy of vascular endothelial (VE)-cadherin gaps in human umbilical endothelial cells (HUVECs). VE-cadherin in endothelial cells was labeled with nonblocking anti–VE-cadherin antibody conjugated with Dylight 488 and cells were treated with control (CTL) (Dulbecco's modified Eagle medium) or 1 μmol/L demecolcine (DCN). Transendothelial migration (TEM) of PMN cells were captured using time-lapse immunofluorescence microscopy in CTL and DCN-treated endothelial cells. A: Time lapse of VE-cadherin Dylight 488 (top rows) and merged differential interference contrast (DIC) and VE-cadherin Dylight 488 images (bottom rows) of PMN cells in the process of transmigration. Images were captured every 10 seconds. Numbers indicated in each still image are elapsed minutes and seconds. Under control conditions, the still images reveal a de novo gap associated with a PMN TEM event (white arrows) close to a preexisting VE-cadherin gap (yellow arrows). In contrast, in the DCN-treated samples, leukocytes adhered, spread, and migrated to a cell-cell junction with preformed VE-cadherin gap (yellow arrows) or without (white arrows) but failed to transmigrate. These images are representative of 100 PMN/endothelial interactions observed in at least three different experiments. B: Quantification of TEM events from three independent experiments with each condition conducted in triplicate and >100 endothelial/PMN interactions. C: At the end of each experiment, samples were fixed and processed for immunofluorescence. D: Insets from representative merged images in C (marked with the dashed box) revealing that microtubules extend to the periphery of the cell in control samples and are still disrupted in DCN-treated endothelial cells. White arrows denote periphery of the cell. Data are expressed as means ± SEM. ∗∗P < 0.01 (t-test). Scale bars: 10 μm (C and D).
Inhibiting VE-Cadherin Gap Formation Inhibits TEM but not Targeted Recycling of the LBRC
Because disrupting LBRC trafficking inhibited VE-cadherin gap formation, targeted recycling of the LBRC may be an upstream event in the process of TEM relative to VE-cadherin gap formation, or the two processes could be cooperative (Figure 1). That is, a small amount of targeted recycling could lead to a bit of VE-cadherin gap formation, clearing the way for more LBRC recruitment, and so on. To distinguish between these possibilities, we examined TEM and targeting of the LBRC under conditions in which VE-cadherin gaps was inhibited. Tyrosines Y658 and Y731 on the cytoplasmic tail of VE-cadherin are the residues to which p120 and either β-catenin or plakoglobin, respectively, bind and link VE-cadherin to the actin cytoskeleton.12, 40, 45 Phosphorylation of these tyrosines abolishes these interactions and leads to destabilization of the adherens junction.40, 45, 46 Mutating Y731 or Y658 to phenylalanine to prevent phosphorylation stabilizes VE-cadherin at the junctions and inhibits TEM by preventing VE-cadherin from being cleared from the site of TEM.40, 47 We transduced endothelial cells with an adenovirus-expressing, GFP-tagged VE-cadherin WT or a DMT (Y658F, Y731F). Forty-eight hours later, expression and localization were assessed by immunofluorescence and Western blotting. Most of the VE-cadherin WT GFP and VE-cadherin DMT tagged protein localized to cell-cell contact sites (Figure 7A). However, in some instances overexpression of VE-cadherin GFP protein resulted in accumulation around the perinuclear region (Figure 8B) in addition to cell-cell junctions. VE-cadherin GFP expression was equivalent to that of endogenous protein (Figure 7B). We first conducted a TEM assay to determine the level of TEM supported by VE-cadherin WT and VE-cadherin DMT monolayers (Figure 7C). In endothelial cells expressing VE-cadherin WT, 80% of monocytes transmigrated, which is comparable to what we observe in nontransduced endothelial cells (Figure 7C). On the other hand, TEM was significantly inhibited in endothelial cells expressing VE-cadherin DMT. Because endogenous VE-cadherin was expressed in these cells to the same extent, the double mutant was acting as a dominant negative inhibitor of TEM. Next, we compared targeting of the LBRC to the site of TEM in endothelial cells expressing VE-cadherin WT versus VE-cadherin DMT (Figure 7D). As expected, VE-cadherin gaps occurred around the transmigrating monocyte in cells expressing VE-cadherin WT, and this was associated with the characteristic enriched immunofluorescence ring of LBRC surrounding the leukocyte at this site (Figure 7D). We postulated that if LBRC trafficking is upstream of VE-cadherin gaps, then inhibiting gap formation by the VE-cadherin DMT should not affect LBRC recruitment. Indeed, in endothelial cells expressing VE-cadherin DMT, monocytes were arrested at the apical surface of endothelial cells and were unable to transmigrate (Figure 7D). There was no gap in VE-cadherin staining, but targeted recycling of the LBRC still occurred (Figure 7E). Together our results indicate LBRC recruitment precedes VE-cadherin gap formation and can occur independently of gap formation.
Figure 7.

Lateral border recycling compartment (LBRC) recruitment still occurs when vascular endothelial (VE)-cadherin cannot leave the cell junction. Endothelial cells were transduced with adenoviral constructs expressing green fluorescent protein (GFP)–tagged VE-cadherin wild-type (WT) or double-mutant (DMT) bearing point mutations at tyrosine residues 658 and 731. A: VE-cadherin WT and DMT localized to cell-cell junctions in endothelial cells as assessed by GFP fluorescence. B: Protein expression of VE-cadherin WT GFP and VE-cadherin DMT GFP was determined by Western blot analysis 48 hours after transduction. C: Quantification of transendothelial migration (TEM) assays reveals that TEM is inhibited in endothelial cells expressing VE-cadherin DMT GFP compared with cells expressing VE-cadherin WT GFP. D: Targeted recycling was visualized. In human umbilical endothelial cells (HUVECs) expressing VE-cadherin WT GFP, LBRC is recruited (red arrows) and a VE-cadherin gap is seen (green arrows) at the site where a monocyte is in the process of transmigrating (XZ view orthogonal). In contrast, in HUVECs expressing VE-cadherin DMT GFP, a monocyte is on a cell-cell junction (merge), no VE-cadherin gap formed (green arrows), and the monocyte does not transmigrate (XZ view orthogonal), but the LBRC is recruited to this site (red arrows). E: Quantification of the targeted recycling assay reveals that in endothelial cells expressing VE-cadherin DMT, the LBRC is recruited independently of a VE-cadherin gap (VE-cadherin staining is intact along the junction). This is in contrast to HUVECs expressing VE-cadherin WT GFP in which LBRC recruitment was associated almost exclusively with VE-cadherin gaps. Data are expressed as means ± SEM from three separate experiments with each condition repeated in triplicates and >100 endothelial/monocyte interactions. ∗∗P < 0.01, ∗∗∗∗P < 0.0001 (t-test). Scale bar = 10 μm (A and D). XZ, orthogonal view displays the XZ plane of a three-dimensional image at a given point.
Figure 8.

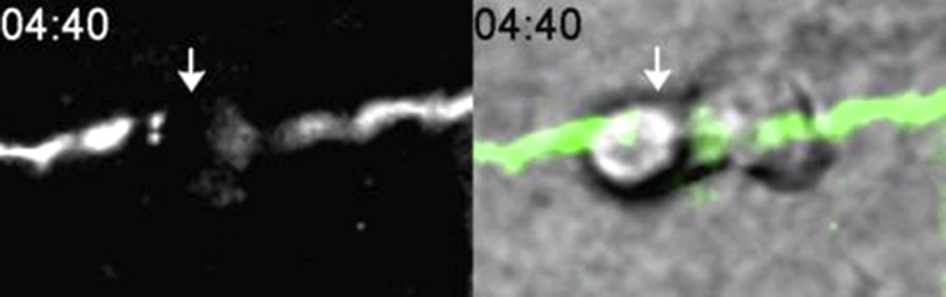
Polymorphonuclear (PMN) transendothelial migration (TEM) is inhibited in endothelial cells expressing vascular endothelial (VE)-cadherin double mutant (DMT). Endothelial cells were transduced with VE-cadherin wild-type (WT) green fluorescent protein (GFP) adenovirus or VE-cadherin DMT GFP. A and B: Time lapse of VE-cadherin GFP (top rows) and merged differential interference contrast and VE-cadherin GFP images (bottom rows) of PMN cells in the process of transmigration. A: In cells expressing VE-cadherin WT GFP, PMN cells locomote to a nearby cell-cell junction (white arrow, time = 00:00, merge). A VE-cadherin gap forms (white arrows, time = 03:40, top row and merge) just as the PMN cell is in the act of transmigrating (white arrows, time = 03:40, merge). TEM occurs within 5 minutes, and VE-cadherin gap reseals shortly after. B: In contrast, cells that express VE-cadherin DMT fail to form a VE-cadherin gap (white arrows, top row) despite the presence of a PMN crawling along a cell-cell junction for up to 20 minutes (white arrows, merge). These images are representative of 100 PMN interactions observed in at least five different experiments. Scale bars: 10 μm.
Live Cell Imaging Reveals that PMN Cells Do Not Transmigrate across Endothelial Cells Expressing VE-Cadherin DMT
Adherens junctions are dynamic structures that are constantly being remodeled, and the association of VE-cadherin with cytoplasmic proteins p120 and β-catenin has been implicated in this process.15, 37 Although the targeted recycling assay captures a snapshot of PMN cells caught in the act of TEM, we wanted to examine the continuous interactions of PMN cells with the endothelium under fluid shear conditions. We expressed VE-cadherin WT and VE-cadherin DMT and captured VE-cadherin gap formation in real time using time-lapse immunofluorescence imaging microscopy. In cells that expressed VE-cadherin WT, we observed de novo gaps that were associated with a transmigration event (Figure 8A and Supplemental Video S3). The gap resealed once transmigration was complete; the overall duration of TEM was 5 minutes. In contrast, in cells that expressed VE-cadherin DMT, VE-cadherin gap formation was inhibited (Figure 8B). Figure 8B and Supplemental Video S4 show a representative PMN cell, crawling along a cell-cell junction of an endothelial cell expressing VE-cadherin DMT GFP. No VE-cadherin gaps formed, and the PMN cells did not undergo TEM throughout the recording (20 minutes).
VE-Cadherin Is Phosphorylated on Leukocyte Adhesion Even when Microtubule Depolymerization Inhibits Targeted Recycling and TEM
In vitro, adhesion of leukocytes to the endothelial monolayer induces an increase in phosphorylation of VE-cadherin under conditions where endothelial cells have been serum starved. It is possible that our treatment with DCN not only inhibited LBRC recruitment but also inhibited VE-cadherin phosphorylation. We therefore determined the phosphorylation state of VE-cadherin in response to monocyte adhesion in control versus DCN-treated endothelial cells. Before conducting these experiments, we assessed the specificity of a commercially available antiphosphorylated VE-cadherin antibody. Endothelial cells were serum-starved overnight in M199 medium supplemented with 0.3% FBS. Cells were treated with control (diluent) or pervanadate, a tyrosine phosphatase inhibitor; under the latter conditions, we expected VE-cadherin to be highly phosphorylated. VE-cadherin was immunoprecipitated with hec1, an anti–VE-cadherin monoclonal antibody,32 or 4G10 antibody, a phosphotyrosine monoclonal antibody, and processed for Western blotting. VE-cadherin immunoprecipitated by hec1 from control and pervanadate-treated cells was detected by hec1, as expected (Figure 9A). In contrast, significant amounts of VE-cadherin were only immunoprecipitated with 4G10 in endothelial cells that had been pervanadate treated (Figure 9, A and B). Similarly, the phosphorylated VE-cadherin antibody detected VE-cadherin immunoprecipitated from endothelial cells that had been treated with pervanadate but not in control conditions (Figure 9B), confirming that this antibody is specific for phosphorylated VE-cadherin.
Figure 9.

Monocyte cell adhesion induces downstream signaling that regulates vascular endothelial (VE)-cadherin phosphorylation. A and B: Human umbilical endothelial cells (HUVECs) were serum starved overnight in M199 that contained 0.3% fetal bovine serum and activated with tumor necrosis factor (TNF)-α for 4 hours. Endothelial cells were treated with control (CTL) (diluent) or 100 μmol/L pervanadate (Perv) for 10 minutes at 37°C. VE-cadherin was immunoprecipitated with hec1, an antiphosphorylated VE-cadherin (anti–pVE-cadherin) antibody, or 4G10, an antiphosphotyrosine monoclonal antibody, as indicated and processed for Western blot analysis. VE-cadherin was detected by hec1 (A) or by pVE-cadherin polyclonal antibody (B). C: Endothelial cells were serum starved as above and were activated with TNF-α for 4 hours. Monocytes were added to HUVEC monolayers for 10 minutes at 37°C as described in Materials and Methods to achieve a ratio of 5:1 (monocytes:endothelial cells). Cells were lyzed and processed for immunoblotting. Blots were probed with the anti–VE-cadherin phosphotyrosine antibody. The asterisk indicates proteolytic fragment detected by the anti–VE-cadherin phosphotyrosine antibody but not anti–VE-cadherin. The same blot was stripped and reprobed with hec1, an anti–VE-cadherin antibody, as indicated. D: Quantification of three separate experiments. E: Endothelial cells were transduced with adenovirus expressing green fluorescent protein (GFP)–tagged VE-cadherin point mutants Y658F, Y731F, and VE-cadherin double mutant (DMT) (Y658F, Y731F). Cells maintained in M199 and 20% human serum were lyzed and processed for immunoblotting. The commercial anti–pVE-cadherin Y731 antibody recognizes native VE-cadherin, VE-cadherin Y658F, and Y731F mutants but fails to recognize VE-cadherin DMT (Y658F, Y731F) GFP. The same blot was stripped and reprobed with hec1, a monoclonal anti–VE-cadherin antibody (bottom blot). F: Schematic diagram of processes required for TEM. Data are presented as means ± SEM (D). ∗∗P < 0.01. DCN, demecolcine; IP, immunoprecipitated; LBRC, lateral border recycling compartment; PBMC, peripheral blood mononuclear cell; TEM, transendothelial migration.
Next, we tested the effects of DCN on the ability of monocyte adhesion to induce VE-cadherin phosphorylation. Endothelial cells were serum-starved overnight in M199 supplemented with 0.3% FBS and treated with TNF-α for 4 hours. Endothelial cells were treated with control (diluent) or DCN. Cells were washed several times, and freshly isolated monocytes were added at a monocyte/endothelial cell ratio of 5:1 and allowed to adhere for 10 minutes at 37°C. Nonadherent cells were washed off, and lysates were prepared as described in Materials and Methods and processed for immuoblotting. The antiphosphorylated VE-cadherin antibody recognized a band of the expected molecular weight for VE-cadherin in control and DCN-treated endothelial cells that had been incubated with monocytes. The immunoblot was stripped and reprobed with antiphosphorylated VE-cadherin antibody. A similar band was detected. The anti-phosphorylated VE-cadherin antibody also detected a lower band that we believe is a proteolytic fragment of VE-cadherin due to the presence of monocytes. VE-cadherin is highly susceptible to proteolysis. In this case, the lower band was not detected by the antiphosphorylated VE-cadherin antibody. The phosphorylated VE-cadherin antibody recognizes the cytoplasmic domain, whereas the antiphosphorylated VE-cadherin antibody is against the extracellular domain. It is possible that the extracellular domain was cleaved off, and thus the antiphosphorylated VE-cadherin antibody is no longer able to recognize this form of VE-cadherin.
There are two residues in VE-cadherin that have been implicated in TEM: residues Y658 and Y731. Endothelial cells were transduced with adenovirus expressing GFP–VE-cadherin mutants: Y658F, Y731F, and DMT (Y658F, Y731F). Under basal conditions in the presence of 20% human serum, there is a basal level of phosphorylated VE-cadherin. Lysates were prepared as described in Materials and Methods and immunoblotted with the antiphosphorylated VE-cadherin antibody. The antiphosphorylated VE-cadherin antibody recognized native VE-cadherin GFP, VE-cadherin Y658F, and VE-cadherin Y731F (Figure 9E). However, it did not recognize the VE-cadherin DMT GFP (Y658F, Y731F). The same blot was stripped and reprobed with antiphosphorylated VE-cadherin antibody. Bands of expected molecular weight for VE-cadherin and VE-cadherin GFP were detected. These results indicate that the antiphosphorylated VE-cadherin recognized phosphorylated VE-cadherin; however, this antibody cannot distinguish between phosphorylated Y658 or Y731.
Our results indicate that adhesion of leukocytes triggers downstream signaling events that result in the phosphorylation of VE-cadherin. This phosphorylation occurs despite the fact that microtubules are depolymerized and LBRC trafficking is inhibited. However, when LBRC trafficking is blocked, VE-cadherin does not move out of the junction, and TEM is inhibited.
Discussion
During paracellular migration, the LBRC is recruited to surround the leukocyte, and VE-cadherin is cleared from that site. Both events are required for TEM.7, 8, 9, 40 Both occur early in the process of transmigration and continue until TEM is complete.6, 8 The association between these processes had previously been unknown. We found that VE-cadherin gap formation was inhibited when LBRC recruitment was inhibited (Figure 2, Figure 3, Figure 4, Figure 5, and 6), even though VE-cadherin was phosphorylated on the residues implicated in its removal from the junction (Figure 9). In contrast, trafficking of the LBRC to the site of TEM could occur under conditions in which VE-cadherin could not leave the junction and gaps in staining did not form (Figures 7 and 8). These observations were seen in our quantitative TEM assays and in live cell imaging studies of TEM. One intriguing aspect of this work is that the LBRC may participate in clearing VE-cadherin from the site of TEM. Our data do not rule out the possibility that it is not the recruitment of LBRC membrane but rather the physical act of the leukocyte moving through the junction that leads to VE-cadherin gap formation. Nevertheless, this would be downstream of and dependent on LBRC recruitment.
Under resting conditions, the VE-specific protein tyrosine phosphatase (VE-PTP) is associated with VE-cadherin via plakoglobin, maintaining it in a hypophosphorylated state. Interaction of activated endothelial cells with leukocytes triggers dissociation of VE-PTP from VE-cadherin, allowing the latter to be phosphorylated.48, 49 Engagement of endothelial ICAM-1 and vascular cell adhesion molecule 1 by leukocyte integrins transduces signals the phosphorylated VE-cadherin and its associated catenins.46, 50, 51, 52 An emerging model is that phosphorylation of VE-cadherin at its cytoplasmic tail results in the weakening of the junctions as VE-cadherin cytoplasmic tail is uncoupled from the actin cytoskeleton. Two key tyrosine residues, Y658 and Y731, on the cytoplasmic tail of VE-cadherin have been implicated in this process.40, 47 (Note that Y658 is distinct from the Y685, which is implicated to have a role in permeability in response to soluble mediators.47) Indeed, when we mutated these two residues in VE-cadherin to phenylalanines, we were able to inhibit leukocyte TEM by approximately 80% in a TEM assay, and VE-cadherin gap formation did not occur (Figures 7 and 8). Wessel et al47 recently found that mice in which VE-cadherin was replaced with the Y731F mutant to mimic the dephosphorylated state of VE-cadherin had a specific defect in lymphocyte transmigration. This finding is similar to what we observed when we expressed VE-cadherin DMT. However, in their system, VE-cadherin Y731 was constitutively phosphorylated, and the investigators proposed that adhesion of leukocytes triggers the dephosphorylation of tyrosine 731 that is required for extravasation to occur via a process that requires internalization of VE-cadherin. Our data (Figure 9) do not distinguish which residue is phosphorylated, Y658 or Y731, because the phosphorylated VE-cadherin antibody recognized both of these sites. Thus, it is possible that Y658 is phosphorylated during TEM and Y731 is dephosphorylated, which would be consistent with the results reported.47 Regardless, all investigators are in agreement that VE-cadherin Y731 is critical for TEM. The overall goal in our study was to inhibit VE-cadherin gap formation to determine whether LBRC trafficking would still occur in the absence of VE-cadherin gaps. We were able to achieve this with our VE-cadherin DMT construct.
VE-cadherin phosphorylation is upstream of PECAM engagement, the first signal known to recruit the LBRC.8, 10 Nonetheless, for both monocytes and neutrophils, under the conditions studied here, VE-cadherin gaps around the leukocyte were inhibited unless the LBRC can recycle (Figure 2, Figure 3, Figure 4, Figure 5, and 6). Phosphorylation of VE-cadherin on tyrosine residues may be necessary, but it is not sufficient, to destabilize adherens junctions. Previous studies using conditions that lead directly to phosphorylation of tyrosines 658, 685, and 731 on VE-cadherin did not weaken barrier function.53 Additional signals (physical or chemical) from the leukocyte and/or endothelial cell must be necessary to weaken the junctions for TEM. Our data suggest that targeted recycling of the LBRC is a major one. The mechanism by which the LBRC participates in clearance of VE-cadherin from the site of TEM is the subject of ongoing investigation.
Although vascular leakage and leukocyte extravasation both occur in inflammation, it is now clear that adherens junctions are differentially regulated for different processes, such as permeability and diapedesis.47, 54 Although disruption of VE-cadherin contacts is required for TEM,7, 40 enhanced permeability does not necessarily lead to increased TEM,54 and TEM can occur without increasing monolayer permeability.43, 55 Furthermore, the mechanism of the response of adherens junctions to global stimuli, such as thrombin or vascular endothelial growth factor may be different from the local stimulation during TEM, although removal of VE-PTP from the junction may be involved in both.48 Endothelial cells respond rapidly to global vascular permeability signals followed by a decrease in VE-cadherin staining at the junctions.56 Endocytosis of VE-cadherin–bearing membrane has been implicated in this process.57 In contrast, the focal and transient (VE-cadherin returns within minutes as the leukocyte transmigrates) (Figures 6 and 8)6, 43 disappearance of VE-cadherin from the site of TEM may not involve endocytosis but rather lateral redistribution in the junctional membrane,7 although this is not universally reported.47
Endocytosis may also be responsible for the spontaneous preformed gaps that are seen in cultured endothelial cells6 (Figures 6 and 8). These have been found to be favored sites of TEM by neutrophils in previous studies.5, 6 Notably, under conditions in which trafficking of the LBRC is inhibited (Figure 6), leukocytes locomote to and over these sites but do not transmigrate.
Our data support a model in which diapedesis is a multistep process that requires the coordination of multiple signaling pathways (Figure 9F). In this model, adhesion of leukocytes to endothelial cells results in phosphorylation of VE-cadherin by one or more signaling pathways.4, 58 Nevertheless, phosphorylation alone is insufficient to induce VE-cadherin gaps but requires PECAM clustering, or a similar signal, that recruits the LBRC to the site of TEM. Recruitment of the LBRC is an early event that occurs before VE-cadherin gap formation. Inhibiting this step reduced VE-cadherin gap formation and TEM.
The physical mechanism(s) by which VE-cadherin is cleared from the site of TEM remains to be determined. However, one possible mechanism is that VE-cadherin is pushed aside from the site where the leukocyte is transmigrating. Phosphorylation of VE-cadherin or associated catenins would result in the uncoupling of VE-cadherin from the actin cytoskeleton. The influx of membrane from the LBRC at the site of TEM could clear VE-cadherin–containing membrane away from this site. Because the LBRC lacks VE-cadherin,10, 30, 31 this region on the adherens junction would appear as a gap. In this case, the LBRC would have a dual function: it could act as a gateway for a leukocyte and provide increased membrane surface area and unligated adhesion and signaling molecules with which the leukocyte must interact, and it would help clear VE-cadherin from this site.
Acknowledgments
We thank Drs. Peter Newman (Blood Center of Wisconsin, Milwaukee, WI) and Geri Kreitzer (Weill Cornell Medical Center, New York, NY) for monoclonal antibody P1.1 ascites and antibody HD, respectively; Dr. Clifford Carpenter for expert technical assistance; and Drs. David Sullivan and Kathy Green for helpful comments on the manuscript.
A.M.G. performed most of the experiments, was responsible for the overall design of the project and interpretation of the data, and wrote the first draft of the manuscript; B.F.C. performed and interpreted the microinjection and neutrophil experiments; and W.A.M. supervised the project, assisted in data interpretation, and edited the manuscript.
Footnotes
Supported by NIH grants R01 HL046849 (W.A.M.), R37 HL064774 (W.A.M.), and F31HL114374 (B.F.C.). Publication costs were supported by the Bessie Eisenberg Memorial Fund.
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.01.010.
Supplemental Data
A de novo vascular endothelial (VE)-cadherin gap forms during polymorphonuclear (PMN) transendothelial migration (TEM). VE-cadherin in endothelial cells was labeled with a nonblocking anti–VE-cadherin antibody conjugated with Dylight 488, and cells were treated with control (dimethyl sulfoxide). TEM of PMN cells was captured using time-lapse immunofluorescence microscopy. Images were captured every 10 seconds. Numbers indicated are elapsed minutes:seconds. Right panel, VE-cadherin Dylight 488 merged differential interference contrast (DIC); left panel, VE-cadherin DyLight 488 images. A de novo gap (white arrow) forms and is associated with a PMN TEM event.
{kind=link}
De novo vascular endothelial (VE)-cadherin gaps do not form when targeting of the lateral border recycling compartment (LBRC) is blocked. VE-cadherin in endothelial cells was labeled with a nonblocking anti–VE-cadherin antibody conjugated with Dylight 488 (as above), and cells were treated with 1 μmol/L of demecolcine. Images were captured using time-lapse immunofluorescence microscopy. Images were captured every 10 seconds. Numbers indicated are elapsed minutes:seconds. Right panel, VE-cadherin Dylight 488 merged differential interference contrast (DIC); left panel, VE-cadherin Dylight 488 images. Polymorphonuclear cells adhere and locomote to a preformed gap but fail to transmigrate under conditions where LBRC trafficking is inhibited.
{kind=link}
Vascular endothelial (VE)-cadherin gap forms in endothelial cells expressing VE-cadherin wild-type (WT) green fluorescent protein (GFP). Endothelial cells were transduced with VE-cadherin WT GFP adenovirus. Images were captured using time-lapse immunofluorescence microscopy. Right panel, VE-cadherin WT GFP merged differential interference contrast (DIC); left panel, VE-cadherin WT GFP images. Numbers indicated are elapsed minutes:seconds. Images were captured every 10 seconds. Polymorphonuclear (PMN) cells adhere and locomote to a junction. A de novo gap forms as the PMN cell is in the process of transmigrating and closes once this process is complete.
{kind=link}
Vascular endothelial (VE)-cadherin gaps do not form in endothelial cells expressing VE-cadherin double mutant (DMT). Endothelial cells were transduced with VE-cadherin DMT green fluorescent protein (GFP) adenovirus. Images were captured using time-lapse immunofluorescence microscopy as before. Numbers indicated are elapsed minutes:seconds. Images were captured every 10 seconds. Left panel, VE-cadherin DMT GFP merged differential interference contrast (DIC); right panel, VE-cadherin DMT GFP channels. Polymorphonuclear (PMN) cells adhere and locomote along cell-cell junctions, but VE-cadherin gaps do not form despite the presence of a PMN cell crawling along the junction for 20 minutes.
{kind=link}
References
- 1.Butcher E.C. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 2.Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 3.Ley K., Laudanna C., Cybulsky M.I., Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 4.Muller W.A. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allport J.R., Muller W.A., Luscinskas F.W. Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J Cell Biol. 2000;148:203–216. doi: 10.1083/jcb.148.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw S.K., Bamba P.S., Perkins B.N., Luscinskas F.W. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol. 2001;167:2323–2330. doi: 10.4049/jimmunol.167.4.2323. [DOI] [PubMed] [Google Scholar]
- 7.Alcaide P., Newton G., Auerbach S., Sehrawat S., Mayadas T.N., Golan D.E., Yacono P., Vincent P., Kowalczyk A., Luscinskas F.W. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood. 2008;112:2770–2779. doi: 10.1182/blood-2008-03-147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mamdouh Z., Chen X., Pierini L.M., Maxfield F.R., Muller W.A. Targeted recycling of PECAM from endothelial cell surface-connected compartments during diapedesis. Nature. 2003;421:748–753. doi: 10.1038/nature01300. [DOI] [PubMed] [Google Scholar]
- 9.Mamdouh Z., Kreitzer G.E., Muller W.A. Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J Exp Med. 2008;205:951–966. doi: 10.1084/jem.20072328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mamdouh Z., Mikhailov A., Muller W.A. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med. 2009;206:2795–2808. doi: 10.1084/jem.20082745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dejana E., Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol. 2012;19:218–223. doi: 10.1097/MOH.0b013e3283523e1c. [DOI] [PubMed] [Google Scholar]
- 12.Dejana E., Orsenigo F., Lampugnani M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 13.Vestweber D., Winderlich M., Cagna G., Nottebaum A.F. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009;19:8–15. doi: 10.1016/j.tcb.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Dejana E., Tournier-Lasserve E., Weinstein B.M. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Gumbiner B.M. Regulation of cadherin adhesive activity. J Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao K., Allison D.F., Buckley K.M., Kottke M.D., Vincent P.A., Faundez V., Kowalczyk A.P. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller W.A., Ratti C.M., McDonnell S.L., Cohn Z.A. A human endothelial cell-restricted, externally disposed plasmalemmal protein enriched in intercellular junctions. J Exp Med. 1989;170:399–414. doi: 10.1084/jem.170.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albelda S.M., Muller W.A., Buck C.A., Newman P.J. Molecular and cellular properties of PECAM-1 [endoCAM/CD31]: a novel vascular cell-cell adhesion molecule. J Cell Biol. 1991;114:1059–1068. doi: 10.1083/jcb.114.5.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller W.A., Weigl S.A., Deng X., Phillips D.M. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman P.J., Berndt M.C., Gorski J., White G.C., 2nd, Lyman S., Paddock C., Muller W.A. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science. 1990;247:1219–1222. doi: 10.1126/science.1690453. [DOI] [PubMed] [Google Scholar]
- 21.Florey O., Durgan J., Muller W. Phosphorylation of leukocyte PECAM and its association with detergent-resistant membranes regulate transendothelial migration. J Immunol. 2010;185:1878–1886. doi: 10.4049/jimmunol.1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller W.A. The use of anti-PECAM reagents in the control of inflammation. Agents Actions Suppl. 1995;46:147–157. doi: 10.1007/978-3-0348-7276-8_15. [DOI] [PubMed] [Google Scholar]
- 23.Liao F., Ali J., Greene T., Muller W.A. Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med. 1997;185:1349–1357. doi: 10.1084/jem.185.7.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogen S., Pak J., Garifallou M., Deng X., Muller W.A. Monoclonal antibody to murine PECAM-1 [CD31] blocks acute inflammation in vivo. J Exp Med. 1994;179:1059–1064. doi: 10.1084/jem.179.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schenkel A.R., Chew T.W., Muller W.A. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J Immunol. 2004;173:6403–6408. doi: 10.4049/jimmunol.173.10.6403. [DOI] [PubMed] [Google Scholar]
- 26.Duncan G.S., Andrew D.P., Takimoto H., Kaufman S.A., Yoshida H., Spellberg J., Luis de la Pompa J., Elia A., Wakeham A., Karan-Tamir B., Muller W.A., Senaldi G., Zukowski M.M., Mak T.W. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol. 1999;162:3022–3030. [PubMed] [Google Scholar]
- 27.Schenkel A.R., Chew T.W., Chlipala E., Harbord M.W., Muller W.A. Different susceptibilities of PECAM-deficient mouse strains to spontaneous idiopathic pneumonitis. Exp Mol Pathol. 2006;81:23–30. doi: 10.1016/j.yexmp.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seidman M.A., Chew T.W., Schenkel A.R., Muller W.A. PECAM-independent thioglycollate peritonitis is associated with a locus on murine chromosome 2. PLoS One. 2009;4:e4316. doi: 10.1371/journal.pone.0004316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muller W.A. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:326–333. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 30.Sullivan D.P., Seidman M.A., Muller W.A. Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am J Pathol. 2013;182:1031–1042. doi: 10.1016/j.ajpath.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng G., Sullivan D.P., Han F., Muller W.A. Segregation of VE-cadherin from the LBRC depends on the ectodomain sequence required for homophilic adhesion. J Cell Sci. 2015;128:576–588. doi: 10.1242/jcs.159053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali J., Liao F., Martens E., Muller W.A. Vascular endothelial cadherin (VE-Cadherin): cloning and role in endothelial cell-cell adhesion. Microcirculation. 1997;4:267–277. doi: 10.3109/10739689709146790. [DOI] [PubMed] [Google Scholar]
- 33.Wright S.D., Rao P.E., Van Voorhis W.C., Craigmyle L.S., Lida K., Talle M.A., Westberg E.F., Goldstein G., Silverstein S.C. Identification of the C3bi receptor of human monocytes and macrophages by using monoclonal antibodies. Proc Natl Acad Sci U S A. 1983;80:5699–5703. doi: 10.1073/pnas.80.18.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller W.A., Luscinskas F.W. Assays of transendothelial migration in vitro. Methods Enzymol. 2008;443:155–176. doi: 10.1016/S0076-6879(08)02009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller W.A., Weigl S. Monocyte-selective transendothelial migration: dissection of the binding and transmigration phases by an in vitro assay. J Exp Med. 1992;176:819–828. doi: 10.1084/jem.176.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vestweber D. VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol. 2008;28:223–232. doi: 10.1161/ATVBAHA.107.158014. [DOI] [PubMed] [Google Scholar]
- 37.Schulte D., Kuppers V., Dartsch N., Broermann A., Li H., Zarbock A., Kamenyeva O., Kiefer F., Khandoga A., Massberg S., Vestweber D. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011;30:4157–4170. doi: 10.1038/emboj.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corada M., Liao F., Lindgren M., Lampugnani M.G., Breviario F., Frank R., Muller W.A., Hicklin D.J., Bohlen P., Dejana E. Monoclonal antibodies directed to different regions of vascular endothelial cadherin extracellular domain affect adhesion and clustering of the protein and modulate endothelial permeability. Blood. 2001;97:1679–1684. doi: 10.1182/blood.v97.6.1679. [DOI] [PubMed] [Google Scholar]
- 39.Corada M., Mariotti M., Thurston G., Smith K., Kunkel R., Brockhaus M., Lampugnani M.G., Martin-Padura I., Stoppacciaro A., Ruco L., McDonald D.M., Ward P., Dejana E. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96:9815–9820. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allingham M.J., van Buul J.D., Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179:4053–4064. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 41.Schenkel A.R., Mamdouh Z., Muller W.A. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- 42.Phillipson M., Heit B., Colarusso P., Liu L., Ballantyne C.M., Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winger R.C., Koblinski J.E., Kanda T., Ransohoff R.M., Muller W.A. Rapid Remodeling of Tight Junctions during Paracellular Diapedesis in a Human Model of the Blood-Brain Barrier. J Immunol. 2014;193:2427–2437. doi: 10.4049/jimmunol.1400700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schenkel A.R., Mamdouh Z., Chen X., Liebman R.M., Muller W.A. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol. 2002;3:143–150. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- 45.Potter M.D., Barbero S., Cheresh D.A. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 46.Turowski P., Martinelli R., Crawford R., Wateridge D., Papageorgiou A.P., Lampugnani M.G., Gamp A.C., Vestweber D., Adamson P., Dejana E., Greenwood J. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci. 2008;121:29–37. doi: 10.1242/jcs.022681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wessel F., Winderlich M., Holm M., Frye M., Rivera-Galdos R., Vockel M., Linnepe R., Ipe U., Stadtmann A., Zarbock A., Nottebaum A.F., Vestweber D. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. 2014;15:223–230. doi: 10.1038/ni.2824. [DOI] [PubMed] [Google Scholar]
- 48.Nottebaum A.F., Cagna G., Winderlich M., Gamp A.C., Linnepe R., Polaschegg C., Filippova K., Lyck R., Engelhardt B., Kamenyeva O., Bixel M.G., Butz S., Vestweber D. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. 2008;205:2929–2945. doi: 10.1084/jem.20080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vockel M., Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood. 2013;122:2512–2522. doi: 10.1182/blood-2013-04-499228. [DOI] [PubMed] [Google Scholar]
- 50.Cook-Mills J.M., Johnson J.D., Deem T.L., Ochi A., Wang L., Zheng Y. Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J. 2004;378:539–547. doi: 10.1042/BJ20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Wetering S., van Buul J.D., Quik S., Mul F.P., Anthony E.C., ten Klooster J.P., Collard J.G., Hordijk P.L. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J Cell Sci. 2002;115:1837–1846. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- 52.van Wetering S., van den Berk N., van Buul J.D., Mul F.P., Lommerse I., Mous R., ten Klooster J.P., Zwaginga J.J., Hordijk P.L. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol. 2003;285:C343–C352. doi: 10.1152/ajpcell.00048.2003. [DOI] [PubMed] [Google Scholar]
- 53.Adam A.P., Sharenko A.L., Pumiglia K., Vincent P.A. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem. 2010;285:7045–7055. doi: 10.1074/jbc.M109.079277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schnoor M., Lai F.P., Zarbock A., Klaver R., Polaschegg C., Schulte D., Weich H.A., Oelkers J.M., Rottner K., Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208:1721–1735. doi: 10.1084/jem.20101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang A.J., Furie M.B., Nicholson S.C., Fischbarg J., Liebovitch L.S., Silverstein S.C. Effects of human neutrophil chemotaxis across human endothelial cell monolayers on the permeability of these monolayers to ions and macromolecules. J Cell Physiol. 1988;135:355–366. doi: 10.1002/jcp.1041350302. [DOI] [PubMed] [Google Scholar]
- 56.Nanes B.A., Chiasson-MacKenzie C., Lowery A.M., Ishiyama N., Faundez V., Ikura M., Vincent P.A., Kowalczyk A.P. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199:365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orsenigo F., Giampietro C., Ferrari A., Corada M., Galaup A., Sigismund S., Ristagno G., Maddaluno L., Koh G.Y., Franco D., Kurtcuoglu V., Poulikakos D., Baluk P., McDonald D., Grazia Lampugnani M., Dejana E. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muller W.A. The regulation of transendothelial migration: new knowledge and new questions. Cardiovasc Res. 2015;107:310–320. doi: 10.1093/cvr/cvv145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A de novo vascular endothelial (VE)-cadherin gap forms during polymorphonuclear (PMN) transendothelial migration (TEM). VE-cadherin in endothelial cells was labeled with a nonblocking anti–VE-cadherin antibody conjugated with Dylight 488, and cells were treated with control (dimethyl sulfoxide). TEM of PMN cells was captured using time-lapse immunofluorescence microscopy. Images were captured every 10 seconds. Numbers indicated are elapsed minutes:seconds. Right panel, VE-cadherin Dylight 488 merged differential interference contrast (DIC); left panel, VE-cadherin DyLight 488 images. A de novo gap (white arrow) forms and is associated with a PMN TEM event.
De novo vascular endothelial (VE)-cadherin gaps do not form when targeting of the lateral border recycling compartment (LBRC) is blocked. VE-cadherin in endothelial cells was labeled with a nonblocking anti–VE-cadherin antibody conjugated with Dylight 488 (as above), and cells were treated with 1 μmol/L of demecolcine. Images were captured using time-lapse immunofluorescence microscopy. Images were captured every 10 seconds. Numbers indicated are elapsed minutes:seconds. Right panel, VE-cadherin Dylight 488 merged differential interference contrast (DIC); left panel, VE-cadherin Dylight 488 images. Polymorphonuclear cells adhere and locomote to a preformed gap but fail to transmigrate under conditions where LBRC trafficking is inhibited.
Vascular endothelial (VE)-cadherin gap forms in endothelial cells expressing VE-cadherin wild-type (WT) green fluorescent protein (GFP). Endothelial cells were transduced with VE-cadherin WT GFP adenovirus. Images were captured using time-lapse immunofluorescence microscopy. Right panel, VE-cadherin WT GFP merged differential interference contrast (DIC); left panel, VE-cadherin WT GFP images. Numbers indicated are elapsed minutes:seconds. Images were captured every 10 seconds. Polymorphonuclear (PMN) cells adhere and locomote to a junction. A de novo gap forms as the PMN cell is in the process of transmigrating and closes once this process is complete.
Vascular endothelial (VE)-cadherin gaps do not form in endothelial cells expressing VE-cadherin double mutant (DMT). Endothelial cells were transduced with VE-cadherin DMT green fluorescent protein (GFP) adenovirus. Images were captured using time-lapse immunofluorescence microscopy as before. Numbers indicated are elapsed minutes:seconds. Images were captured every 10 seconds. Left panel, VE-cadherin DMT GFP merged differential interference contrast (DIC); right panel, VE-cadherin DMT GFP channels. Polymorphonuclear (PMN) cells adhere and locomote along cell-cell junctions, but VE-cadherin gaps do not form despite the presence of a PMN cell crawling along the junction for 20 minutes.