

Abstract
Background
Transcriptional gene regulation is a fundamental process in nature, and the experimental and computational investigation of DNA binding motifs and their binding sites is a prerequisite for elucidating this process. ChIP-seq has become the major technology to uncover genomic regions containing those binding sites, but motifs predicted by traditional computational approaches using these data are distorted by a ubiquitous binding-affinity bias. Here, we present an approach for detecting and correcting this bias using inter-species information.
Results
We find that the binding-affinity bias caused by the ChIP-seq experiment in the reference species is stronger than the indirect binding-affinity bias in orthologous regions from phylogenetically related species. We use this difference to develop a phylogenetic footprinting model that is capable of detecting and correcting the binding-affinity bias. We find that this model improves motif prediction and that the corrected motifs are typically softer than those predicted by traditional approaches.
Conclusions
These findings indicate that motifs published in databases and in the literature are artificially sharpened compared to the native motifs. These findings also indicate that our current understanding of transcriptional gene regulation might be blurred, but that it is possible to advance this understanding by taking into account inter-species information available today and even more in the future.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2682-6) contains supplementary material, which is available to authorized users.
Keywords: Binding-affinity bias, ChIP-seq, Phylogenetic footprinting, Evolution, Transcription factor binding sites, Gene regulation
Background
Predicting transcription factor binding sites and their motifs is essential for understanding transcriptional gene regulation and thus of importance in almost all areas of modern biology, medicine, and biodiversity research [1, 2]. Countless approaches exist for predicting motifs from these genomic regions [3–6], but predicting motifs from ChIP-seq data and similar experimental data is hampered by the contamination with false positive genomic regions as well as the enrichment of high-affinity binding sites [7–9].
The contamination with false positive genomic regions is caused by at least three reasons. First, the transcription factor or other DNA binding protein pulled down by immunoprecipitation may not bind directly to the binding site [10]. Second, ChIP-seq target regions may not contain a binding site due to experimental settings such as sequencing depth or DNA fragment length [11, 12]. Third, false positive regions may be predicted in the subsequent ChIP-seq data analysis due to never perfect analysis pipelines and too low signal cutoff thresholds [8]. These three effects may lead to the selection of false positive ChIP-seq regions that do not contain at least one binding site.
The enrichment of high-affinity binding sites is caused by at least two reasons. First, most antibodies have a preference of binding high-affinity binding sites with a higher probability than low-affinity binding sites, causing the set of binding sites bound in the ChIP-seq experiment to be partially different from the set of binding sites bound in vivo [13, 14]. Second, true positive regions with low-affinity binding sites are rejected due to too high signal cutoff thresholds [5, 8]. These two effects may lead to an under-representation of low-affinity binding sites and an over-representation of high-affinity binding sites in ChIP-seq regions.
Taken together, the contamination with false positive genomic regions leads to the contamination bias [15] and thus to the prediction of artificially softened motifs, whereas the enrichment of sequences with high-affinity binding sites leads to the binding-affinity bias [16] and thus to the prediction of artificially sharpened motifs. Neglecting these effects leads to distorted motifs and could potentially affect all downstream analyses [17–20]. Existing approaches for predicting motifs are capable of detecting and correcting the contamination bias, which has been found to increase the quality of motif prediction considerably [8, 21, 22], and here we investigate the possibility of detecting and correcting the binding-affinity bias.
Detecting the binding-affinity bias seems impossible based on sequence data from one species alone, but it seems possible based on inter-species information. This is possible due to the fact that the binding-affinity bias is stronger in the target regions of the ChIP-seq experiment in the reference species than in orthologous regions of phylogenetically related species. This stronger binding-affinity bias yields more biased motifs in the reference species than in phylogenetically related species, and this difference may be used for detecting and potentially correcting the binding-affinity bias.
Phylogenetic footprinting models typically (i) take into account ChIP-seq data of only one species and (ii) do not take into account heterogeneous substitution rates among different DNA regions, heterotachious evolution of DNA regions, and loss-of-function mutations in binding sites. The consideration of (i) ChIP-seq data of more than one species and (ii) heterogeneity, heterotachy, and loss-of-function mutations are likely to improve both phylogenetic footprinting as well as the detection and correction of the binding-affinity bias, but in this work we investigate if the detection and correction of this bias is possible based on (i) ChIP-seq data of only one species and (ii) a simple phylogenetic footprinting model that neglects heterogeneity, heterotachy, and loss-of-function mutations.
We first investigate if the effect of observing more biased motifs in the reference species than in phylogenetically related species is measurable beyond statistical noise in target regions of five ChIP-seq data sets of human and in orthologous regions of monkey, dog, cow, and horse. We then develop a phylogenetic footprinting model that incorporates the binding-affinity bias, investigate if this model improves or deteriorates motif prediction compared to traditional models that do not incorporate it, and compare the motifs predicted with and without the correction of the binding-affinity bias.
Results and discussion
In subsection “Using sequence-information of phylogenetically related species to detect the binding-affinity bias”, we describe the basic idea of how the binding-affinity bias could be detected based on inter-species information using a toy example. In the remaining subsections we perform three studies based on ChIP-seq data sets of five transcription factors and on multiple alignments of the human ChIP-seq target regions with orthologous regions from monkey, dog, cow, and horse. In subsection “Decrease of information contents in motifs from phylogenetically related species” we investigate if the effect of observing more biased motifs in the reference species than in phylogenetically related species is measurable in these five data sets. In subsection “Modeling the binding-affinity bias increases classification performance”, we investigate if a correction of the binding-affinity bias leads to an improvement or a deterioration of the classification performance. In subsection “Modeling the binding-affinity bias leads to softened motifs”, we compare the sequence motifs predicted with and without the correction of the binding-affinity bias.
Using sequence-information of phylogenetically related species to detect the binding-affinity bias
Detecting and correcting the binding-affinity bias might be possible because the binding-affinity bias inherent to the ChIP-seq experiment in the reference species (Fig. 1a) is stronger than the indirect binding-affinity bias in orthologous regions from phylogenetically related species. Under this assumption, the information content of the predicted motifs [23] should decrease with the phylogenetic distance from the reference species due to the increasing number of mutations.
Fig. 1.
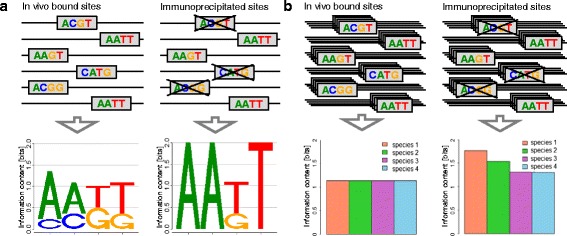
Influence of binding-affinity bias on information content. a Binding-affinity bias in the reference species. The left column shows binding sites bound in vivo as well as the sequence logo. In the right column, enrichment of high-affinity binding sites by chromatin immunoprecipitation leads to a different motif with higher information content. b Binding-affinity bias in the reference species and three phylogenetically related species. The left column shows binding sites bound in vivo and the information content of the species–specific motifs. In the right column, the enrichment of high-affinity binding sites in the reference species and the other three species leads to different motifs with different information content in each species. The effect of this enrichment decreases with the phylogenetic distance from the reference species as reflected by decreasing information contents. Please find the sequences of all species in Table 1
To illustrate this idea, we present a toy example consisting of six binding sites from four phylogenetically related species in Fig. 1b and Table 1. In this toy example, we assume an exaggerated binding-affinity bias of three high-affinity binding sites captured by the ChIP-seq experiment and three low-affinity binding sites not captured by the ChIP-seq experiment. In real world applications the native motif is unknown and the motif predicted on the available data is biased to an unknown degree. In the presented toy example, however, the native motif is considered to be known so that the effect of the binding-affinity bias on the motifs of the reference species (species 1) and the phylogenetically related species (species 2, 3, and 4) can be illustrated.
Table 1.
Influence of binding-affinity bias on information content. We illustrate the effect of binding-affinity bias with the given toy example of a ChIP-seq experiment for six binding sites in four species. Due to low binding-affinity, red binding sites are insufficiently bound. This results in the absence of red binding sites in the measured data which we denote binding-affinity bias. Binding sites with low binding-affinity typically comprise dissimilar bases in contrast to black binding sites with high affinity and common bases. The absence of red binding sites leads to a sharpening of the resulting motif, which we indicate using the information content. The information content without binding-affinity bias is equal in all species, whereas the information content with binding-affinity bias increases in all species. The vital point is that the effect of binding-affinity bias decreases with phylogenetic distance, which involves an increasing number of mutations. Please find a visualization of this toy example in Fig. 1 b
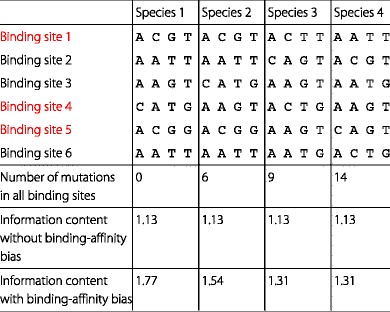
The motif predicted from the three target regions containing high-affinity binding sites is strongly biased in reference species 1, and it is impossible to predict the native motif from only those three target regions. However, a shadow of this strong binding-affinity bias also exists in orthologous regions of species 2, 3, and 4, so the motifs predicted from these orthologous regions in species 2, 3, and 4 are biased, too. This bias in species 2, 3, and 4, however, is weaker than the bias in reference species 1, and this difference can be exploited for detecting and correcting the binding-affinity bias and for predicting the native motif from the three target regions of high-affinity binding sites in reference species 1 and their orthologous regions in species 2, 3, and 4.
Specifically, the binding-affinity bias introduced by the ChIP-seq experiment in reference species 1 causes a strong increase of the information content of the predicted motif (1.77 bit) compared to the native motif (1.13 bit). The shadow of the binding-affinity bias in species 2, 3, and 4 also causes an increase of the information contents of the motifs predicted in species 2 (1.54 bit), species 3 (1.31 bit), and species 4 (1.31 bit), but this increase in species 2, 3, and 4 is smaller than in reference species 1 (Table 1 and Fig. 1b). The increase of information content decreases with the number of observed mutations and thus the phylogenetic distance of species 2, 3, and 4 to reference species 1 in which the ChIP-seq experiment has been performed. Hence, the observation of a decreased information content of motifs predicted in orthologous regions of phylogenetically related species compared to the information content of the motif predicted in the reference species could indicate the presence of a binding-affinity bias and possibly allow the correction of that bias.
Decrease of information contents in motifs from phylogenetically related species
We investigate this hypothesis on human ChIP-seq data of five transcription factors [10, 24] and multiple alignments of the human ChIP-seq target regions with orthologous regions from monkey, dog, cow, and horse [25] (“Data” Methods). We calculate the information contents of motifs from human (reference species), monkey, dog, cow, and horse for each of the five data sets (“Decrease of information contents in motifs from related species” Methods) and present the results in Fig. 2. We find for each of the five data sets that the information content of the motif from the reference species is significantly higher (p<1.83×10−14, Wilcoxon Signed-Rank Test, Additional file 1: Table S1) compared to the information contents of the motifs from monkey, dog, cow, and horse.
Fig. 2.
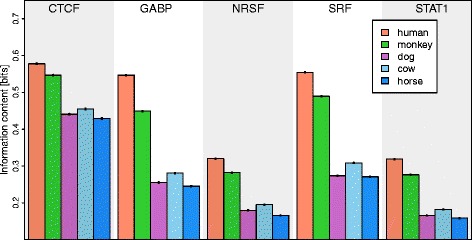
Mean information content and standard error for motifs of five transcription factors in five species. The information content of motifs in the reference species (human) is significantly higher compared to the four phylogenetically related species (p<1.8×10−14). The information content typically decreases with the phylogenetic distance from the reference species
Modeling the binding-affinity bias increases classification performance
Motivated by this observation, we develop a phylogenetic footprinting model capable of taking into account the contamination bias (), the binding-affinity bias (), neither one or the other , or both () (“Modeling the binding-affinity bias” Methods and Additional file 1: Section 1). In order to study to which degree these models are capable of modeling multiple alignments originating from ChIP-seq data, we consider the principle of parsimony [26], which states that the simplest of competing explanations is the most likely to be correct. As the new model is more complex than the traditional model , we should accept it only if it provides a more accurate representation of the data. A standard approach for measuring how accurately a model represents a data set is to measure its performance of classifying, in this case, motif-bearing and non-motif-bearing alignments, and a standard approach for measuring classification performance is stratified repeated random sub-sampling validation (“Measuring classification performance” Methods, Fig. 5).
Fig. 5.
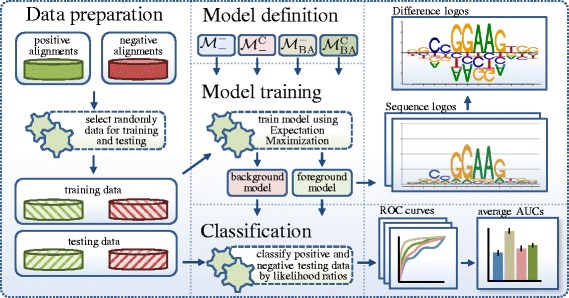
Overview of the workflow presented in this manuscript. In the data preparation step, we randomly compile disjoint training data and testing data each with positive alignments and negative alignments for each of the transcription factors CTCF, GABP, NRSF, SRF, and STAT1. In the model training step, we train each of the four presented foreground models as well as a background model by expectation maximization with 150 restarts. We choose the foreground model and the background model with maximum likelihood, classify the testing data using a likelihood-ratio classifier, and extract different characteristics such as the ROC curve, the PR curve, the inverse temperature, and the inferred motif. We repeat the described procedure 100 times and calculate mean values and standard errors for several quantities such as the areas under the ROC curves or the PR curves
Using this approach we measure the performance of the four models , , , and to classify each of the five data sets against the other four. Fig. 3a shows that yields a higher classification performance than in all five data sets (p<2.3×10−17, Wilcoxon Signed-Rank Test, Additional file 1: Table S2), indicating that the new model is more realistic than the traditional model . We also find that yields a significantly higher classification performance than in all five data sets (p<1.8×10−17, Wilcoxon Signed-Rank Test), which indicates that taking into account the binding-affinity bias has a larger impact on the classification performance than taking into account the contamination bias (Additional file 1: Figure S1, Figure S2, Figure S10, Figure S11, Figure S12, Figure S13, Figure S14, Figure S15 and Figure S16).
Fig. 3.
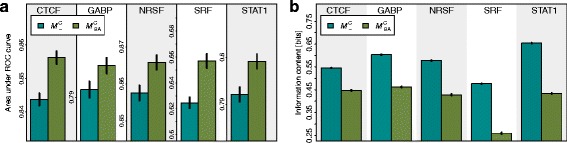
Comparison of models and . a Mean classification performance and standard error of the two models and quantified by the area under the receiver operating characteristic curve. We find for each of the five data sets a significantly increased classification performance for compared to . Examples for ROC curves are shown in Additional file 1: Figure S10, Figure S11, Figure S12, Figure S13, Figure S14 and Figure S15. b Mean information content and standard error of the motifs predicted by the two models and . We find for each of the five data sets a significantly decreased information content in motifs predicted by compared to (p<4.0×10−18)
Modeling the binding-affinity bias leads to softened motifs
Next, we investigate the information contents of the corrected motifs predicted by models and that take into account the binding-affinity bias and the traditional motifs predicted by models and that neglect this bias. Fig. 3b shows that the information contents of motifs predicted by are significantly higher than the information contents of motifs predicted by (p<4.0×10−18, Wilcoxon Signed-Rank Test). We also find that the information contents of motifs predicted by are higher than the information contents of motifs predicted by (p<4.0×10−18, Wilcoxon Signed-Rank Test, Additional file 1: Table S4), stating that the binding-affinity bias is stronger than the contamination bias. Equivalently, this states that the joint effect of both biases leads to an artificial sharpening of the motifs and an artificial overestimation of the binding affinities (Additional file 1: Figure S3, Figure S4, Figure S17, Figure S18).
Finally, we inspect the differences of the corrected motifs predicted by and and the traditional motifs predicted by and . Fig. 4 shows the differences between the base distributions of pairs of motifs for and by difference logos (“Visualizing motif differences with DiffLogo” Methods). We find for each of the five data sets that the corrected motifs are softer than the traditional motifs distorted by the binding-affinity bias. Specifically, we find that the amount of decrease of the most abundant bases in the corrected motifs compared to the traditional motifs is roughly proportional to the base abundance, whereas the increase of the remaining bases is not proportional to the base abundance. Hence, the corrected motifs are not simply a uniformly softened version of the traditional motifs, but motifs with different degrees of dissimilarity at different positions (Additional file 1: Figure S5, Figure S6,Figure S7, Figure S8 and Figure S9).
Fig. 4.
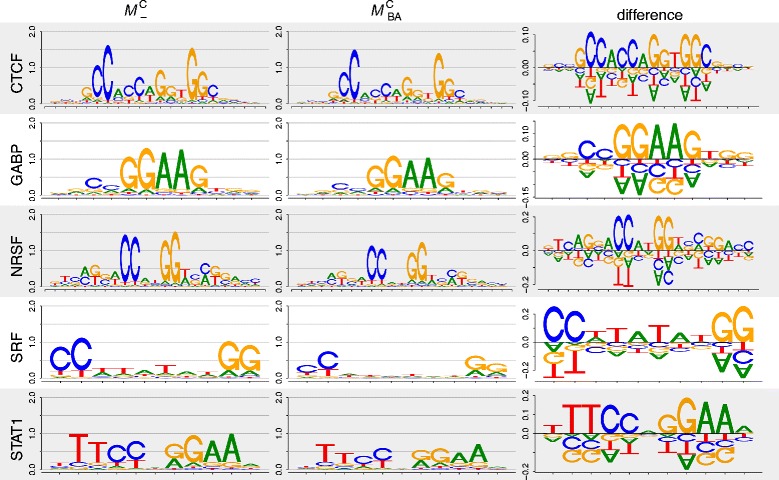
Differences of motifs predicted by and . The height of the base stacks is quantified by the Jensen-Shannon divergence, where high base stacks represent high motif differences. We find significant motif differences exceeding 0.1 bit for all five data sets (Additional file 1: Figure S5, Figure S6, Figure S7, Figure S8 and Figure S9)
Conclusions
We studied the possibility of detecting and correcting the binding-affinity bias in ChIP-seq data using inter-species information. We found that the fact that this bias is stronger in target regions of the reference species than its shadow in orthologous regions of phylogenetically related species enables the detection and correction of this bias. We proposed a phylogenetic footprinting model capable of taking into account the binding-affinity bias in addition to the contamination bias, and we applied this model and its three special cases that neglect one of the two biases or both to five ChIP-seq data sets. We found by stratified repeated random sub-sampling validation that taking into account the binding-affinity bias always improves motif prediction, that the motif binding-affinity bias leads to a distortion of motifs that is even stronger than the distortion caused by the contamination bias, and that the corrected motifs are typically softer than those predicted by traditional approaches. The comparison of corrected and traditional motifs showed small but noteworthy differences, suggesting that the refinement of traditional motifs from databases and from the literature might lead to the prediction of novel binding sites, cis-regulatory modules, or gene-regulatory networks and might thus advance our attempt of understanding transcriptional gene regulation as a whole.
Methods
In this section we describe “Decrease of information contents in motifs from related species” (i) the determination of the information contents of motifs in the reference species and phylogenetically related species, “Modeling the binding-affinity bias” (ii) the phylogenetic footprinting model that can take into account the binding-affinity bias, the contamination bias, neither one or the other, or both, “Measuring classification performance” (iii) the measurement of the classification performance of these four phylogenetic footprinting models using stratified repeated random sub-sampling validation, and “Visualizing motif differences with DiffLogo” (iv) the visualisation of differences between the corrected and the traditional motifs.
Decrease of information contents in motifs from related species
We determine the information content I(P) of a motif P as described in [23]:
| 1 |
where is the alphabet, pℓ,a is the probability of base a at position ℓ in motif P, and Hℓ(P) denotes the information content of position ℓ in motif P.
We measure the information contents of motifs in five species using repeated random sub-sampling as follows. Initially, we choose one motif for each of the transcription factors CTCF, GABP, NRSF, SRF, and STAT1 from the JASPAR database, namely MA0139.1 for CTCF, MA0062.2 for GABP, MA0138.2 for NRSF, MA0083.2 for SRF, and MA0137.3 for STAT1 [27]. In the first step, we generate a test set from the set of positive alignments (Table 2) by removing randomly 200 alignments. In the second step, we predict for each transcription factor one binding site per target region in all target regions of the reference species (human) in the corresponding test data set, extract the predicted binding sites from the reference species as well as the binding sites at the same positions in the orthologous regions, and calculate for each species the information content of the resulting motif as specified above. We perform both steps 100 times and report the mean and standard error of the information content for each of the five species.
Table 2.
Data set statistics for human ChIP-seq data. For each of the five transcription factors (TFs) CTCF, GABP, NRSF, SRF, and STAT1, we specify the (i) average length of transcription factor binding site (TFBS), the (ii) number of alignments, and the (iii) average length of alignments
| TF | TFBS length | Number of alignments | Avg. length |
|---|---|---|---|
| CTCF | 20 bp | 467 | 213 bp |
| GABP | 12 bp | 451 | 236 bp |
| NRSF | 21 bp | 460 | 245 bp |
| SRF | 12 bp | 394 | 242 bp |
| STAT1 | 11 bp | 360 | 244 bp |
Modeling the binding-affinity bias
In this section we describe the probabilistic model for modeling the binding-affinity bias as a data generating process. A derivation of the log-likelihood for motif-bearing and non-motif-bearing alignments can be found in Additional file 1: Section 1.
Let O be the number of species. A data set comprises N independent multiple sequence alignments. We use Xn to refer to the n-th sequence alignment. Every alignment is formed by O sequences. The o-th sequence is denoted by . By convention, the reference species (that in which the selection process has taken place) is species 1. Each sequence of alignment Xn is composed of Ln nucleotides. We denote by the u-th nucleotide of the o-th sequence of the n-th alignment. All nucleotides are presented by the set .
We assume the existence of a common ancestor of all of O species. The sequence of the common ancestor of the n-th alignment is a hidden variable Yn, with representing its u-th nucleotide. The substitution probability that nucleotide is substituted by the nucleotide is denoted by the variable γo.
An alignment Xn may contain a binding site or not. This is denoted by the variable Mn. The length of the binding site is denoted by the variable W and the position of the binding site in alignment Xn is denoted by the variable ℓn.
The n-th alignment Xn is sampled as follows. The first decision to be made is whether or not the alignment contains a binding site. This is denoted by variable Mn which follows a Bernoulli distribution with parameter 1−α. Thus, whenever variable Mn is equal to 1 (), the alignment contains a binding site and when Mn is equal to 0 (), it does not.
Thus, parameter α is the probability that alignment Xn contains no binding site. If α equals 0, the sampled data is uncontaminated, because all alignments contain a copy of the binding site. The larger the value of α, the higher the percentage of non motif-bearing alignments in the sampled data. A value of α equal to 1 models a data set where no binding sites are present.
Next we introduce the data generating process for non-motif-bearing alignments and later we explain that for motif-bearing alignments.
Sample the primordial sequence as follows: For each position u of the sequence sample nucleotide from the background equilibrium distribution π0 independent of the previous nucleotides.
For each of the descent species o∈{1,…,O}, sample its sequence given the primordial sequence as follows: To sample nucleotide u of the descent species o, we apply to nucleotide u of the primordial sequence the F81 [28] mutation model with the background equilibrium distribution π0 and the substitution probability γo.
The generating process for motif-bearing sequences is slightly more complex, since it has to deal both with the generation of the binding site and with the selection process. First, we describe how to sample an alignment without taking into account the selection process. Second, we show how to modify this procedure so that the selection process is considered.
Sample a motif-bearing alignment Xn as follows:
Sample the start position of the binding site ℓn from the uniform distribution.
Sample the primordial sequence. For each position u of the sequence outside the binding site, we sample nucleotide from the background equilibrium distribution π0. For each position u of the binding site, we sample nucleotide from the equilibrium distribution .
For each of the descent species o∈{1,…,O}, sample its sequence as follows: For each position u of the descent species o outside the binding site, apply to nucleotide of the primordial sequence the F81 mutation model taking as equilibrium distribution π0. For each position u of the descent species o inside the binding site, apply to nucleotide of the primordial sequence the F81 mutation model taking as equilibrium distribution .
Finally, to model the selection process, we introduce the variable β. β is used to quantify the degree of the binding-affinity bias in the reference species. We assume that a transcription factor binds binding site B with a probability proportional to p(B|π)β−1. As B occurs in vivo with probability p(B|π), it occurs in the set of immunoprecipitated sequences with a probability proportional to p(B|π)·p(B|π)β−1=p(B|π)β.
We can interpret the meaning of β as follows: If β is greater than one, low-affinity binding sites are more frequently rejected with respect to p(B) and high-affinity binding sites are less frequently rejected with respect to p(B). This leads to an under-representation of low-affinity binding sites and an over-representation of high-affinity binding sites in the ChIP-seq data set, thus modeling a data set that is affected by the binding-affinity bias. If β is equal to one, low-affinity binding sites are rejected as frequently as high-affinity binding sites, leading to a representative set of binding sites in the ChIP-seq data set, which is not affected by the binding-affinity bias.
Based on that selection model, sample a motif-bearing alignment that has passed the selection process as follows:
Sample a motif-bearing alignment disregarding the selection process following the procedure specified above.
Decide whether the alignment is accepted or rejected based on the probability of acceptance of the binding site found at the reference species. If the alignment is rejected, go to step 1.
Thus, we denote (i) the model with α=0 and β=1 by , (ii) the model with with α>0 and β=1 by , (iii) the model with α=0 and β>1 by , and (iv) the model with α>0 and . can neither handle the contamination bias nor the binding-affinity bias. can only handle the contamination bias, but not the binding-affinity bias. can only handle the binding-affinity bias, but not the contamination bias. And can handle both the contamination bias and the binding-affinity bias.
We call , , , and foreground models. For modeling the background alignments, we use the model with α=1 and β=1, which we call background model and which we denote by .
Measuring classification performance
For measuring the classification performance of the four models , , , and we perform stratified repeated random sub-sampling validation as illustrated in Fig. 5 using data sets of the five human transcription factors CTCF, GABP, NRSF, SRF, and STAT1 that have been used for benchmarking the phylogenetic footprinting program MotEvo [25].
In step 1, we generate two training sets and two disjoint test sets for each of the five transcription factors as follows. We randomly select 200 alignments from the set of alignments (Table 2) of a particular transcription factor as positive training set, and we choose the set of the remaining alignments as positive test set. We randomly select 500 alignments from the set of alignments of the four remaining transcription factors as negative training set and another disjoint set of 500 alignments as negative test set.
In step 2, we train a foreground model (, , , or ) on the positive training set and a background model () on the negative training set by expectation maximization [29] using a numerical optimization procedure in the maximization step.
We restart the expectation maximization algorithm, which is deterministic for a given data set and a given initialization, 150 times with different initializations and choose the foreground model and the background model with the maximum likelihood on the positive training data and the negative training data, respectively, for classification. We use a likelihood-ratio classifier of the two chosen foreground and background models, apply this classifier to the disjoint positive and negative test sets, and calculate the receiver operating characteristics curve, the precision recall curve, and the area under both curves as measures of classification performance.
We repeat both steps 100 times and determine (i) the mean area under the receiver operating characteristic curve and its standard error and (ii) the mean area under the precision recall curve and its standard error.
Data
The data used in this work originate from human ChIP-seq data of the five human transcription factors CTCF, GABP, NRSF, SRF, and STAT1, where the ChIP-seq data for GABP and SRF published in [10] are available from the QuEST web page [30], and the ChIP-seq data for CTCF, NRSF, and STAT1 published in [24] are available from the SISSRs web page [31]. All five data sets have been filtered for high-quality reads and mapped to a reference genome [10, 24], and peak calling has been performed by MACS [32]. Peaks have been extended or cropped to 400 bp, binding regions that potentially comprise more than one of the five transcription factors have been removed, and the 900 binding regions with the highest MACS score have been retained [25]. Orthologous regions from mouse, dog, cow, monkey, horse, and opossum have been extracted from the UCSC database [33], multiple alignments of these orthologous regions have been obtained using T-Coffee [34], and these multiple alignments are kindly provided by [25].
To prepare ungapped alignments from these gapped data sets of the five transcription factors CTCF, GABP, NRSF, SRF, and STAT1, we perform the following three steps. (i) Remove the species that cause the highest number of gaps in all alignments. Accordingly, we remove mouse and opossum and keep orthologous regions from human, monkey, cow, dog, and horse. (ii) Remove all columns in each of the alignments that contain at least one gap to obtain ungapped alignments. (iii) Remove all ungapped alignments that are shorter than 21 bp, which is the length of the longest motif (NRSF) in the performed studies. Table 2 shows details about the resulting data. All data are available as Additional file 2.
Visualizing motif differences with DiffLogo
We used the R package DiffLogo [35] to depict the differences between the predicted motifs of the models , , , and . DiffLogo is an open source software that is capable of depicting the differences between multiple motifs [35]. This is realized by visualizing all pair-wise differences in an N×N–grid with an empty diagonal. Each entry in the grid is called difference logo. The degree of difference of two motifs is calculated by the sum of all stack heights in the corresponding difference logo and is indicated by the background color from red (most dissimilar among all motif pairs) to green (most similar among all motif pairs). The individual sequence logos of the motifs are shown above the table.
A single difference logo depicts the position-specific differences between the base distributions of two sequence motifs. Differences are visualized using a stack of bases for each motif position. The height of each base stack is calculated by the Jensen-Shannon divergence, which is proportional to the degree of base distribution dissimilarity. The Jensen-Shannon divergence is zero if both base distributions are identical, increases with increasing difference of the two base distributions, and reaches a maximum of 2 bit if the two base distributions are maximally different, i.e., if two bases occur only in one of the two motifs each with a probability of 1/2 and the other two bases occur only in the other motif each with a probability of 1/2. The height of each base within a stack is given by the difference of abundance. Thus, the height of a base is proportional to the degree of differential symbol abundance. Bases with a positive height indicate a gain of abundance and bases with a negative height indicate a loss of abundance. The stack height in the positive direction must be equal to the stack height in the negative direction, because the sum of base abundance gain must be equal to the sum of base abundance loss.
Acknowledgements
We thank Lothar Altschmied, Helmut Bäumlein, Sven-Erik Behrens, Karin Breunig, Jan Grau, Katrin Hoffmann, Robert Paxton, Patrice Peterson, and Marcel Quint for valuable discussions and DFG (grant no. GR3526/1), Gencat (2014 SGR 118), and Collectiveware (TIN2015-66863-C2-1-R) for financial support.
Additional files
In section 1, Modeling the binding-affinity bias, we describe how to determine the likelihood of non-motif-bearing and motif-bearing alignments modeling the contamination bias and the binding-affinity bias.
In section 2, Example interpretation of difference logos, we give an exemplary interpretation of some difference logos.
Section 3, Supplementary Figures, contains supplementary Figures S1-S18.
Section 4, Supplementary Tables, contains supplementary Tables S1-S10. (PDF 3492 kb)
Sequence data. This archive contains data files of gap-free alignments of the ChIP-seq positive regions for each of the transcription factors CTCF, GABP, NRSF, SRF, and STAT1 in FASTA format. (ZIP 645 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MN and IG developed the key idea. MN and JC developed the computational methods. MN and HT performed the studies. All authors wrote, read, and approved the final manuscript.
References
- 1.Nowrousian M. Next-generation sequencing techniques for eukaryotic microorganisms: sequencing-based solutions to biological problems. Eukaryot Cell. 2010;9(9):1300–10. doi: 10.1128/EC.00123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villar D, Flicek P, Odom DT. Evolution of transcription factor binding in metazoans - mechanisms and functional implications. Nat Rev Genet. 2014;15(4):221–33. doi: 10.1038/nrg3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park PJ. Chip–seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furey TS. Chip–seq and beyond: new and improved methodologies to detect and characterize protein–dna interactions. Nat Rev Genet. 2012;13(12):840–52. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. Chip-seq guidelines and practices of the encode and modencode consortia. Genome Res. 2012;22(9):1813–31. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alipanahi B, Delong A, Weirauch MT, Frey BJ. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol. 2015;33(8):831–8. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins J, Grant C, Noble WS, Bailey TL. Assessing phylogenetic motif models for predicting transcription factor binding sites. Bioinformatics. 2009;25(12):339–47. doi: 10.1093/bioinformatics/btp201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomes AL, Abeel T, Peterson M, Azizi E, Lyubetskaya A, Carvalho L, Galagan J. Decoding chip-seq with a double-binding signal refines binding peaks to single-nucleotides and predicts cooperative interaction. Genome Res. 2014;24(10):1686–97. doi: 10.1101/gr.161711.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain D, Baldi S, Zabel A, Straub T, Becker PB. Active promoters give rise to false positive ‘Phantom Peaks’ in ChIP-seq experiments. Nucleic Acids Res. 2015;43(14):6959–68. doi: 10.1093/nar/gkv637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valouev A, Johnson A, David S and. Sundquist. Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods. 2008;5(9):829–34. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rye MB, Sætrom P, Drabløs F. A manually curated chip-seq benchmark demonstrates room for improvement in current peak-finder programs. Nucleic Acids Res. 2011;39(4):e25. doi: 10.1093/nar/gkq1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung YL, Luquette LJ, Ho JWK, Ferrari F, Tolstorukov M, Minoda A, Issner R, Epstein CB, Karpen GH, Kuroda MI, Park PJ. Impact of sequencing depth in ChIP-seq experiments. Nucleic Acids Res. 2014;42(9):178–4. doi: 10.1093/nar/gku178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of chip-seq (macs) Genome Biol. 2008;9(9):137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nix DA, Courdy SJ, Boucher KM. Empirical methods for controlling false positives and estimating confidence in chip-seq peaks. BMC Bioinformatics. 2008;9(1):523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey TL, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, Madrigal P, Taslim C, Zhang J. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput Biol. 2013;9(11):e1003326. doi: 10.1371/journal.pcbi.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Håndstad T, Rye MB, Drabløs F, Sætrom P. A ChIP-Seq Benchmark Shows That Sequence Conservation Mainly Improves Detection of Strong Transcription Factor Binding Sites. PLoS ONE. 2011;6(4):18430. doi: 10.1371/journal.pone.0018430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14(5):51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park D, Lee Y, Bhupindersingh G, Iyer VR. Widespread Misinterpretable ChIP-seq Bias in Yeast. PloS One. 2013;8(12):83506. doi: 10.1371/journal.pone.0083506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc Nat Acad Sci. 2013;110(46):18602–7. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott JH, Grimshaw J, Altman R, Bero L, Goodman SN, Henry D, Macleod M, Tovey D, Tugwell P, White H, Sim I. Informatics: Make sense of health data. Nature. 2015;527:31–2. doi: 10.1038/527031a. [DOI] [PubMed] [Google Scholar]
- 21.Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Ismb. 1995;3:21–9. [PubMed] [Google Scholar]
- 22.Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in chip-seq peak detection. PloS one. 2010;5(7):11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188(3):415–31. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 24.Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucl Acids Res. 2008;36(16):5221–31. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold P, Erb I, Pachkov M, Molina N, van Nimwegen E. MotEvo: integrated Bayesian probabilistic methods for inferring regulatory sites and motifs on multiple alignments of DNA sequences. Bioinformatics. 2012;28(4):487–94. doi: 10.1093/bioinformatics/btr695. [DOI] [PubMed] [Google Scholar]
- 26.Sober E. The principle of parsimony. Brit J Philos Sci. 1981;32(2):145–56. doi: 10.1093/bjps/32.2.145. [DOI] [Google Scholar]
- 27.Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen C-YY, Chou A, Ienasescu H, Lim J, Shyr C, Tan G, Zhou M, Lenhard B, Sandelin A, Wasserman WW. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42(Database issue):142–7. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17(6):368–76. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence CE, Reilly AA. An expectation maximization (em) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins: Struct Funct Bioinformatics. 1990;7(1):41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- 30.Quantitative Enrichment of Sequence Tags: QuEST. http://mendel.stanford.edu/sidowlab/downloads/quest/. Accessed 29 Mar 2016.
- 31.ChIP-Seq Data Analysis: Identification of Protein–DNA Binding Sites with SISSRs Peak-Finder. http://dir.nhlbi.nih.gov/papers/lmi/epigenomes/sissrs/. Accessed 29 Mar 2016. [DOI] [PMC free article] [PubMed]
- 32.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9(9):137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, Kober KM, Miller W, Pedersen JS, Pohl A, Raney BJ, Rhead B, Rosenbloom KR, Smith KE, Stanke M, Thakkapallayil A, Trumbower H, Wang T, Zweig AS, Haussler D, Kent WJ. The UCSC genome browser database: 2008 update. Nucleic Acids Res. 2008;36(suppl 1):773–9. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 35.Nettling M, Treutler H, Grau J, Keilwagen J, Posch S, Grosse I. DiffLogo: a comparative visualization of sequence motifs. BMC Bioinf. 2015;16(1):1. doi: 10.1186/s12859-015-0767-x. [DOI] [PMC free article] [PubMed] [Google Scholar]