

Abstract
Allelic exchange is an efficient method of bacterial genome engineering. This protocol describes the use of this technique to make gene knockouts and knockins, as well as single nucleotide insertions, deletions and substitutions in Pseudomonas aeruginosa. Unlike other approaches to allelic exchange, this protocol does not require heterologous recombinases to insert or excise selective markers from the target chromosome. Rather, positive and negative selection are enabled solely by suicide vector-encoded functions and host cell proteins. Here, mutant alleles, which are flanked by regions of homology to the recipient chromosome, are synthesized in vitro and then cloned into allelic exchange vectors using standard procedures. These suicide vectors are then introduced into recipient cells by conjugation. Homologous recombination then results in antibiotic resistant single-crossover mutants in which the plasmid has integrated site-specifically into the chromosome. Subsequently, unmarked double-crossover mutants are isolated directly using sucrose-mediated counter-selection. This two-step process yields seamless mutations that are precise to a single base pair of DNA. The entire procedure requires ~2 weeks.
Keywords: Pseudomonas aeruginosa, gene replacement, mutagenesis
INTRODUCTION
The bacterium Pseudomonas aeruginosa is one of the most prevalent hospital pathogens in all developed countries1–3. This microorganism is an important laboratory model and a mainstay for studying motility and biofilm formation, as well as cell signaling, virulence and drug resistance in Gram-negative bacteria. Hundreds of P. aeruginosa genomes have been sequenced, and there is an ever-expanding number of genomic resources available for this organism. This includes transposon mutant libraries4–6, an open reading frame (ORF) collection7, microarrays8, massively-parallel sequencing workflows for RNA-seq9–13, ChIP-seq13–15, Tn-seq16, and transcription start site mapping10, as well as a genome database with community-driven annotation17.
Yet despite being a highly studied model bacterium, only 18% of the protein coding regions in the P. aeruginosa PAO1 genome have experimentally demonstrated biological roles, and merely an additional ~21% are highly similar to those with a known function in other organisms. The remaining genes have either only generic, homology-derived functional attributes (~22%), or no discernible physiological purpose (39%)17. The low level characterization of these ORFs underscores a continued need for facile techniques to engineer P. aeruginosa strains with precisely defined genotypes. A key technique used for this purpose is allelic exchange, which uses homologous recombination to target an endogenous gene and replace it with a mutant allele.
Allelic exchange has been used to engineer site-directed mutations in a wide range of bacterial species18–21. This method was first applied to P. aeruginosa genetics in the mid-1980s22,23. Over the past 30 years, advances in allelic exchange for P. aeruginosa have been driven by the invention of suicide vectors22, the application of conditional lethal genes to counter-selection24, and the use of heterologous recombinases (such as Flp20 and Cre25) to remove antibiotic resistance markers after gene replacement. Allelic exchange methods have also benefitted from innovations in cloning technology, including yeast recombineering26 and Gateway technology27–31. Nevertheless, because the execution of two-step allelic exchange is versatile and varied, application of this method can be daunting even for experienced geneticists. Here we explain the molecular biology of allelic exchange, and provide a comprehensive and up-to-date protocol for engineering mutations into the P. aeruginosa genome with single-nucleotide precision28,31–35.
The principle of two-step allelic exchange
Most published methods of allelic exchange in P. aeruginosa depend on plasmids with a ColE1 origin of replication, which replicate in Escherichia coli but not in Pseudomonas species. An assortment of these so-called suicide vectors has been built with suitable antibiotic resistance cassettes and counter-selection markers for manipulating P. aeruginosa (Table 1). To facilitate allelic replacement, an allelic exchange vector is engineered with a copy of the gene of interest, which contains the desired mutation, as well as flanking regions of DNA that are homologous to the recipient chromosome. After the construction of this suicide delivery vector, allelic exchange involves two steps (Fig. 1).
TABLE 1.
Allelic exchange vectors for Pseudomonas aeruginosa
| Vectora, b | Description and pertinent features | Accession Noc | Reference |
|---|---|---|---|
| Restriction cloning | |||
| pEX18Apa, b, d | Apr/CbroriT, sacB, lacZα, MCS from pUC18 | AF004910 | 20 |
| pEX18Gma, b | GmroriT, sacB, lacZα, MCS from pUC18 | AF047518 | 20 |
| pEX18Tca, b | TcroriT, sacB, lacZα, MCS from pUC18 | AF047519 | 20 |
| pEX19Gmb | GmroriT, sacB, lacZα, MCS from pUC19 | KM887142c | 20,99 |
| pEXG2 | Gmrmob, sacB, lacZα | KM887143c | 100 |
| pEX19EYFP | GmroriT, sacB, lacZα, MCS from pUC19, yfp | --- | 81 |
| pEX19ECFP | GmroriT, sacB, lacZα, MCS from pUC19, cfp | --- | 81 |
| Saccharomyces cerevisiae-based recombineering | |||
| pMQ30 | GmroriT, sacB, lacZα, URA3 | DQ230317 | 26 |
| pMQ75 | GmroriT, sacB, lacZα, URA3 | DQ230319 | 74 |
| Gateway® cloning - donor vectors | |||
| pDONRPEX18Apa | Cmr and Apr/Cbr, pEX18Ap with Gateway donor site | KM880129c | This study |
| pDONRPEX18Gma | Cmr and Gmr, pEX18Gm with Gateway donor site | KM880128c | This study |
| pDONRPEX18Tca | Cmr and Tcr, pEX18Tc with Gateway donor site | KM880130c | This study |
| pDONRX | Cmr and Gmr, pMK2010 with Gateway donor site | --- | 30 |
| Gateway® cloning - destination vectorse | |||
| pEX18ApGWa, b | Cmr and Apr/Cbr, pEX18Ap with Gateway destination site | AY928469 | 27 |
| pEX18GmGWa | Cmr and Gmr, pEX18Gm with Gateway destination site | KM880127c | 29 |
| pEXG2GW | Cmr and Gmr, pEXG2 with Gateway destination site | --- | 30 |
Plasmids available by request to Joe J. Harrison (jjharris@ucalgary.ca).
Plasmids available by request to Herbert Schweizer (hschweizer@ufl.edu).
Sequencing primers used to obtain DNA sequence deposited in Genbank are listed in Table S1.
Orignially named pEX18T.
Destination vectors can be designated either by a ‘pDEST’ prefix or ‘GW’ suffix. Both designations are equivalent and utilized in the literature. Antibiotic selection (E. coli/P. aeruginosa): Ap, ampicillin; Cb, carbenicillin; Cm, chloramphenicol, Gm, gentamicin; Tc, tetracycline. oriT, origin of transfer; sacB, levansucrase; lacZα, α-peptide of β-galactosidase.
Figure 1.

Two-step allelic exchange. Host cell proteins drive homologous recombination between the allelic exchange vector and the recipient chromosome. The asterisk denotes any insertion, deletion or base substitution, which may be large (>10 kb for deletions) or as small as a single nucleotide. Dashed lines indicate the outcomes of homologous recombination that are selected at each of the two key steps of allelic exchange. After first-crossover, which occurs shortly after conjugation, antibiotic selection (Step 36) is used to select for merodiploids. Subsequently, sucrose counter-selection (Step 37) is used to select for double crossovers. Depending on the physical locus of the second crossover, recombination will either restore the wild type allele or fix the mutant allele in the bacterial chromosome. A third but rare possible outcome is counter-selection escape. This outcome produces a sucrose- and antibiotic-resistant merodiploid, resulting from an inactivating mutation in sacB.
In the first-step of allelic exchange, the suicide vector is introduced into P. aeruginosa. This may be accomplished by electroporation36,37. However, most allelic exchange vectors contain the origin of transfer (oriT) from the broad-host range plasmid RP4 (Table 1); therefore, these vectors are most often mobilized by conjugation from E. coli into P. aeruginosa, which is more efficient than electroporation. A site-specific chromosomal integration event, which is mediated by homologous (or Campbell-type38) recombination, must occur in order to obtain resistance to the antibiotic marker encoded on the suicide vector because the ColE1-type plasmids cannot replicate in P. aeruginosa (Fig. 1). This event is termed a single-crossover. Recombination frequency increases with homologous sequence lengths; therefore, the number of antibiotic resistant merodiploids produced at this first step increases with increasing lengths of vector-encoded homologous sequences.
In the second step of allelic exchange, the plasmid backbone is excised from the chromosome through a rare second homologous recombination event, which is termed a double-crossover. This double-crossover mutant is isolated by counter-selection. This is frequently accomplished by selecting for the loss of a gene that confers sensitivity to an agent in the growth medium. Although there are many means of counter-selection39, a large number of allelic exchange vectors for Gram-negative bacteria, including those used for manipulating P. aeruginosa, encode the Bacillus subtilis sacB gene (Table 1). The sacB gene encodes levansucrase. This enzyme catalyzes the hydrolysis of sucrose and synthesis of levan polysaccharides, which are high-molecular weight fructose polymers. Although the molecular basis of toxicity remains unclear, levansucrase activity is localized primarily to the periplasm in Gram-negative bacteria40 and presumably also in Gram positive bacteria, such as coryneforms and mycobacteria, for which the sacB system also works as a conditional lethal gene41,42. This activity confers acute sucrose sensitivity40,43,44, which is dependent on incubation temperature as well as sodium chloride concentration in the growth medium45. Hence, double-crossover mutants can be selected by growing the first-crossover mutants on sucrose, as only those bacteria that have lost sacB gene expression can grow under this condition. In principle, therefore, sucrose counter-selection results in the loss of either the wild type or mutant allele from the chromosome. In practice, however, sucrose resistant merodiploids, which have acquired inactivating mutations in sacB, also appear on occasion (Fig. 1). This latter phenomenon is termed counter-selection escape46. In some bacteria, this may be caused by high-frequency insertion of transposable IS elements into the sacB gene44,47–49. However, while IS elements from some P. aeruginosa isolates have been studied in detail50,51, it is not clear that all strains of P. aeruginosa (such as PAO1) encode functional IS elements. Thus loss-of-function mutations in sacB occur with relatively low frequency in laboratory strains such as PAO1 (see Anticipated Results), because these mutations presumably result from random insertion, deletion, or substitution mutations in the sacB gene. Regardless of the underlying causality, all sucrose resistant merodiploids are easily distinguished from double-crossover mutants because they remain antibiotic resistant (Fig. 1).
Advantages of two-step allelic exchange
There are many published procedures for creating mutant alleles in vitro; however, the protocol presented here utilizes splicing-by-overlap extension (SOE) polymerase chain reaction (PCR) to construct contiguous mutant alleles that are uninterrupted by antibiotic resistance markers. A similar strategy has been used for engineering unmarked Escherichia coli and Vibrio cholerae strains for nearly 20 years18,52,53. This allows for precise, unscarred and unmarked mutations to be selected for at the stage of double-crossover (Fig. 1). This eliminates the need for subsequent Flp-FRT or Cre-lox recombination steps to remove an antibiotic resistance cassette20,25,27, saving labour, reducing the chance of generating a polar mutation, enabling genome engineering with single-base precision, and eliminating risks of off-target recombination54.
By contrast to this protocol, a primary advantage of creating marked mutations is the ability to apply positive selection for mutants at the stage of second crossover27. This can reduce the amount PCR required to identify mutant strains. However, the subsequent excision of antibiotic resistance cassettes using exogenous recombinases such as Flp20,27 requires about 5 to 7 days of additional work55, and leaves a scarred deletion. An empirical comparison of the technique presented in this protocol to an established Flp-dependent method27 is provided at the end of this introduction. For further information on gene replacement techniques that require Flp-FRT recombination, we direct the reader to a description elsewhere20,27.
The primary alternative to allelic exchange is PCR-mediated gene replacement with the λ Red recombinase system. Red recombineering has been used extensively to engineer E. coli and Salmonella enterica56–60 and has been adapted for use with P. aeruginosa61,62. By contrast to the enteric bacteria, however, current protocols for λ Red recombination in P. aeruginosa require very long primers or multi-fragment splicing-by-overlap-extension (SOE)-PCR to generate mutant alleles62. Also, recipient cells must express the λ Red recombination genes gam, bet and exo, and therefore, an initial step is required to transform the recipient strain with a vector that expresses these genes. An advantage of two-step allelic exchange is that once a suicide vector is constructed, it can be used directly on many genetic backgrounds of P. aeruginosa. Thus it is simple to introduce the desired mutation into many strains in parallel or to create multiple mutations in a single strain sequentially. Moreover, there is no need for additional PCR to produce mutant alleles or transformations to create suitable recipient strains.
Finally, this protocol has been troubleshot extensively, being deployed into 9 Pseudomonas laboratories in 4 countries during its development. At the time of this publication, we collectively estimate that our groups have created more than a thousand mutants of laboratory strains PAO1 and PA14 with this protocol. Also, this protocol has been used to engineer strains from a library of environmental34 and clinical isolates34,63 – including those from eye, ear, blood, lung and urinary tract infections – with a success rate of about 50%34. Altogether, the manipulated isolates belong to the two most abundant phylogenetic groups of P. aeruginosa strains, which represent >95% of all genome-sequenced strains of this species.
Modifications, applications and limitations
Expert users may find a variety of standard molecular cloning techniques preferable alternatives to those described here. However, deviations from media compositions and growth conditions provided in this protocol for donor E. coli and recipient P. aeruginosa strains may result in suboptimal results, a reduction in efficiency, and even failure to generate mutants.
With the use of vectors encoding appropriate selection and counter-selection functions, this protocol is broadly applicable to engineering the genomes of other bacteria, especially other Pseudomonas and Burkholderia species64. In addition to the allelic exchange vectors useful for P. aeruginosa genome engineering (Table 1), a variety of pEX-based plasmids encoding alternative resistance cassettes (including kanamycin, trimethoprim and tellurite resistance genes) have been put to use in a broad range of Gram-negative bacteria65,66. Additionally, there are many alternative strategies for counter-selection, which may be crucial for manipulating bacteria that are insensitive to SacB-mediated sucrose toxicity46,67,68.
The reader should note that there are several key limitations of allelic exchange:
Allelic exchange depends on host cell recombination proteins, and therefore, this protocol cannot be used to manipulate bacteria that are recombination deficient (for example, ΔrecA strains). Also, this protocol may not be equally efficient for all strains because recombination frequencies can vary considerably between strains of the same species.
If there is shared synteny between strains, it may be possible to use a mutant allele generated for one strain to target the orthologous gene in another strain34,63. However, due natural genetic variation between strains, it is likely that this will introduce additional single nucleotide polymorphisms (SNPs) in the adjacent regions of homology.
Allelic exchange will not be successful at inactivating those genes that are essential for growth under the conditions provided. In P. aeruginosa, the number of essential genes is estimated to be 200–4005,69.
Complementation analysis should be carried out to verify the phenotypes of null mutants. The primary purpose of complementation is to rule out polarity or secondary site mutations as the possible cause of a mutant phenotype. Many genetic tools have been developed for this purpose, including shuttle vectors29,70, site-specific transposons55,71 and integration-proficient plasmids72. We direct the reader to descriptions of these tools elsewhere29,55,70–72.
Experimental design
A basic knowledge of molecular and microbiological techniques is required to execute the genome engineering protocols described here. These basic techniques are described in standard laboratory manuals for molecular cloning73,74. Novice users should note that examples of enzymes and cloning strategies are provided as part of this protocol; however, expert users will find that a variety of reagents, enzymes, and kits may be substituted with appropriate alternatives. Novice users will find additional assistance for generating mutant alleles in the Supplementary Tutorial. Although the Supplementary Tutorial is focused on making a deletion mutation, it illustrates design principles and protocol steps that are shared among all variations of the allelic exchange method.
The process of allelic exchange can be divided into six stages (with a total of 49 short Procedure steps), which are illustrated in the general flow chart (Fig. 2):
-
(1)
Vector selection and primer design (Steps 1–3).
-
(2)
PCR to synthesize mutant alleles in vitro (Steps 4–12).
-
(3)
Cloning the mutant allele into an allelic exchange vector (Steps 13–19).
-
(4)
PCR identification and sequencing of insertions in allelic exchange vectors (Steps 20–35).
-
(5)
Introduction of the mutant allele into a P. aeruginosa recipient and merodiploid selection (Step 36).
-
(6)
Counter-selection, PCR identification and sequencing of mutations in P. aeruginosa cells (Steps 37–49).
Figure 2.

Workflow for making precision-engineered mutant strains of P. aeruginosa. This protocol requires ~40 h over two weeks. Multiple allelic exchange vectors that target different genes can be created in parallel, providing significant economy of time. Once an allelic exchange vector is created, it may be used again in the future (starting from Step 36Aii) to generate the mutation in another cell line in as little as 9 d.
Vector selection, primer design and using PCR to synthesize mutant alleles in vitro (Steps 1–12)
The initial step for engineering the P. aeruginosa genome is to choose a suitable allelic exchange vector (Table 1). The foremost concern is to choose a resistance marker for an antibiotic to which the recipient strain is sensitive. Next a vector is chosen that allows for one’s preferred cloning strategy to insert the mutant allele into the plasmid (Table 1). Many allelic exchange vectors contain a multiple cloning site (MCS) as well as the lacZα fragment for blue-white selection in suitable host strains expressing lacZΔM15 β-galactosidase (Table 1). In principle, these same vectors are also compatible with standard ligation-independent and Gibson cloning75 techniques. Additionally, an elegant system for assembling allelic exchange vectors using in vivo recombineering in Saccharomyces cerevisiae has been developed26. Finally, a growing number of vectors have been built with donor (attP) or destination (attR) sites that are compatible with Gateway® technology (Table 1). Because of its quick execution time, cloning efficiency and cost-effectiveness, this protocol relies on the use of Gateway® recombineering to create plasmids for allelic exchange (Box 1). Note that all primers must be synthesized with the appropriate attachment or restriction sites as well as homology extensions that enable cloning and assembly of the PCR products (Box 2).
BOX 1. MOLECULAR CLONING WITH IN VITRO RECOMBINEERING.
A wide variety of standard and commercial cloning techniques are available to molecular biologists. While it is possible to use any of these approaches here, our group28,32 and others27,29,30 have found that Gateway® in vitro recombineering has many benefits for cloning mutant alleles and creating allelic exchange vectors. Gateway® technology utilizes the recombination proteins of bacteriophage λ to drive site-specific recombination between attachment (att) sites101,102. Such sites (attB1 and attB2) are synthesized at the 5’-ends of forward and reverse PCR primers (Box 2), such that recombination of PCR products with corresponding target sites (attP1 and attP2) in a donor (“pDONR”) plasmid is directional. This recombination process, which is mediated by BP Clonase® (Invitrogen), produces an entry (“pENTR”) vector in which the inserted PCR product is flanked with different attachment sites (attL1 and attL2). Subsequently, the entry vector can be recombined with a destination (“pDEST” or “–GW”) vector, which encodes another set of target sites (attR1 and attR2). The recombination of an entry and destination vector is mediated by LR Clonase® (Invitrogen), and produces an “expression” vector in which the insert is flanked by attB1 and attB2 sites. Thus expression vectors can be recombined with donor vectors using BP Clonase®. Gateway® reactions require 1 h and are highly efficient because a typical reaction produces at least 500–1000 independent clones103. Additionally, because Gateway® vectors exploit ccdB-based counterselection at the transformation step102, these reactions yield E. coli clones in which 75–99% of the isolated colonies have the desired insert. Several Gateway® compatible allelic exchange vectors have been created for the purpose of manipulating Gram-negative bacteria, including P. aeruginosa (Table 1). A key benefit to this technology is that mutant alleles can be shuttled between donor and destination allelic exchange vectors without the need for addition PCR. This can be invaluable for troubleshooting unforeseen challenges with antibiotic selection, particularly for clinical isolates. Gateway® recombinational technology, therefore, can substantially accelerate the process of gene replacement. The use of Gateway® technology is inexpensive (currently, a half-volume Clonase® reaction, which is used in this protocol, costs ~$5US) and creates additional economy, relative to restriction-and-ligation, by significantly reducing hands-on time and increasing cloning efficiency.
BOX 2. GUIDELINES FOR PRIMER DESIGN.
Primer sequences are dictated by the target amplicon; however, there may be some flexibility in the target. Whenever possible, choose a region of target DNA where the GC content is <70%, and primers should have no more than a 3 to 1 GC:AT ratio or vice versa. The GC content of the P. aeruginosa genome is ~66.6%104, but ideally, primers will have a GC content of between ~55 and ~68%.
Calculate primer melting temperature using an algorithm that corrects for salt concentration. Oligocalc (http://www.basic.northwestern.edu/biotools/oligocalc.html) is a free web tool that can be used to calculate salt-adjusted melting temperatures105.
Primers should have a salt-adjusted melting point of 62 ± 2 °C for the region of homology to the target amplicon.
Regions of reverse complementarity to another DNA fragment, which are used to facilitate assembly of PCR products during SOE-PCR, should have a salt-adjusted melting point of 70 ± 1 °C.
Sequencing primers are best as 17- to 20-mers with melting points of 50 to 60 °C (i.e. the “Seq-F” and “Seq-R” primers in Fig. 3).
Primers must not have long regions (i.e. >6 bp) of self-complementarity that could be predicted to form secondary structures. Also, ensure that primers do not self-anneal, or have complementarity to their partner – this helps to avoid the formation of primer dimers.
-
Primers can be flanked by any desired sequence, including an attachment sequence (such as an attB sequence for Gateway® cloning) or a restriction site. Some frequently used restriction and attachment sites (shaded regions correspond to the DNA sequence recognized by the enzyme):
5′-ATC CGG AAG CTT-3′ HindIII 5′-ATC CGG CTG CAG-3′ PstI 5′-ATC CGG GGA TCC-3′ BamHI 5′-ATC CGG GAA TTC-3′ EcoRI 5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC A-3′ attB1 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTA-3′ attB2 Δ CRITICAL Do not include these extra sequences in the calculation of melting temperature.
Δ CRITICAL An extra 4 and 6 bp have been added at the end of a primer with an attachment or a restriction site, respectively. The additional DNA is required to facilitate enzyme binding to the substrate.
Building deletion alleles
Building a deletion allele and confirming a chromosomal “knockout” requires three primer pairs. The first primer pair (“Up-F” and “Up-R”) targets a genomic region upstream of the desired deletion point, and the second (“Down-F” and “Down-R”) targets the genomic region downstream of the desired deletion point. Lastly, a third set of primers (“Seq-F” and “Seq-R”) is designed to sequence through and confirm the deletion of the site for the putative mutation on the chromosome (Fig. 3a). These primers are typically designed outside the targeted upstream and downstream regions of homology (Fig. 3a). However, it is possible to nest these primers within the regions of homology, especially if cloning the targeted PCR amplicon has proven to be difficult. In lieu of this third set of primers, it is possible to use the “Up-F” and “Down-R” primers that were used to clone the upstream and downstream fragments, respectively, to identify deletion mutants at the last stage of this protocol. However, because these primers are tailed with attachment or restriction sites, the “Up-F” and “Down-R” primers will not always be suitable for sequencing the region of the deletion following counter-selection. An effective deletion strategy typically leaves a miniature ORF that encodes a short polypeptide, and the goal is to delete 95 – 99% of the target gene. To create a deletion mutation that is in-frame, the “Up-R” and “Down-R” primers are designed so that their 5’-ends are a multiple of 3 nucleotides from the start and stop codons, respectively (Fig. 3a). These stipulations for creating in-frame deletions, however, still allow primers to be designed with some flexibility.
Figure 3.

How to design and build synthetic mutant alleles. (a) Deletion alleles. (b) Insertion alleles. (c) Alleles with single nucleotide polymorphisms (SNPs), short deletions or insertions (~25 bp or less). Note that “Seq-F” and “Seq-R” primers may be nested within regions of homology if the target amplicon is difficult to clone by PCR.
Similar to most other prokaryotes, the average ORF length in the P. aeruginosa PAO1 genome is ~1.0 kb76. To create a deletion of this size, a sequence length of ~400–500 bp for each of the upstream and downstream homologous regions is sufficient to drive recombination with the P. aeruginosa chromosome at readily selectable frequencies. The size of these regions of homology should be increased for larger deletions, and including an additional ~50–100 bp for each additional 1 kb of DNA being deleted from the chromosome is recommended. For example, this protocol was used to delete an 11.5 kb region from the P. aeruginosa PAO1 R2 pyocin gene cluster (PA0615-PA0628) using a construct that contained a total of ~1.8 kb homology to the chromosome28,77. To create a deletion allele, the two PCR products encoding regions flanking the deletion point are assembled by SOE-PCR78. To facilitate assembly of the two fragments, one of the primers (“Up-R” or “Down-F”) is designed with an extension that has reverse complementarity to the other fragment (Fig. 3a and Box 2).
The reader should note that target genes may contain promoter elements, ribosomal binding sites (RBSs) or start codons for adjacent ORFs. It is also possible that targeted sequences may be expressed as non-coding RNA (ncRNA) from the opposing strand or contain a site for epigenetic modification. Mutagenesis of these target genes can be carried out in ways that are predicted to reduce or eliminate polar effects that may affect expression of downstream genes or operons. For instance, it is possible to use genome sequence data to exclude predicted RBSs and start codons from chromosomal regions targeted for deletion. Additionally, it is possible to introduce a nonsense mutation into a target gene that contains overlapping promoter elements. Such a strategy could be used to create a truncated ORF that encodes a short polypeptide, but leaves transcriptional elements of the adjacent gene intact (see Building alleles and gene fragments with SNPs). In P. aeruginosa, for example, this approach has been used to truncate the fleN ORF, which contains a promoter for the adjacent fliA gene79.
Building insertion alleles
This protocol may be used to introduce an exogenous genetic element into the chromosome. Building an insertion allele and confirming a chromosomal “knockin” requires at least four primer pairs. The first primer pair (“Up-F” and “Up-R”) targets a genomic region upstream of the desired insertion point, and the second (“Down-F” and “Down-R”) targets the genomic region downstream of the desired insertion point. The third primer pair (“Ins-F” and “Ins-R”) targets the exogenous genetic element to be inserted into the genome. Finally, a fourth set of primers (“Seq-F” and “Seq-R”) is designed to read through the putative insertion site on the chromosome (Fig. 3b). To facilitate assembly of the three fragments, two of the primers (“Up-R” or “Ins-F” and “Ins-R” or “Down-F”) are designed with extensions that have reverse complementarity to one of the other fragments (Fig. 3b). The individual fragments are amplified in a first round of PCR, then all three are combined and fused in a second round of SOE-PCR. In principle, it may be possible to assemble more than three PCR fragments together at this step.
The ability to create precise insertion mutations with allelic exchange has many potential applications. For example, it is possible to create an unscarred knockin of a transcriptional regulator and inducible promoter element (for example, E. coli araC-PBAD28,35,80) upstream of a gene or operon in order to gain artificial control over its expression. Other useful applications include the insertion of epitope tags into proteins, or the fusion of an ORF with a fluorescent protein81–83.
Building alleles and gene fragments with single nucleotide polymorphisms (SNPs)
Constructing a de novo mutant allele with a SNP, short deletion or insertion mutation may be carried out using well-established protocols for site-directed mutagenesis by SOE-PCR84. Briefly, this approach uses two pairs of primers, each of which targets a 400–500 bp segment of the genome; however, the internal primers generate overlapping, complementary 3' ends on the intermediate segments and introduce nucleotide substitutions (Fig. 3c). The SOE-PCR product is thus a ~800–1000 bp DNA fragment in which the point mutation is approximately centered within a contiguous region of homology to the target chromosome. Note that central position of the point mutation is critical: if the point mutation is near either end of the homologous region, then this will significantly reduce the probability that a second-crossover event will incorporate the mutation into the chromosome. Alternatively to SOE-PCR, a mutant allele, or a fragment of that allele, can be cloned directly by PCR from a strain bearing the desired mutation. Such an approach may be useful for understanding the fitness benefits of SNPs that have evolved in laboratory strains or clinical isolates77,85. Finally, a set of primers (“Seq-F” and “Seq-R”) is designed to read through the putative mutation site on the chromosome (Fig. 3c). Importantly, these primers are used for Sanger sequencing to identify the P. aeruginosa recipients that have acquired the desired mutation at the final stage of this protocol.
Cloning the mutant allele into an allelic exchange vector (Steps 13–19)
Any cloning method can be used to create an allelic exchange vector containing the synthesized mutant allele; however, this can be accomplished rapidly with a single, hour-long enzymatic step that utilizes Gateway® technology (Box 1). In this protocol, attB1 and attB2 flanked PCR products are transferred into a suitable donor allelic exchange vector using BP Clonase® II (Invitrogen). Alternatively, the attB1- and attB2-flanked SOE-PCR product can be cloned first into another entry vector, such as pDONR221 (Kmr, Invitrogen) or pDONR223 (Spr)86. The reaction is then transformed into E. coli DH5α using standard chemical methods or electroporation (Box 3). In the case where the reader has elected to clone into pDONR221 or pDONR223 as a first step, the resulting entry clone, which is identified by screening for insertions and then Sanger sequenced, is recombined with a suitable destination allelic exchange vector (Table 1) in a second recombination step (Box 4). Subsequently, the process of screening for plasmid insertions and Sanger sequencing is repeated. However, this two-step Gateway® cloning approach adds at least 4 days to the protocol (Box 4). Thus the most efficient way of Gateway® cloning is to insert the mutant allele directly into a donor allelic exchange vector (Table 1). Subsequently – and only if required – a destination allelic exchange vector, which can be created by LR Clonase®-mediated recombination between the destination and entry allelic exchange vectors, can be produced if there is a need for an alternate antibiotic marker for merodiploid selection (Box 4).
BOX 3. PREPARATION OF ELECTROCOMPETENT E. coli ● TIMING ~1 d.
-
Pick an isolated colony from an LB agar plate and grow in 3 ml of NSLB at 37 °C and 250 rpm to an OD600 of 0.50–0.60.
Δ CRITICAL STEP E. coli cells should be harvested in exponential-growth phase.
-
Transfer 2.0 ml of the culture to a 2.0 ml microcentrifuge tube. Spin at ≥10,000 × g for 2 min at 4 °C.
Δ CRITICAL STEP E. coli cells must be kept ice cold.
Remove the supernatant. Resuspend the cell pellet in 1.0 ml of ice cold 10% glycerol. Spin at ≥10,000 × g for 1 min at 4 °C.
Wash the cells once more with 1.0 ml ice cold 10% glycerol and resuspend the cell pellet in 20 µl of ice cold 10% glycerol.
Mix 1 µl of Clonase® reaction or 1 µl of ligation reaction (Step 13), or ~100–200 ng of purified, plasmid DNA (Step 31) with the electrocompetent cells. Transfer the cells to a pre-chilled 0.1-cm cuvette.
Introduce the DNA into the cells by electroporation (1.8 kV, 25 µF capacitance and 200 Ω resistance). After electroporation, immediately add 0.5 ml of SOC and transfer the cells to a sterile culture tube.
Incubate the cells for 1 h at 37 °C and 250 rpm.
Plate 50 µl and 450 µl aliquots on LB agar plates with the appropriate antibiotic selection and incubate for 20–24 h at 37 °C.
BOX 4. TRANSFERRING MUTANT ALLELES BETWEEN GATEWAY® VECTORS ● TIMING 4 d.
A mutant allele can be transferred from an entry vector (with attL sites) into a destination allelic exchange vector (with attR sites) in a single reaction. Such a transfer is crucial, for example, when using pDONR221 or pDONR223 as a first step in Gateway® cloning. Additionally, transferability between vectors encoding alternate antibiotic resistance cassettes is also highly useful when optimizing allelic exchange to engineer drug resistant P. aeruginosa isolates. The following protocol has been adapted from the Gateway® Technology Manual (Life Technologies™), and uses half of the recommended LR Clonase® Enzyme Mix. This offers considerable savings to the user.
-
Set up a half-volume reaction by adding the following components to a 0.2 ml PCR tube or a well of a PCR microplate:
0.50 µl of Gateway® entry vector (100 ng/µl)
0.50 µl of Gateway® destination vector (100 ng/µl)
Bring the volume to 4.0 µl with TE buffer, pH 8.0
-
Thaw the LR Clonase® II Enzyme Mix on ice for 2 min. Vortex the mix briefly twice (for 2 s each time). Add 1.0 µl of LR Clonase® II Enzyme Mix to the reaction. Carefully and thoroughly mix the reaction using a micropipette.
Δ CRITICAL STEP If the reaction mixture has dispersed in the PCR tube, spin in a minicentrifuge to collect the contents at the bottom of the tube.
Incubate the Gateway® cloning reaction for 1 h at room temperature.
Add 0.5 µl of the Proteinase K (provided with the LR Clonase II Enzyme mix) to the reaction mixture. Incubate for 10 min at 37 °C.
Transform E. coli DH5α with the recombination mixture as described in Step 14.
Screen for insertions in the destination allelic exchange vector and archive the sequence-verified construct by repeating Steps 15–30.
PCR identification and sequencing of insertions in allelic exchange vectors (Steps 20–35)
Colony PCR is used to quickly and directly screen E. coli colonies to identify those clones bearing allelic exchange vectors in which the mutant allele has been inserted. If any approach other than Gateway® cloning has been used, then this process may be guided by standard practices for blue-white selection19,20. By contrast, blue-white selection is not applicable to Gateway® cloning, and instead, this approach utilizes ccdB counter-selection to select for clones with inserts. CcdB is a gyrase-subunit A modifying enzyme that poisons DNA gyrase in E. coli cells and blocks bacterial growth87. Gateway® donor and destination vectors encode ccdB on the interior of their att sites, and therefore, these plasmids cannot be propagated in E. coli DH5α (these plasmids are maintained in strains, such as DB3.1 or ccdB Survival 2™ cells from Invitrogen, that have mutations in gyrA that confer resistance to CcdB). However, a successful recombination event causes the ccdB gene to be lost from Gateway® vectors, providing positive selection for recombinants in the DH5α strain. Thus >75% of the Gateway® clones typically have the insert at this stage without need for further selection. All pEX18- and pEX19-based vectors contain M13F and M13R primer binding sites, and thus all clones may be screened by PCR for inserts, and even sequenced, using the M13 universal primers (Supplementary Table 1). PCR occasionally introduces unwanted mutations into the synthesized alleles, and therefore, two independent clones that test positive for the insert by PCR are streaked for isolation. Next, plasmid DNA is purified from each clone and confirmed via Sanger sequencing. Gateway® vectors are difficult templates for Sanger sequencing, and therefore, it is crucial to inform your service provider about the nature of the template so that the sequencing protocol can be modified accordingly.
Introduction of the mutant allele into a P. aeruginosa recipient and merodiploid selection (Step 36)
Although allelic exchange vectors can be transformed into P. aeruginosa cells by electroporation37, we have found that the average number of merodiploids recovered from biparental mating is at least 10- to 100-fold greater: electroporation produces on the order of 101 to 102 merodiploids37, whereas biparental mating produces on the order of 102 to 104 merodiploids (see Anticipated Results). Therefore, this protocol uses conjugation to transfer suicide plasmids from an E. coli donor to the P. aeruginosa recipient and we direct the reader to a description of electroporation elsewhere37. A useful tool for biparental mating is a reusable syringe filter apparatus (see Equipment Setup), which presumably brings donor and recipient cells into close physical proximity to one another (Fig. 4). In our hands, this “mating” filter is a quick, reliable and alternative means of recovering merodiploids that is comparable in efficiency to carefully executed “puddle” mating (see Anticipated Results). We provide options for biparental “filter” and “puddle” mating techniques, and either may be used at the preference of the reader.
Figure 4.

How to build and use a mating filter apparatus.
Here we advocate the use of nutritional counter-selection as opposed to antibiotics to prevent the growth of the E. coli donor strain during the isolation of P. aeruginosa merodiploids after mating. Vogel-Bonner minimal medium (VBMM) agar, which contains 10 mM citrate as a carbon source, can be used to select against E. coli88. Because E. coli does not express a citrate transporter during aerobic culture89, it cannot use citrate as carbon source, and therefore, E. coli will not grow on VBMM in an aerated incubator. The recovery of P. aeruginosa merodiploids on VBMM agar may require up to 48 h of incubation at 37 °C. By contrast, this recovery process may take less than 24 h at 37 °C on nutrient rich lysogeny broth (LB) agar containing either irgasan (also called triclosan) and/or chloramphenicol. However, we have observed that the use of VBMM results in the recovery of ≥10-fold more merodiploids than LB agar with either irgasan or chloramphenicol-based counter-selection (Supplementary Fig.1). This discrepancy in recovery rate is not understood. However, because this phenomenon occasionally leads to protocol failure (Supplementary Fig.1), we recommend the use of VBMM agar for the isolation of merodiploids. Alternatively, we also recommend the use of auxotrophic E. coli donor strains, such as ST1890 or RHO366, which require the addition of 5-aminolevulinic acid (ALA) or diaminopimelic acid (DAPA) to the LB agar medium, respectively, in order to grow. In the case of this latter substitution, biparental mating with ST18 or RHO3 is carried out on LB agar containing ALA or DAPA, respectively. Subsequently, merodiploids can be selected directly on LB agar containing only antibiotic selection for the P. aeruginosa recipient.
Counter-selection, PCR identification and sequencing of mutations in P. aeruginosa cells (Steps 37–49)
The final stage of allelic exchange is to select against merodiploids and to identify the desired mutants. This is accomplished by streaking isolated merodiploids on no salt lysogeny broth (LB) agar that contains 15% (w/v) sucrose. In the case of large deletions or insertions, single colonies isolated on sucrose agar are screened for the desired mutation by PCR using “Seq-F” and “Seq-R” primers (Fig. 3). These individual colonies are spotted onto selective media. PCR products, which correspond to the size of the mutant allele and that were generated from sucrose resistant and antibiotic sensitive colonies, are subsequently sent for Sanger sequencing to confirm the mutation. In the case of small deletions and insertion mutations as well as SNPs, PCR products from 8–16 of the sucrose resistant and antibiotic sensitive colonies are sent for Sanger sequencing. Subsequently, the desired mutant is identified by sequence analysis.
Procedural controls
In general, positive and negative controls for routine PCR, cloning reactions or transformations – such as those that contain control templates, no DNA or no enzyme – are non-informative for most steps, and moreover, may be redundant because all constructs and strains are sequence verified as part of this protocol. Also, positive controls involving the use of control plasmids to verify Gateway Clonase® reactions (as recommended by the manufacturer) may add considerable expense to the protocol while yielding no additional value. Thus, readers must exercise judgment with procedural controls, and we recommend applying these specifically when repeating failed steps (see TROUBLESHOOTING). However, users may find it highly beneficial to test selective media either when fresh batches are made, or by including appropriate antibiotic resistant and sensitive strains at key selection steps (for example, Step 39, see Supplementary Tutorial). We also recommend including a control with no template (i.e. boiled colony) when using PCR to identify newly generated strains with an unmarked mutation (Steps 38–45).
Comparison to Flp-dependent protocols for marked mutant generation
We compared the present protocol to an established method for generating marked mutations that requires subsequent excision of a gentamcin resisance marker (aacC1) by Flp-FRT recombination27. To begin, we used the present protocol to engineer precise, unmarked deletions at 25 gene loci encoding components of the P. aeruginosa type IV pilus (see Anticipated Results). Next, we utilized the allelic exchange vector pDONRPEX18Ap (Table 1, encodes β-lactamase) to engineer marked ΔpilC::aacC1, ΔpilR::aacC1 and ΔpilN::aacC1 mutant strains (see Fig. 5). For the sake of comparison, deletion alleles marked with aacC1 were created with homology regions identical to those used to generate ΔpilC, ΔpilR and ΔpilN alleles. Not surprisingly, merodiploids were recovered at a frequency equivalent to that of unmarked mutants (Fig. 6A). Similar to the present protocol (Fig. 6B), counter-selection on NSLB + 15% sucrose + 60 µg/ml Gm yielded double-crossover mutants at a frequency of <10−5. However, 100% of the carbenicillin-sensitive, gentamicin-resistant colonies contained the desired aacC1-marked mutation (compared with 46% for the present protocol, see Anticipated Results). While this is a beneficial aspect of generating marked mutations, subsequent excision of the aacC1 gene by established methods27 required an additional 6 days of work and resulted in a scarred mutation. By contrast, the “Flp-free” technique presented in this protocol is at an advantage because this last step is not required. This saved nearly a week of work – and resulted in mutations that were seamless.
Figure 5.

Systematic disruption of type IV pilus gene loci in P. aeruginosa PAO1. (A) Organization of T4P genes in the P. aeruginosa PAO1 chromosome. (B) Effect of gene deletion on twitching motility. A total of 25 precise, unscarred, unmarked mutations were constructed using the present protocol. By contrast, 3 marked mutations were generated using the established method of Choi and Schweizer27. This latter method requires an additional Flp-FRT recombination step to excise the aminoglycoside acetyltransferase (aacC1) antibiotic resistance cassette, leaving a scarred mutation. In both panels, blue represents a gene deletion that was loss-of-function, vermillion represents a gene deletion that had no effect on diameter of the twitching motility zone, and white represents a gene that was not subject to mutation. This genetic analysis produced motility phenotypes that were consistent with previous reports95, which includes no change in motility zone diameter for ΔfimT, ΔpilY2, ΔpilH and ΔchpB strains95,96. Because pilY2, pilH and chpB occur within operons, this analysis illustrates the precision of the allelic exchange technique. Moreover, the two methods of mutation construction gave similar results.
Figure 6.

Outcomes of merodiploid selection and sucrose counter-selection using the two-step allelic exchange protocol. (A) Numbers of merodiploids recovered after biparental mating (Step 36). Each datum point represents an independent mating with pEX18Gm, pDONRPEX18Gm, pDONRPEX18Ap or pEX18GmGW vectors, each containing one of 37 different mutant alleles. Data have been normalized to the length of the regions of homology to the recipient chromosome. Solid lines and error bars represent the mean number and standard deviation of P. aeruginosa merodiploids recovered after conjugation. *P ≤ 0.05 with Student’s t-test. Elevated incubation temperatures after conjugation (Step 36Ax) increase the number of merodiploids recovered in the subsequent procedure step (Step 36Axi). However, the lower temperature (30 °C) is used to minimize clonal expansion. There is no difference in the number of merodiploids recovered after use of the filter or puddle mating techniques (Step 36A versus 36B). Finally, similar numbers of merodiploids are generated by this protocol and established methods27 for generating marked mutations. (B) Frequency of sucrose-mediated counter-selection of double crossover mutants (~10−5 on no salt lysogeny broth [NSLB] + 15% sucrose) and counter-selection escape (<10−7, on NSLB +15% sucrose + 60 µg/ml gentamicin (Gm), not detectable in this assay). Selection frequency for double crossover mutants and counter-selection escape is shown for three independent experiments.
MATERIALS
REAGENTS
Agarose (161–3101, Bio-Rad)
Ampicillin (Ap) (A1593, Sigma-Aldrich)
Carbenicilin (Cb) (C9231, Sigma-Aldrich)
Chloramphenicol (Cm) (C0857, Sigma-Aldrich)
Kanamycin (Km) (60615, Sigma-Aldrich)
Gentamicin (Gm) (G1914, Sigma-Aldrich)
Spectinomycin (Sp) (S9007, Sigma-Aldrich)
Tetracycline (Tc) (T4062, Sigma-Aldrich)
Bacto-agar (214010, BD Difco)
Bacto-tryptone (211705, BD Difco)
Citric acid (A940-500, Fisher Scientific)
BP Clonase II Enzyme mix (11789-020, Life Technologies)
LR Clonase II Enzyme mix (11791-020, Life Technologies)
Chemically competent or electrocompetent E. coli cells:
DNA ladder, 100 bp (N3231, New England Biolabs)
DNA ladder, 1 kb (N3232, New England Biolabs)
dNTP mixture, 10 mM each (N0447S, New England Biolabs)
Glycerol (G5516, Sigma-Aldrich)
Gel loading dye (6 ×) (New England Biolabs)
GoTaq Flexi DNA polymerase (PR-M8291, Promega)
Guanosine (G6752, Sigma-Aldrich)
Genomic DNA isolation kit (69504, Qiagen DNeasy Blood & Tissue Kit)
Gel extraction kit to purify DNA from agarose gels (22021, Qiagen QIAEX II Gel Extraction Kit; or 28704, Qiagen QIAquick Gel Extraction Kit)
Plasmid DNA isolation kit (27104, Qiagen QIAprep Spin Miniprep kit)
PCR clean-up kit (28104, Qiagen QIAquick PCR Purification Kit)
Noble agar (214230, BD Difco)
Nucleic acid gel stain (21141, Intron Biotechnology RedSafe Nucleic Acid Staining Solution)
Phosphate buffered saline (PBS), sterile (E703-1L, Amresco)
Phusion High-Fidelity DNA polymerase with proofreading ability (F-530L, Thermo Scientific)
Pseudomonas Isolation Agar (PIA) (R454392, Fisher Scientific)
Restriction enzymes (as required, New England Biolabs)
SOC media (B9020S, New England Biolabs)
Sucrose, molecular biology grade (J65148, Alfa Aesar)
Tris-EDTA (TE) buffer, pH 8.0, nuclease free (AM9849, Life Technologies)
Tris-HCl buffer, pH 8.5 (19086, Qiagen Buffer EB)
Tris-acetate-EDTA (TAE), 10 × solution (BP1335-4, Fisher Scientific)
Water, PCR grade, nuclease-free (10977-015, Life Technologies)
Water, Milli-Q (or other “ultrapure” water source)
Yeast extract (211929, BD BBL)
K2HPO4 (P288–500, Fisher Scientific)
NaCl (BP358-212, Fisher Scientific)
NaNH4HPO4•4H2O (S218–500, Fisher Scientific)
MgSO4•7H20 (M63–500, Fisher Scientific)
Primers (Integrated DNA Technologies): oligomers required for constructing the mutant allele and detecting the mutation in the Pseudomonas chromosome (Designed in Steps 1–3, see Supplementary Table 1 for examples), M13F(−21) primer (5’-GTAAAACGACGGCCAG-3’), and M13R primer (5’-CAGGAAACAGCTATGAC-3’)
Genomic DNA (gDNA) from the recipient P. aeruginosa strain
Purified allelic exchange vector (see Table 1)
Competent E. coli DH5α cells (T3009, Zymo Research Mix & Go Competent Cells)
Competent E. coli S17.1, S17.1 (λ pir+), SM10, SM10 (λ pir+), ST18 or RHO3 or another suitable donor strain that has been prepared by standard protocols73
EQUIPMENT
Gel electrophoresis apparatus and power supply (Bio-Rad)
Electroporator (165–2100, MicroPulser, Bio-Rad)
Electroporation cuvettes with 0.1 cm gap width (40–100, Genesee Scientific)
Standard laboratory incubators (11 690 638D, Isotemp, Fisher Scientific)
Incubator shaker with culture tube rack (model I24, New Brunswick Scientific)
Inoculating loops, 10 µl, sterile, disposable (12000-810, VWR)
Insulated ice bucket (188482001, Bel-Art)
Lazy “L” cell spreaders (89042-018, VWR)
Filter holder (Swinnex, SX00 025 00, Millipore)
Gel documentation system (Alphaimager Mini System, Protein Simple)
Magnetic hotplate stirrer (97042-674, VWR)
Membrane Filters, PVDF, 0.45 µm (Durapore HVLP 025 00, Millipore)
Microcentrifuge (75002436, Sorvall Legend Micro 21, Thermo Scientific)
Microcentrifuge tubes - 1.5 ml and 2.0 ml (MT-0150-BCS and MT-0200-BCS, Biotix)
Microcentrifuge tubes, screw-capped, 2.0 ml (72.694.006, Sarstedt)
Micropipettes - P2, P20, P200, and P1000 (L-STARTXLS+, Rainin)
Micropipette tips, sterile (Rainin)
Multichannel micropipette – P10 (L8–10XLS+, Rainin)
PCR microplates and 8-tube strips (Bio-Rad)
Petri dishes (100 mm x 15 mm) (SB93-03, Fisher Scientific)
Polyester swabs, sterile (25–806 1WD, Puritan)
Glass culture tubes, sterile (47729-580, VWR)
Polypropylene caps for culture tubes (16199-007, VWR)
Thermal cycler, gradient, and accessories for PCR (T1000 Touch, Bio-Rad)
Ultra-low temperature freezer (U700, New Brunswick)
UV spectrophotometer (ND-2000, NanoDrop)
UV transilluminator (95–0449-01, UVP)
Syringe, 10 ml, with Luer lock (148232A, Fisher Scientific)
Falcon tube, 50 ml (14–959-49A, Fisher Scientific)
Dry block heater with block for glass culture tubes (12621-084, VWR)
REAGENT SETUP
Δ CRITICAL Unless noted otherwise, all buffers and sterile broth media may be prepared ahead of time stored for at least 6 months at room temperature. Antibiotic stock agents should be added to broth media immediately prior to use. Petri plates with solidified agar media prepared with or without antibiotics may be wrapped in plastic sleeves and stored upside down at 4 °C for at least 4 months. Agar media containing Tc must be wrapped with aluminum foil to protect it from light during storage.
Antibiotics
Prepare stock agents at 100 × (Tc) or 1000 × concentration (Table 2) in the appropriate solvent. Sterilize water soluble antibiotics by filtration. Divide all stock solutions into 1.0 ml aliquots and store at −20 °C until needed. Δ CRITICAL Antibiotic concentrations must be normalized for activity, which varies from lot-to-lot. Activity, which is determined by bioassay by the manufacturer, is usually written on the label or on the certificate of analysis for the lot. Note that Cm and Tc are light sensitive and must be stored in the dark.
TABLE 2.
Antibiotics and concentrations (µg ml−1) used in media for selection of the specified bacteria
| Antibiotic | Abbreviation | Solubility | E. coli | P. aeruginosa |
|---|---|---|---|---|
| Ampicillin | Ap | Water | 100 | --- |
| Carbenicilin | Cb | Water | 50 | 300 |
| Chloramphenicol | Cm | Ethanol | 10 | --- |
| Kanamycin | Km | Water | 25 | --- |
| Gentamicin | Gm | Water | 10 | 60 |
| Spectinomycin | Sp | Water | 50 | --- |
| Tetracycline | Tc | Ethanol | 10 | 100 |
Lysogeny broth (LB) media
10 g of Bacto-tryptone, 5.0 g of yeast extract and 5.0 g of NaCl, bring up to 1.0 l with ultrapure water. Adjust to pH 7.2. Sterilize by autoclaving at 121 °C. Allow to cool and add antibiotics as required.
LB agar
10 g of Bacto-tryptone, 5.0 g of yeast extract, 5.0 g of NaCl and 15.0 g Bacto-agar, bring up to 1.0 l with ultrapure water. Adjust to pH 7.2. Sterilize by autoclaving at 121 °C. Allow agar to cool to 60 °C. Add antibiotics as required. Pour ~20–25 ml aliquots into Petri dishes and allow agar to solidify.
Sucrose (50% w/v) solution
500 g sucrose and bring to 1.0 1 with ultrapure H2O. Sterilize by filtration.
Sucrose (300 mM) solution
102.7 g sucrose and bring to 1.0 l with ultrapure H2O. Sterilize by filtration.
No salt LB (NSLB)
10 g of Bacto-tryptone and 5.0 g of yeast extract, bring to 1.0 l with ultrapure water. Adjust to pH 7.2. Sterilize by autoclaving at 121 °C.
NSLB agar with 15% sucrose
10 g of Bacto-tryptone, 5.0 g of yeast extract and 15.0 g of Bacto-agar, bring to 700 ml with ultrapure water. Adjust to pH 7.2. Sterilize by autoclaving at 121 °C. Allow agar to cool to 60 °C, then add 300 ml sucrose (50% w/v) solution. Pour ~20–25 ml aliquots into Petri dishes and allow agar to solidify.
TAE + G buffer
Add 100 ml of 10 × TAE to 900 ml of ultrapure water. Add 0.28 g of guanosine and dissolve using a magnetic stirrer. Guanosine (1 mM) is added to shield DNA from UV light during preparative gel documentation and DNA excision. This increases cloning efficiency91.
Vogel-Bonner minimal media, 10 × stock (10 × VBMM)
2.0 g MgSO4•7H2O, 20 g citric acid, 100 g K2HPO4, 35 g NaNH4HPO4•4H2O, bring to 1.0 l with ultrapure water. Adjust to pH 7.0. Sterilize by filtration.Δ CRITICAL While 10 × VBMM may be stored at room temperature for at least three months, a fresh batch should be prepared if crystalline precipitates appear in the stock.
VBMM agar
Add 15 g of Noble agar to 900 ml ultrapure water. Sterilize by autoclaving at 121 °C. Allow agar to cool to 60 °C. Add 100 ml of 10 × VBMM. Add antibiotics as required. Pour ~20–25 ml aliquots into Petri dishes and allow agar to solidify.
EQUIPMENT SETUP
Mating filter apparatus
Assemble the filter holder, O-ring and membrane filter as illustrated in Fig. 4. Wrap assembled filters in aluminum foil. Sterilize by autoclaving at 121 °C with an additional drying cycle. Δ CRITICAL Membrane filters must have a pore size of 0.45 µm, because filters with smaller pores sizes will clog and filters with larger pore sizes will not retain bacterial cells. Additionally, many filters are sold in packages with paper inserts that separate the individual PVDF membranes. It is crucial to ensure that these paper inserts are removed and not inadvertently inserted into the filter holder.
PROCEDURE
Vector selection and primer design ● TIMING 1 h on 1 d
-
Precisely design your desired mutant allele, allelic exchange vector and required primers in silico (see Experimental Design). This is greatly facilitated by the use of DNA sequence analysis software such as Geneious (Biomatters), Vector NTI (Life Technologies) or ApE (http://biologylabs.utah.edu/jorgensen/wayned/ape/). Primer sequences may be generated using PCR primer design algorithms included with the sequence analysis software, or alternatively, with a text editor and online tools by following a simple set of guidelines (Box 2). The genome sequences of the laboratory P. aeruginosa strains are available from the Pseudomonas genome database (http://www.pseudomonas.com).
Δ CRITICAL STEP Be sure to include the 5’ primer extensions that encode restriction sites, attachment (att) sites, or regions of complementarity that are used for the chosen method of cloning (Box 2).
Order or synthesize the required primers (ssDNA oligomers) as salt-free, but otherwise unpurified.
Dissolve lyophilized primer DNA in TE buffer, pH 8.0 to produce a primer stock solution with an ssDNA concentration of 100 µM (i.e. add 10 µl TE buffer per nanomole of DNA).
PCR to build mutant alleles in vitro. ●; TIMING 6 h on 1 d
-
4Set up a 5- or 10-reaction high-fidelity PCR master mix in order to amplify the target DNA fragments, as follows:
Component Volume (µl) 1 reaction 5 reactions 10 reactions HF buffer (5 ×) 4.00 20.0 40.0 dNTPs (10 mM) 0.40 2.0 4.0 DMSO, 100% 0.60 3.0 6.0 F primer (100 µM) 0.05 0.25 0.50 R primer (100 µM) 0.05 0.25 0.50 Template DNA* x 5x 10x Ultrapure water 14.7 – x 73.5 – 5x 147 – 10x Phusion® DNA polymerase 0.20 1.0 2.0 Total volume 20.00 100.0 200.0 *Add a volume that provides 10–20 ng gDNA or 5–10 ng of plasmid DNA per reaction.Δ CRITICAL STEP High-fidelity polymerases have a proofreading (3’→5’) exonuclease activity that can degrade PCR primers. If a delay is anticipated between the addition of the polymerase and the start of the thermal cycler, then the PCR reactions should be kept on ice (4 °C).
-
5
Aliquot 20.0 µl of the master mix into PCR tubes or wells of a PCR plate.
Δ CRITICAL STEP Arrange aliquots of each master mix in the PCR plate so that they are oriented parallel to the temperature gradient of the thermal cycler.
-
6Move the PCR reactions to the thermal cycler and run the PCR program as follows (based on optimal conditions for Phusion® DNA Polymerase):
Cycle Description Temperature (°C) Time (s) 1 Initial denature 98 30 2 – 35 Denature 98 10 Anneal Gradient (55 – 72) 15 Extend 72 20 per 1 kb of the amplicon 36 Final extend 72 300 37 Hold 10 infinite Δ CRITICAL STEP There is no need to hold the temperature below 10 °C for the routine cloning of DNA. Holding at 10 °C minimizes wear on Peltier heating systems, which are in most thermal cyclers, and can significantly extend the lifespan of the instrument.
■ PAUSE POINT PCR reactions may be stored at 4 °C for at least 1 week and at −20 °C for at least 1 month.
-
7
Pour a 1.0% TAE + G gel and add the nucleic acid gel stain directly to the gel. Load 10 µl aliquots of the PCR products with 6 × gel loading dye into the wells of the gel along with an appropriately sized DNA ladder. Run the gel and then image it using an ultraviolet (UV) transilluminator and digital camera.
? TROUBLESHOOTING
-
8
Using a new razor blade, cut out DNA bands of the appropriate size and place each slice in a separate 2.0 ml microcentrifuge tube.
■ PAUSE POINT Gel slices containing DNA may be stored at 4 °C for up to 1 week.
-
9
Purify the DNA using a gel purification kit. Elute the DNA into Tris-HCl buffer using buffer volumes recommended by the manufacturer of the gel purification kit.
Δ CRITICAL STEP Ensure that the silica-membrane column (eg. QIAquick Gel Extraction Kit) or silica particles (eg. QIAEX II Gel Extraction Kit) are completely dry before eluting the DNA into Tris-HCl buffer.
-
10
Determine the concentration and purity of the eluted DNA by measuring the A260 and A280 on a UV spectrophotometer.
■ PAUSE POINT Purified DNA may be stored short-term (<1 week) at 4°C or long-term (>1 week) at −20 °C.
-
11Set up a 5- or 10- reaction high-fidelity SOE-PCR master mix in order to assemble and amplify the desired mutant allele as follows:
Component Volume (µl) 1 reaction 5 reactions 10 reactions HF buffer (5 ×) 4.00 20.0 40.0 dNTPs (10 mM) 0.40 2.0 4.0 DMSO, 100% 0.60 3.0 6.0 F primer (100 µM) 0.05 0.25 0.50 R primer (100 µM) 0.05 0.25 0.50 DNA fragments* x + y + (z) 5[x + y + (z)] 10[x + y + (z)] Ultrapure water 14.7 – [x + y + (z)] 73.5 – 5[x + y + (z)] 147 – 10[x + y + (z)] Phusion® DNA polymerase 0.20 1.0 2.0 Total volume 20.00 100.0 200.0 *Add a volume that provides 2–5 ng of DNA fragment per reaction. These master mixes can be used for two (x, y) or three (x, y, z) fragment assemblies.Δ CRITICAL STEP If a delay is anticipated between the addition of the polymerase and the start of the thermal cycler, then the PCR reactions should be kept on ice (4 °C).
-
12
Repeat Steps 5 to 10 to amplify, image, purify and quantify the SOE-PCR product, increasing the extension time of the PCR reaction (as described in Step 6) to allow for the amplification of a longer amplicon.
Cloning the mutant allele into an allelic exchange vector. ● TIMING 3 h on 1 d
-
13
To insert the assembled mutant allele from Step 12 into the chosen allelic exchange vector using Gateway® technology, set up a half-volume reaction by adding the following components to a 0.2 ml PCR tube or a well of a PCR microplate: 0.50 µl of Gateway® donor vector (100 ng/µl), a volume of purified SOE-PCR product corresponding to 10–25 ng of DNA and bring the volume to 4.0 µl with TE buffer, pH 8.0.
Δ CRITICAL STEP The PCR product can alternatively be cloned into the allelic exchange vector using another standard technique (as described in Experimental Design).
-
14
Thaw the BP Clonase® II Enzyme Mix on ice for 2 min. Vortex the mix briefly twice (for 2 s each time). Add 1.0 µl of BP Clonase® II Enzyme Mix to the reaction. Carefully and thoroughly mix the reaction using a micropipette.
Δ CRITICAL STEP If the reaction mixture has dispersed in the PCR tube, spin in a minicentrifuge to collect the contents at the bottom of the tube.
-
15.
Incubate the Gateway® cloning reaction for 1–3 h at room temperature. For increased efficiency, this reaction may be left overnight on the bench top.
-
16.
Add 0.5 µl of the Proteinase K (provided with the BP Clonase II Enzyme mix) to the reaction mixture. Incubate for 10 min at 37 °C.
-
17.
To transform chemically competent E. coli DH5α (eg. Zymo5α Mix & Go Competent Cells; high transformation efficiency, i.e. >108 transformants per µg of pUC19 DNA) add 2.5 µl of the recombination, ligation or assembly mix to a 50 µl aliquot of thawed competent cells, Incubate on ice for 5–45 min.
Δ CRITICAL STEP Note that there is no need to desalt the Gateway®, ligation or assembly reaction before proceeding.
Δ CRITICAL STEP Electrocompetent E. coli DH5 cells can be used instead as described in Box 3.
-
18.
Add 0.5 ml of SOC and transfer the transformed cells to a sterile culture tube. Incubate the cells for 1 h at 37 °C and 250 r.p.m..
CRITICAL STEP: Note that it is not necessary to outgrow E. coli cells that have been transformed with a vector encoding a β-lactamase, which confers resistance to Ap or Cb.
-
19.
Plate 50 µl and 450 µl aliquots on LB agar with the appropriate antibiotic selection and incubate for 20–24 h at 37 °C.
? TROUBLESHOOTING
Screening for insertions in allelic exchange vectors and Sanger sequencing ● TIMING 7 h over 3 d
-
20.
Using a P20 micropipette, collect a tiny amount of a bacterial colony from the selective agar plate from Step 19 and suspend it in 50 µl of ultrapure water. Typically, 4 colonies are screened for each plasmid being constructed.
Δ CRITICAL STEP The bacterial suspension should be only faintly turbid. Using too much starting material can inhibit the downstream PCR reaction.
-
21.
Using a multichannel pipette, transfer 3.0 µl of each colony in suspension onto a selective agar plate. Let these “spot” plates dry and then incubate at 37 °C overnight. Note that each 3 µl droplet put onto the plate will grow into a colony that can be streaked out for isolation (as described in Step 28) once the clone with the desired insert has been identified by PCR (as detailed in Steps 22–27).
-
22.
Boil the remaining cells in suspension using a thermal cycler set at 100 °C for 5 min. When finished, add 103 µl of ultrapure water to each well in order to dilute the plasmid DNA.
Δ CRITICAL STEP If the template concentration is too high, DNA polymerase can be inhibited.
-
23.Create a colony PCR master mix using GoTaq® Flexi DNA polymerase as follows:
Component Volume (µl) 1 reaction 5 reactions 10 reactions Green GoTaq buffer (5×) 4.00 20.0 40.0 MgCl2 (25mM) 2.00 10.0 20.0 dNTPs (10 mM) 0.40 2.00 4.00 DMSO, 100% 0.60 3.00 6.00 M13F primer (100 µM) 0.05 0.25 0.50 M13R primer (100 µM) 0.05 0.25 0.50 Ultrapure water 11.8 59.0 118 GoTaq® polymerase 0.10 0.50 1.00 Total volume 19.00 95.0 190.0 -
24.
Aliquot 19.0 µl of the master mix into each PCR tube or well of a PCR microplate.
-
25.
Add 1.0 µl of a boiled and diluted bacterial colony to each PCR reaction from Step 22.
-
26.Move the PCR reactions to the thermal cycler and use the following PCR program (optimized for GoTaq® Flexi DNA polymerase):
Cycle Description Temperature (°C) Time (s) 1 Initial denature 95 180 2 – 30 Denature 95 10 Anneal* Tm 30 Extend 72 60 per 1 kb of the amplicon 31 Final extend 72 300 32 Hold 10 infinite *M13F(−21) and M13R primers have a Tm of 47 °C; for other sequencing primers use the Tm calculated with Oligocalc (Box 2).Δ CRITICAL STEP To time the PCR reactions with the growth of bacterial colonies prepared in Step 21, it is possible to run the thermal cycler overnight.
-
27.
Pour a 1.0% TAE + G gel. Load 10 µl aliquots of the PCR products with 6 × gel loading dye into the wells of the gel along with an appropriately sized DNA ladder. Run the gel and then image it using an ultraviolet (UV) transilluminator and digital camera.
? TROUBLESHOOTING
-
28.
Use the results of colony PCR to identify clones with the desired insert. This identification is based on the size of the band in the gel. Clones containing the desired insert will produce a PCR product that is equal to the size of the SOE-PCR product plus the distance between M13F and M13R priming sites (or the target sites of alternative sequencing primers used in Step 26) and the cloned insert (see Supplementary Tutorial). Based on this identification, pick a colony from the corresponding spot plate prepared in Step 21. Streak this colony out on LB agar containing appropriate antibiotic selection. Pick and streak out a second colony from the same plate using the same criteria. Incubate these plates at 37 °C overnight.
-
29.
For each streak plate grown in the previous step, transfer 1.0 ml of sterile PBS into a 2.0 ml microcentrifuge tube. Use a sterile polyester-tipped swab to collect a 3–4 cm2 lawn of bacteria from the surface of each agar streak plate. Suspend the collected bacteria in sterile PBS.
Δ CRITICAL STEP Retain the streak plates at 4 °C for up to 1 week.
-
30.
Collect the cells by centrifugation at ≥10,000 × g for 1 minute. Discard the supernatant.
-
31.
Use a plasmid DNA isolation kit (eg. QIAprep Spin Miniprep kit) to isolate and purify the plasmids, following the manufacturer’s instructions. Elute the plasmids in 50 µl of Tris-HCl, pH 8.5.
-
32.
Determine the concentration and purity of the eluted DNA by measuring the A260 and A280 on a UV spectrophotometer. The plasmid preps should yield ~5–20 µg of DNA each with an A260/A280 ratio of 1.8 to 2.0.
-
33.
Send the purified plasmids for Sanger sequencing using a preferred outsource. M13F(−21) and M13R universal primers, which are provided free of charge by many Sanger sequencing providers, may be used for sequencing the mutant allele in any pEX18-based plasmid (Table 1), or in pDONR221- or pDONR223-based plasmids.
Δ CRITICAL STEP Gateway® vectors are often considered difficult templates for the purposes of Sanger sequencing; however, adjustments can be made to the sequencing protocol to accommodate these vectors if the sequencing provider is informed at the time of sample submission.
-
34.
Analyze the sequence of the plasmid using BLASTN92 (http://www.pseudomonas.com) or CLUSTALW93 (http://www.genome.jp/tools/clustalw/) tools. Choose one plasmid that has a 100% match to the predicted sequence for use as the allelic exchange vector.
? TROUBLESHOOTING
-
35.
Prepare a glycerol stock of the E. coli DH5α strain harboring the plasmid of interest. To do this, add 1.0 ml of LB and 0.5 ml of sterile glycerol (50% w/v) to a screw-capped 2.0 ml tube. Next, use a polyester-tipped swab to collect a 3–4 cm2 lawn of bacteria from the surface of the agar streak plate (retained at Step 29) corresponding to the plasmid that has a 100% match to the desired sequence. Suspend the bacteria in the LB-glycerol solution. Freeze the glycerol stock at −80 °C.
■ PAUSE POINT E. coli DH5α cell lines may be stored indefinitely at −80 °C.
Introduction of the mutant allele into a P. aeruginosa recipient and merodiploid selection ● TIMING 9 h over 4 d
-
36.
Transform a donor E. coli strain, and then use conjugation to introduce the allelic exchange vector into P. aeruginosa using either a mating filter apparatus (option A) or standard ‘puddle mating’ (option B):.
-
Mating filter apparatus
Transform the allelic exchange vector from Step 34 into a suitable donor E. coli strain, such as S17.1 (λ pir+) or SM10 (λ pir+), using a standard chemical transformation or electroporation protocol (Box 3). On the same day, streak out the recipient P. aeruginosa strain on LB agar. Grow overnight at 37 °C.
Pick a single colony of the transformed donor E. coli strain and streak out on LB agar with appropriate antibiotic selection. Incubate the agar plate overnight at 37 °C. On the same day, pick a single colony of the recipient P. aeruginosa strain and inoculate 3 ml LB. Grow overnight at 37 °C and 250 r.p.m.
-
Prepare a glycerol stock of the E. coli donor strain (see Step 35). Freeze the glycerol stock at −80 °C.
■ PAUSE POINT E. coli donor cell lines may be stored indefinitely at −80 °C. It is possible to resume this protocol at Step 36Ai. with a fresh streak plate of the E. coli donor taken from cryogenic storage.
-
Add 3.0 ml of fresh LB to the overnight culture of the recipient P. aeruginosa strain from Step 36Aii. Incubate at 42 °C for a minimum of 3 h or until the donor strain has reached an OD600 value of 0.5–0.6 (as detailed in next Step).
Δ CRITICAL STEP Incubation at 42 °C reduces some of the antibacterial effectors produced by P. aeruginosa and makes it more amenable to mating.
At the same time that the recipient strain is diluted, pick a single colony of the transformed donor E. coli strain from Step 36Aii and inoculate 3 ml LB. Grow at 37 °C and 250 r.p.m. to an OD600 of 0.5–0.6 (~3–4 h).
Pre-warm an LB agar plate to 30 °C.
-
Assemble a mating filter apparatus (Fig. 4). Pull the plunger out of a sterile 10 ml syringe. Join the 10 ml syringe to sterile filter holder.
Δ CRITICAL STEP Ensure that O-ring is in place and that the Luer lock joints and the filter chamber are sealed tightly.
Place the assembled apparatus over the open top of a 50 ml Falcon tube. Add 3.0 ml of the donor E. coli strain from Step 36Av and 1.0 ml of the recipient P. aeruginosa strain from Step 36Aiv to the syringe. Insert the plunger into the syringe and press the bacterial culture through the mating filter apparatus. The bacteria will collect in a thin layer on the top of the filter.
-
Remove the syringe and discard it in a biohazard waste container. Using aseptic technique, unscrew the top of the filter holder. Use flamed tweezers to transfer the filter to the surface of the pre-warmed LB agar plate.
Δ CRITICAL STEP Ensure that the bacteria on the surface of the filter are facing upwards. Do not sandwich the bacteria between the agar surface and the filter.
Incubate the LB agar plate with the mating filter overnight at 30 °C.
Use a sterile plastic inoculation loop to scrape the bacterial lawn off of the filter and suspend in 1.0 ml of sterile PBS. Mix the suspension by pipetting up and down with a P1000 micropipette. Plate 10, 100 µl and 500 µl aliquots on VBMM agar plates with the appropriate antibiotic selection and incubate for 48 h at 37 °C.
-
Puddle mating
Transform a suitable E. coli donor strain and grow up the E. coli donor and the P. aeruginosa recipient as described in Steps 36Ai to 36Avi.
Into each of two 2.0 ml microcentrifuge tubes add 1.5 ml of the E. coli donor strain and 0.5 ml of the P. aeruginosa recipient strain. Collect the cells by centrifugation at 10,000 × g for 5 min. Discard the supernatant. Resuspend each cell pellet in 50 µl LB and combine. Transfer this cell mixture onto the middle of the pre-warmed LB agar plate. Allow the “puddle” to dry on the agar surface. Incubate the LB agar plate overnight at 30 °C.
Using a sterile plastic inoculation loop to scrape the bacterial “puddle” off of the surface of LB agar plate, repeat Step 36Axi.
? TROUBLESHOOTING
-
Counter-selection, PCR identification and sequencing of mutations in P. aeruginosa cells ● TIMING 9 h over 6 d
-
37.
Pick an isolated single colony from the surface of one of the VBMM agar plates prepared in Step 36Axi (or Step 36Biii) with an inoculation loop. Use the inoculation loop to streak the colony onto NSLB + 15% sucrose agar. Incubate this plate for 36 to 48 h at 30 °C.
? TROUBLESHOOTING
-
38.
Using a P20 micropipette, collect a tiny amount of a bacterial colony from the NSLB + 15% sucrose agar plate and suspend it in 50 µl of ultrapure water. Typically, 8 colonies from each NSLB + 15% sucrose plate are screened for the desired mutation.
Δ CRITICAL STEP When in suspension, the bacteria should be only faintly turbid. Using too much starting material can inhibit the downstream PCR reaction.
-
39.
Using a multichannel pipette, transfer 3.0 µl of each colony in suspension onto 3 types of agar media: LB agar, Pseudomonas isolation agar (PIA) and LB agar containing the antibiotic used for merodipoloid selection. Let these plates dry and then incubate overnight at 37 °C.
Δ CRITICAL STEP Retain these plates at 4 °C. Note that spots on LB or PIA agar will grow into a colony that can be streaked out for isolation (in Step 49) once the clone with the desired mutation has been identified by PCR and/or Sanger sequencing (as described in Steps 40–48).
-
40.
Boil the remaining colonies in suspension using a thermocycler set at 100 °C for 5 min. When finished, add 109 µl of ultrapure water to each well in order to dilute the starting concentration of gDNA.
Δ CRITICAL STEP If the template concentration is too high, DNA polymerase can be inhibited.
-
41.Create a colony PCR master mix using GoTaq® Flexi DNA polymerase as follows:
Component Volume (µl) 1 reaction 5 reactions 10 reactions Green GoTaq buffer (5×) 6.0 30.0 60.0 MgCl2 (25mM) 3.0 15.0 30.0 dNTPs (10 mM) 0.6 3.0 6.0 DMSO, 100% 0.9 4.5 9.0 Seq-F primer (100 µM) 0.1 0.5 1.0 Seq-R primer (100 µM) 0.1 0.5 1.0 Ultrapure water 18.15 90.75 181.5 GoTaq® polymerase 0.15 0.75 1.5 Total volume 29.0 145.0 290.0 Δ CRITICAL STEP If different mutations are being screened in parallel, prepare multiple master mixes using the primer pair appropriate to each targeted mutation.
-
42.
Aliquot 29.0 µl of the master mix into each PCR tube or well of a PCR microplate.
-
43.
Add 1.0 µl of a boiled bacterial colony from Step 40 to each PCR reaction.
-
44.
Repeat Steps 26 and 27, retaining the remainder of the PCR reactions at 4 °C for sequencing in Steps 45 and 46.
Δ CRITICAL STEP When choosing an extension time, use a time suitable to amplify the largest expected target.
Δ CRITICAL STEP To time the PCR reactions with the growth of bacterial colonies prepared in Step 39, it is possible to run the thermal cycler overnight.
-
45.
Identify the unmarked mutants from the PCR reactions using option A if the goal is to produce a large (≥25 bp) deletion or insertion, or by sequencing using option B if the goal is to produce a SNP or short (<25 bp) deletion or insertion mutation.
Δ CRITICAL STEP Merodiploids frequently produce multiple bands in colony PCR reactions and will be positive for growth on LB agar containing antibiotic selection. Any contaminating E. coli from the conjugation will grow on LB agar, but will not grow on PIA.
-
Identifying mutants with large (≥25 bp) deletion or insertion mutations
Identify the PCR reactions corresponding to the desired mutants from the gel images taken in Step 44. Double-cross over mutants will have one of two potential PCR products: a band corresponding to a wild type allele, or another band corresponding to the mutant allele. Additionally, these mutants will have grown on LB and PIA agar, and will not have grown on LB agar containing antibiotic selection.
Purify the PCR products corresponding to two of the engineered mutants selected at the previous step. If the PCR reaction produced a single band on the gel (corresponding to the target region of the genome), clean up the reaction using a PCR clean-up kit (eg. QIAquick PCR Purification Kit), following manufacturer’s instructions. Alternatively, if the PCR reaction produced multiple bands on the gel, use a razor blade to cut out the DNA band of the expected size. Purify the DNA using a gel extraction kit (eg. QIAEX II Gel Extraction Kit) and elute the DNA in ultrapure water, following manufacturer’s instructions.
Determine the concentration and purity of the eluted DNA by measuring the A260 and A280 on a UV spectrophotometer.
-
Identifying mutants with SNPs or small (<25 bp) insertions and deletions.
As it may not be possible to identify SNPs or small insertion or deletion mutations from the gel image (see Experimental Design), Sanger sequencing may be used for this purpose. Purify the PCR products from Step 44 for all of the colonies arrayed in Step 39 that are sucrose resistant and antibiotic sensitive, as described in Step 45Aii-iii.
? TROUBLESHOOTING
-
-
46
Send the purified PCR products from Step 45Aii and 45Bi for Sanger sequencing. Use the primers “Seq-F” and “Seq-R” in each of two different sequencing reactions in order to sequence each of the PCR products.
-
47.
Analyze the sequence of the PCR products using the BLASTN algorithm against the target P. aeruginosa genome (http://www.pseudomonas.com). If the goal was to produce a deletion mutation, the alignment will produce a match to two regions in the bacterial chromosome, one upstream and the other downstream of mutated region of DNA. For an insertion mutation, the BLASTN alignment will produce a match to one or two regions of the bacterial chromosome, depending on both the insertion and Sanger sequencing read length. For a SNP or small insertion or deletion mutation, an alignment for one region with a mismatch or gap will be reported by the algorithm.
? TROUBLESHOOTING
-
48.
For every precisely engineered mutant, pick two colonies from Step 39 corresponding to the desired unmarked mutation. Streak each of these out on LB agar and incubate these plates at 37 °C overnight.
-
49.
Prepare a glycerol stock for each of the sequence-verified P. aeruginosa mutants isolated in Step 48. Freeze the glycerol stock at −80 °C. Glycerol stocks may be stored indefinitely at −80 °C.
TIMING
Steps 1–3, 1 h, day 1, vector selection and primer design
Steps 4–12, 6 h, day 2, PCR to build mutant alleles in vitro
Steps 13–19, 3 h, day 3, cloning the mutant allele into an allelic exchange vector
Steps 20–28, 4 h, day 4, PCR to screen for insertions in allelic exchange vectors
Steps 29–33, 2 h, day 5, purification of allelic exchange vectors and Sanger sequencing
Steps 34–35,1 h, day 6, sequence analysis and archiving
Steps 36A or 36B, 2 h, day 6, transform donor E. coli strain with allelic exchange vector; 1 h, day 7, grow donor and recipient strains; 30 min hands-on time over 3–6 h, day 8, biparental mating; 10 min, day 9, merodiploid selection
Step 37, 5 min, day 11, sucrose counter-selection
Steps 38–45, 4 h, day 13, PCR to identify mutants and growth on selective media
Steps 46, 2 h, day 14, PCR product sequencing to verify mutations
Steps 47–48, 2 h, day 15, sequence analysis and strain selection
Step 49, 30 min, day 16, archiving
Box 3, 15 min, 1 d, electroporation of E. coli (as required)
Box 4, (optional, occurs on 4 additional days), 2 h, day 1, recombine entry and destination vector; 4 h, day 2, PCR to screen for insertions in new allelic exchange vector; 1.5h, day 3, purification of new allelic exchange vector and Sanger sequencing; 1 h, day 4, sequence analysis and archiving
TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
TABLE 3.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 7, 27 | No bands were visible on gel – not even the DNA ladder |
No nucleic acid gel stain was added |
Soak gel in appropriate nucleic acid gel stain |
| No PCR products were produced |
Primers | Double-check primer design and, if required, resynthesize primers |
|
| Too much template | Dilute template or boiled colonies an additional 5-fold |
||
| Difficult template | Increase denaturation times; Use an alternate buffer or polymerase; Redesign primers |
||
| 12 | No SOE-PCR product | Error in primer design | Double-check primers, especially regions of reverse complementarity |
| 19 | A low number of colonies were recovered |
Inhibited Clonase® reaction | Ensure that residual ethanol has evaporated before eluting SOE- PCR product |
| Low transformation efficiency of E. coli cells |
Prepare a fresh batch of competent cells; Buy competent cells from a preferred supplier |
||
| 34 | Cloned alleles have point mutations |
Random errors, PCR “jackpot,” Bona fide SNP in genome of target strain |
Use a high-fidelity polymerase for allele synthesis; Sequence additional clones; Repeat cloning process |
| Cloned alleles have a point mutation in a region targeted by primer |
Error in design or synthesis of primers |
Check primer design; Resynthesize primers |
|
| 36A or B | Cannot transform donor E coli cell line with allelic exchange vector |
Restriction incompatibility with cloning strain used at Step 17 |
Use E. coli DH5α for cloning |
| No merodiploids were isolated at selection. |
Plasmid integration is low frequency |
Repeat electroporation or biparental mating |
|
| Use of incorrect selective media | Double-check antibiotics used in selective medium |
||
| Target gene may be essential | No simple solution using the present protocol |
||
| Lawn of bacteria on selective plates (i.e. selection escape) |
Selective media was incorrectly prepared or degraded; recipient cell is antibiotic resistant; inoculum effect |
Double check media preparation and antibiotic concentrations; choose a different antibiotic marker; dilute cultures an additional 10- to 100-fold before plating merodiploids on selective agar |
|
| 37 | Lawn of bacteria on NSLB + 15% sucrose agar (i.e. counter-selection escape) |
Counter-selection escape; merodiploid has acquired inactivating mutation in sacB; allelic exchange vector has inactivating mutation in sacB; counter-selective media was incorrectly prepared |
Repeat electroporation or conjugation with corrected merodiploid selection; repeat counter-selection; double check sequence of sacB in allelic exchange vector; double-check composition of counter-selective media |
| 45, 47 | Multiple bands on gel were produced by PCR reaction |
Non-specific DNA amplification |
Modify PCR protocol to increase its stringency; if multiple PCR products cannot be separated effectively by electrophoresis, use a nested sequencing primer for the mutant allele; redesign “Seq-F” and “Seq-R” primers |
| No large insertion or deletion mutation was detected |
Small sample size | Screen an additional 8 colonies isolated from NSLB + 15% sucrose agar |
|
| Sequencing failed to identify a mutant with a SNP or short deletion or insertion mutation |
Small sample size | Screen an additional 8 colonies isolated from NSLB + 15% sucrose agar |
|
| No insertion or deletion mutation is detected after protocol is repeated multiple times |
Target gene may be essential | No simple solution using the present protocol |
ANTICIPATED RESULTS
As a demonstration, we have used the present protocol to generate a set of 25 mutants with deletions in genes encoding structural and regulatory components of the type IV pilus (T4P). The P. aeruginosa T4P is well-studied for its role in twitching motility, a form of bacterial translocation across moist surfaces. Twitching motility is easy to quantify by staining bacteria and measuring the motility zone at the plastic-agar interface in standard Petri plates94,95. Many genes required for T4P biosynthesis and function are arranged in operons (Fig. 5A), yet inactivating mutations in several of these genes do not abolish twitching motility. This includes, for example, fimT, pilY2, pilH and chpB95,96 (Fig. 5B). Genes of the T4P range in size from 357 bp (pilZ) to 7419 bp (chpA). Thus, systematic disruption of T4P genes illustrates the success of this protocol at making precise deletions (both large and small) at a variety of genome loci. Also, this demonstration has provided benchmark data for anticipated outcomes that are discussed below, but note that Anticipated Results for primer design, PCR and cloning steps are presented in the Supplementary Tutorial, and Supplementary Figs. 2 and 3. Lastly, all primers, strains, and plasmids used in this demonstration and in the Supplementary Tutorial are summarized in Supplementary Tables 1, 2 and 3, respectively.
Merodiploid selection (Step 36)
Using the present protocol, approximately 5.0 × 102 merodiploids were obtained per biparental mating per 1 kb of vector homology to the recipient chromosome (Step 36, Fig. 6A). Similar numbers of merodiploids were produced using filter and puddle mating techniques (Step 36A vs. Step 36B, Fig. 6A). Novice users often point out that increasing incubation temperature from 30 to 37 °C during biparental mating increases the yield of merodiploids (Step 36Aix and Step 36Bii, Fig. 6A). However, merodiploids are routinely cultured at 30 °C to minimize clonal expansion. In principle, this decreases the number of bacterial generations and likelihood of random secondary site mutations in mutant strains. We have noted no difference in the number of merodiploids recovered from biparental mating of P. aeruginosa with either E. coli S17.1 or E. coli SM10 donor strains (Supplementary Fig. 4). Using standard procedures for serial dilution and viable cell counting97, we have estimated that merodiploids occur at a frequency of ~10−7 in the bacterial population.
Counter-selection and mutant detection (Steps 37, 39, 45 and 48)
Single merodiploid colonies streaked out on NSLB + 15% sucrose typically produce ~ 1.0 × 102 colonies (Step 37). Double-crossovers occurred at a frequency of <10−5 (Fig. 6B). From a random sample of 52 antibiotic-sensitive colonies isolated on sucrose (representing 8 targeted loci: fimT, pilC, pilF, pilI, pilN, pilR, pilS, and pilU), we were able to detect 24 (46%) with the desired double-crossover mutation by PCR (Step 45A). A total of 16 mutants were selected (2 for each targeted locus), and all deletions were successfully confirmed by sequencing PCR products with nested sequencing primers (Step 48).
Mutations that cause counter-selection escape occur at a frequency of less than 10−7 (Fig. 6B). However, isolation of colonies that have escaped counter-selection is highly variable, which is typical of random mutation98. Based on our experience, ~5% of biparental matings produce merodiploid colonies that are antibiotic- and sucrose-resistant. However, we have observed that sucrose-resistant merodiploids may sometimes represent the vast majority of colonies isolated on sucrose from a single biparental mating (Step 39). It is tempting to speculate that this may be due to the timing at which a random mutation occurred (i.e. mutants emerging in early generations of bacterial cultures will expand to a large proportion of the population by late generations98).
Supplementary Material
{kind=link}
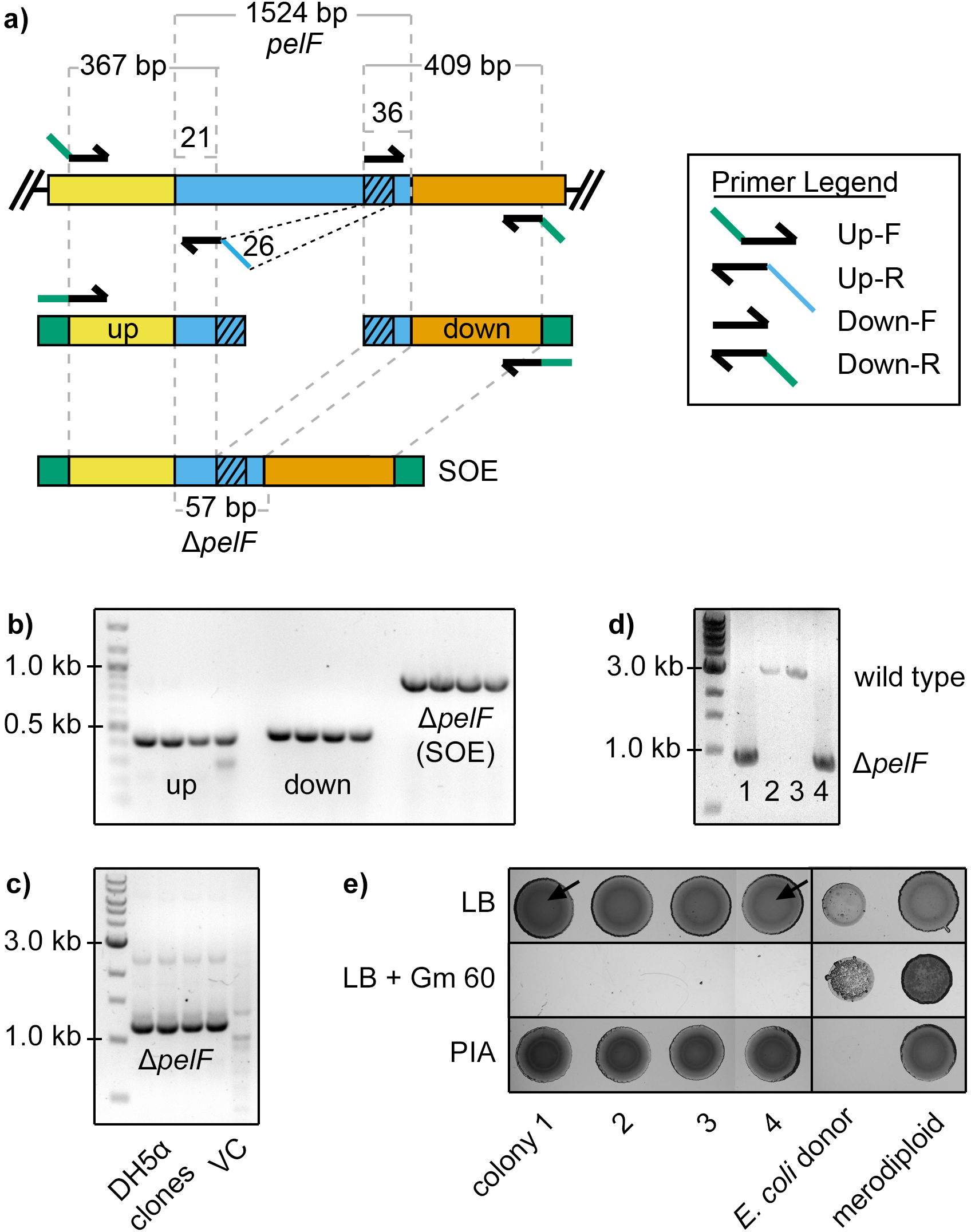{kind=link}
{kind=link}
{kind=link}
EDITORIAL SUMMARY.
Here, the authors describe genetically engineering the Pseudomonas genome by two-step allelic exchange. Suicide vector-encoded alleles are used to generate mutations by homologous recombination at the single base pair level.
Acknowledgments
The authors would like to thank Michael F. Hynes, Michael Wilton, Dave van Ditmarsch, Jacquelyn D. Rich and Geoff Winsor for useful discussions and feedback on this manuscript. J.J.H. is supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) and a Canada Research Chair (Tier II) from the Canadian Institutes for Health Research (CIHR). Infrastructure has been provided to J.J.H. through support from the Canada Foundation for Innovation (CFI).
Footnotes
Note: Supplementary information is available via the HTML version of this article.
AUTHOR CONTRIBUTIONS
L.R.H. and J.J.H. wrote the manuscript; L.R.H., B.R.B., H.A., Y.I., T.E.R., M.E.L., J.J.Y., B.S.T, K.M.S., R.J.S., P.L.H., P.K.S., T.T.-N., M.R.P, H.R.P. and J.J.H. designed, tested and/or troubleshot the protocol; L.R.H, H.A., T.E.R., M.E.L., C.L. and J.J.H. created strains and plasmids; T.E.R, M.E.L, C.L. and J.J.H. generated data presented in Anticipated Results and Supplementary Information. All authors revised and proofread the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Giske CG, Monnet DL, Cars O, Carmeli Y ReAct-Action on Antibiotic, R. Clinical and economic impact of common multidrug-resistant gram-negative bacilli. Antimicrob. Agents Chemother. 2008;52:813–821. doi: 10.1128/AAC.01169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Review of Anti-infective Therapy. 2013;11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- 3.Boucher HW, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 4.Held K, Ramage E, Jacobs M, Gallagher L, Manoil C. Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J. Bacteriol. 2012;194:6387–6389. doi: 10.1128/JB.01479-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobs MA, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liberati NT, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labaer J, et al. The Pseudomonas aeruginosa PA01 gene collection. Genome Res. 2004;14:2190–2200. doi: 10.1101/gr.2482804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whiteley M, et al. Gene expression in Pseudomonas aeruginosa biofilms. Nature. 2001;413:860–864. doi: 10.1038/35101627. [DOI] [PubMed] [Google Scholar]
- 9.Chugani S, et al. Strain-dependent diversity in the Pseudomonas aeruginosa quorum-sensing regulon. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E2823–E2831. doi: 10.1073/pnas.1214128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wurtzel O, et al. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Path. 2012;8:e1002945. doi: 10.1371/journal.ppat.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dotsch A, et al. The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS One. 2012;7:e31092. doi: 10.1371/journal.pone.0031092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomez-Lozano M, Marvig RL, Molin S, Long KS. Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ. Microbiol. 2012;14:2006–2016. doi: 10.1111/j.1462-2920.2012.02759.x. [DOI] [PubMed] [Google Scholar]
- 13.Jones CJ, et al. ChIP-Seq and RNA-Seq reveal an AmrZ-mediated mechanism for cyclic di-GMP synthesis and biofilm development by Pseudomonas aeruginosa. PLoS Path. 2014;10:e1003984. doi: 10.1371/journal.ppat.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanka A, et al. Identification of the alternative sigma factor SigX regulon and its implications for Pseudomonas aeruginosa pathogenicity. J. Bacteriol. 2014;196:345–356. doi: 10.1128/JB.01034-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balasubramanian D, et al. Deep sequencing analyses expands the Pseudomonas aeruginosa AmpR regulon to include small RNA-mediated regulation of iron acquisition, heat shock and oxidative stress response. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallagher LA, Shendure J, Manoil C. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. mBio. 2011;2:e00315–e00310. doi: 10.1128/mBio.00315-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winsor GL, et al. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 1997;179:6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quandt J, Hynes MF. Versatile suicide vectors which allow direction seletion for gene replacement in Gram-negative bacteria. Gene. 1993;127:15–21. doi: 10.1016/0378-1119(93)90611-6. [DOI] [PubMed] [Google Scholar]
- 20.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 21.Ruvkun GB, Ausubel FM. A general method for site-directed mutagenesis in prokaryotes. Nature. 1981;289:85–88. doi: 10.1038/289085a0. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg JB, Ohman DE. Construction and characterization of Pseudomonas aeruginosa algB mutants: role of algB in high-level production of alginate. J. Bacteriol. 1987;169:1593–1602. doi: 10.1128/jb.169.4.1593-1602.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flynn JL, Ohman DE. Use of a gene replacement cosmid vector for cloning alginate conversion genes from mucoid and nonmucoid Pseudomonas aeruginosa strains: algS controls expression of algT. J. Bacteriol. 1988;170:3228–3236. doi: 10.1128/jb.170.7.3228-3236.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schweizer HP. Alielic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol. 1992;6:1195–1204. doi: 10.1111/j.1365-2958.1992.tb01558.x. [DOI] [PubMed] [Google Scholar]
- 25.Quenee L, Lamotte D, Polack B. Combined sacB-based negative selection and Cre-lox antibiotic marker recycling for efficient gene deletion in Pseudomonas aeruginosa. Biotechniques. 2005;38:63–67. doi: 10.2144/05381ST01. [DOI] [PubMed] [Google Scholar]
- 26.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl. Environ. Microbiol. 2006;72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi KH, Schweizer HP. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 2005;5:30. doi: 10.1186/1471-2180-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tseng BS, et al. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol. 2013;15:2865–2878. doi: 10.1111/1462-2920.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfgang MC, Lee VT, Gilmore ME, Lory S. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Developmental Cell. 2003;4:253–263. doi: 10.1016/s1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 30.Fulcher NB, Holliday PM, Klem E, Cann MJ, Wolfgang MC. The Pseudomonas aeruginosa Chp chemosensory system regulates intracellular cAMP levels by modulating adenylate cyclase activity. Mol. Microbiol. 2010;76:889–904. doi: 10.1111/j.1365-2958.2010.07135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almblad H, et al. The cyclic AMP-Vfr signaling pathway in Pseudomonas aeruginosa is inhibited by cyclic di-GMP. J. Bacteriol. 2015;197:2190–2200. doi: 10.1128/JB.00193-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao K, et al. Psl trails guide exploration and microcolony formation in Pseudomonas aeruginosa biofilms. Nature. 2013;497:388–391. doi: 10.1038/nature12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen D, et al. Oligoribonuclease is a central feature of cyclic diguanylate signaling in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 2015 doi: 10.1073/pnas.1421450112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colvin KM, et al. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 2012;14:1913–1928. doi: 10.1111/j.1462-2920.2011.02657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colvin KM, et al. The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Path. 2011;7:e1001264. doi: 10.1371/journal.ppat.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zimmermann A, Reimmann C, Galimand M, Haas D. Anaerobic growth and cyanide synthesis of Pseudomonas aeruginosa depend on anr, a regulatory gene homologous with fnr of Escherichia coli. Mol. Microbiol. 1991;5:1483–1490. doi: 10.1111/j.1365-2958.1991.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 37.Choi KH, Kumar A, Schweizer HP. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells. J. Microbiol. Methods. 2006;64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Campbell AM. Episomes. Adv. Genet. 1963;11:101–145. [Google Scholar]
- 39.Bochner BR, Huang HC, Schieven GL, Ames BN. Positive selection for loss of tetracycline resistance. J. Bacteriol. 1980;143:926–933. doi: 10.1128/jb.143.2.926-933.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ried JL, Collmer A. An nptI, sacB, sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 1987;57:239–246. doi: 10.1016/0378-1119(87)90127-2. [DOI] [PubMed] [Google Scholar]
- 41.Schafer A, et al. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 42.Pelicic V, Reyrat JM, Gicquel B. Expression of the Bacillus subtilis sacB gene confers sucrose sensitivity on Mycobacteria. J. Bacteriol. 1996;178:1197–1199. doi: 10.1128/jb.178.4.1197-1199.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinmetz M, Le Coq D, Djemia HB, Gay P. Analyse génétique de sacB, gène de structure d’une enzyme secrétée, la lévane-saccharase de Bacillus subtilis Marburg. Mol. Gen. Genet. 1983;191:138–144. doi: 10.1007/BF00330901. [DOI] [PubMed] [Google Scholar]
- 44.Gay P, Le Coq D, Steinmetz M, Berkelman T, Kado CI. Positive selection procedure for entrapment of insertion sequence elements in Gram-negative bacteria. J. Bacteriol. 1985;164:918–921. doi: 10.1128/jb.164.2.918-921.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blomfield IC, Vaughn V, Rest RF, Eisenstein BI. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol. 1991;5:1447–1457. doi: 10.1111/j.1365-2958.1991.tb00791.x. [DOI] [PubMed] [Google Scholar]
- 46.Gregg CJ, et al. Rational optimization of tolC as a powerful dual selectable marker for genome engineering. Nucleic Acids Res. 2014;42:4779–4790. doi: 10.1093/nar/gkt1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jager W, Schafer A, Kalinowski J, Puhler A. Isolation of insertion elements from gram-positive Brevibacterium, Corynebacterium and Rhodococcus strains using the Bacillus subtilis sacB gene as a positive selection marker. FEMS Microbiol. Lett. 1995;126:1–6. doi: 10.1111/j.1574-6968.1995.tb07381.x. [DOI] [PubMed] [Google Scholar]
- 48.Fu R, Voordouw G. Targeted gene-replacement mutagenesis of dcrA, encoding an oxygen sensor of the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Microbiology. 1997;143(Pt 6):1815–1826. doi: 10.1099/00221287-143-6-1815. [DOI] [PubMed] [Google Scholar]
- 49.Simon R, Hotte B, Klauke B, Kosier B. Isolation and characterization of insertion sequence elements from gram-negative bacteria by using new broad-host-range, positive selection vectors. J. Bacteriol. 1991;173:1502–1508. doi: 10.1128/jb.173.4.1502-1508.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kung VL, Ozer EA, Hauser AR. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2010;74:621–641. doi: 10.1128/MMBR.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fowler RC, Hanson ND. Emergence of carbapenem resistance due to the novel insertion sequence ISPa8 in Pseudomonas aeruginosa. PLoS One. 2014;9:e91299. doi: 10.1371/journal.pone.0091299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klose KE, Mekalanos JJ. Distinct roles of an alternative sigma factor during both free-swimming and colonizing phases of the Vibrio cholerae pathogenic cycle. Mol. Microbiol. 1998;28:501–520. doi: 10.1046/j.1365-2958.1998.00809.x. [DOI] [PubMed] [Google Scholar]
- 53.Metcalf WW, et al. Conditionally replicative and conjugative plasmids carrying lacZα for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 54.Barekzi N, et al. High-frequency Flp recombinase-mediated inversions of the oriC-containing region of the Pseudomonas aeruginosa genome. J. Bacteriol. 2000;182:7070–7074. doi: 10.1128/jb.182.24.7070-7074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi K-H, Schweizer HP. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 2006;1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 56.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. 2006;40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 58.Karlinsey JE. lambda-Red genetic engineering in Salmonella enterica serovar Typhimurium. Methods Enzymol. 2007;421:199–209. doi: 10.1016/S0076-6879(06)21016-4. [DOI] [PubMed] [Google Scholar]
- 59.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy KC, Campellone KG, Poteete AR. PCR-mediated gene replacement in Escherichia coli. Gene. 2000;246:321–330. doi: 10.1016/s0378-1119(00)00071-8. [DOI] [PubMed] [Google Scholar]
- 61.Liang R, Liu J. Scarless and sequential gene modification in Pseudomonas using PCR product flanked by short homology regions. BMC Microbiol. 2010;10:209. doi: 10.1186/1471-2180-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lesic B, Rahme LG. Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol. Biol. 2008;9:20. doi: 10.1186/1471-2199-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Starkey M, et al. Pseudomonas aeruginosa rugose small colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 2009;191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fazli M, Harrison JJ, Gambino M, Givskov M, Tolker-Nielsen T. In-frame and unmarked gene deletions in Burkholderia cenocepacia via an allelic exchange system compatible with Gateway technology. Appl. Environ. Microbiol. 2015;81:3623–3630. doi: 10.1128/AEM.03909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barrett AR, et al. Genetic tools for allelic replacement in Burkholderia species. Appl. Environ. Microbiol. 2008;74:4498–4508. doi: 10.1128/AEM.00531-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lopez CM, Rholl DA, Trunck LA, Schweizer HP. Versatile dual-technology system for markerless allele replacement in Burkholderia, pseudomallei. Appl. Environ. Microbiol. 2009;75:6496–6503. doi: 10.1128/AEM.01669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reyrat JM, Pelicic V, Gicquel B, Rappuoli R. Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect. Immun. 1998;66:4011–4017. doi: 10.1128/iai.66.9.4011-4017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schweizer H. Bacterial genetics: past achievements, present state of the field, and future challenges. Biotechniques. 2008;44:633–634. doi: 10.2144/000112807. 636–641. [DOI] [PubMed] [Google Scholar]
- 69.Lee SA, et al. General and condition-specific essential functions of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 2015 doi: 10.1073/pnas.1422186112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schweizer HP. Escherichia, Pseudomonas shuttle vectors derived from pUC18/19. Gene. 1991;97:109–112. doi: 10.1016/0378-1119(91)90016-5. [DOI] [PubMed] [Google Scholar]
- 71.Choi K-H, et al. A Tn7-based broad-range bacterial cloning and expression system. Nature Methods. 2005;2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 72.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. Integration proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid. 2000;43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 73.Green MR, Sambrook J. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- 74.Ausubel FM, et al. Short protocols in molecular biology: A compendium of methods from Current protocols in molecular biology. 5th. John Wiley & Sons, Inc; 2002. [Google Scholar]
- 75.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 76.Xu L, et al. Average gene length is highly conserved in prokaryotes and eukaryotes and diverges only between the two kingdoms. Mol. Biol. Evol. 2006;23:1107–1108. doi: 10.1093/molbev/msk019. [DOI] [PubMed] [Google Scholar]
- 77.Penterman J, et al. Rapid Evolution of Culture-Impaired Bacteria during Adaptation to Biofilm Growth. Cell Rep. 2014;6:293–300. doi: 10.1016/j.celrep.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 79.Baraquet C, Murakami K, Parsek MR, Harwood CS. The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res. 2012;40:7207–7218. doi: 10.1093/nar/gks384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma L, et al. Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Path. 2009;5:e1000354. doi: 10.1371/journal.ppat.1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Güvener ZT, Tifrea DF, Harwood CS. Two different Pseudomonas aeruginosa chemosensory signal transduction complexes localize to cell poles and form and remould in stationary phase. Mol. Microbiol. 2006;61:106–118. doi: 10.1111/j.1365-2958.2006.05218.x. [DOI] [PubMed] [Google Scholar]
- 82.Huangyutitham V, Guvener ZT, Harwood CS. Subcellular clustering of the phosphorylated WspR response regulator protein stimulates its diguanylate cyclase activity. mBio. 2013;4 doi: 10.1128/mBio.00242-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guvener ZT, Harwood CS. Subcellular location characteristics of the Pseudomonas aeruginosa GGDEF protein, WspR, indicate that it produces cyclic-di-GMP in response to growth on surfaces. Mol. Microbiol. 2007;66:1459–1473. doi: 10.1111/j.1365-2958.2007.06008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2007;2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 85.van Ditmarsch D, et al. Convergent evolution of hyperswarming leads to impaired biofilm formation in pathogenic bacteria. Cell Rep. 2013;4:697–708. doi: 10.1016/j.celrep.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rual J-F, et al. Human ORFeome Version 1.1: A platform for reverse proteomics. Genome Res. 2004;14:2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bernard P, Gabant P, Bahassi EM, Couturier M. Positive-selection vectors using the F plasmid ccdB killer gene. Gene. 1994;148:71–74. doi: 10.1016/0378-1119(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 88.Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: Partial purification and some properties. J. Biol. Chem. 1956;218:97–106. [PubMed] [Google Scholar]
- 89.Blount ZD, Barrick JE, Davidson CJ, Lenski RE. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature. 2012;489:513–518. doi: 10.1038/nature11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thoma S, Schobert M. An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol. Lett. 2009;294:127–132. doi: 10.1111/j.1574-6968.2009.01556.x. [DOI] [PubMed] [Google Scholar]
- 91.Grundemann D, Schomig E. Protection of DNA during preparative agarose gel electrophoresis against damage induced by ultraviolet light. Biotechniques. 1996;21:898–903. doi: 10.2144/96215rr02. [DOI] [PubMed] [Google Scholar]
- 92.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215 doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 93.Chenna R, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leighton TL, Buensuceso R, Howell PL, Burrows LL. Biogenesis of Pseudomonas aeruginosa type IV pili and regulation of their function. Environ. Microbiol. 2015 doi: 10.1111/1462-2920.12849. in press. [DOI] [PubMed] [Google Scholar]
- 95.Burrows LL. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu. Rev. Microbiol. 2012;66:493–520. doi: 10.1146/annurev-micro-092611-150055. [DOI] [PubMed] [Google Scholar]
- 96.Darzins A. Characterization of a Pseudomonas aeruginosa gene cluster involved in pilus biosynthesis and twitching motility: sequence similarity to the chemotaxis proteins of enterics and the gliding bacterium Myxococcus xanthus. Mol. Microbiol. 1994;11:137–153. doi: 10.1111/j.1365-2958.1994.tb00296.x. [DOI] [PubMed] [Google Scholar]
- 97.Harrison JJ, et al. Microtiter susceptibility testing of microbes growing on peg lids: a miniaturized biofilm model for high-throughput screening. Nat. Protoc. 2010;5:1236–1254. doi: 10.1038/nprot.2010.71. [DOI] [PubMed] [Google Scholar]
- 98.Luria SE, Delbruck M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 2005;102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hartley JL, Temple GF, Brasch MA. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Katzen F. Gateway recombinational cloning: a biological operating system. Expert opinion on drug discovery. 2007;2:571–589. doi: 10.1517/17460441.2.4.571. [DOI] [PubMed] [Google Scholar]
- 103.Liang X, Peng L, Baek CH, Katzen F. Single step BP/LR combined Gateway reactions. Biotechniques. 2013;55:265–268. doi: 10.2144/000114101. [DOI] [PubMed] [Google Scholar]
- 104.Stover CK, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 105.Kibbe WA. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:W43–W46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.