

Abstract
Introduction
Enlargement of a pre-existing intracranial aneurysm is a well-established risk factor of rupture. Excessive low wall shear stress concomitant with turbulent flow in the dome of an aneurysm may contribute to progression and rupture. However, how stress conditions regulate enlargement of a pre-existing aneurysm remains to be elucidated.
Results
Wall shear stress was calculated with 3D-computational fluid dynamics simulation using three cases of unruptured intracranial aneurysm. The resulting value, 0.017 Pa at the dome, was much lower than that in the parent artery. We loaded wall shear stress corresponding to the value and also turbulent flow to the primary culture of endothelial cells. We then obtained gene expression profiles by RNA sequence analysis. RNA sequence analysis detected hundreds of differentially expressed genes among groups. Gene ontology and pathway analysis identified signaling related with cell division/proliferation as overrepresented in the low wall shear stress–loaded group, which was further augmented by the addition of turbulent flow. Moreover, expression of some chemoattractants for inflammatory cells, including MCP-1, was upregulated under low wall shear stress with concomitant turbulent flow. We further examined the temporal sequence of expressions of factors identified in an in vitro study using a rat model. No proliferative cells were detected, but MCP-1 expression was induced and sustained in the endothelial cell layer.
Conclusions
Low wall shear stress concomitant with turbulent flow contributes to sustained expression of MCP-1 in endothelial cells and presumably plays a role in facilitating macrophage infiltration and exacerbating inflammation, which leads to enlargement or rupture.
Introduction
As medical treatments have advanced, the outcomes for patients with various diseases have greatly improved. However, in cases of subarachnoid hemorrhage, this is not necessarily true. Subarachnoid hemorrhage often causes sudden death without a chance for therapeutic intervention, even in young patients, and its prognosis is still quite poor regardless of improvements in neurocritical care [1]. Intracranial aneurysm (IA), histologically characterized as a lesion with disrupted internal elastic lamina and excessive degeneration of media, is a major cause of subarachnoid hemorrhage [2, 3]. Considering the poor outcomes once after subarachnoid hemorrhage occurs [1] and the high incidence of IA in the general population [2], appropriate treatment to prevent rupture is crucial. However, there is no practicable medical treatment available for patients with IAs to prevent enlargement or rupture. The primary reason underlying this reality is that the detailed mechanisms regulating IA formation and progression remain to be elucidated.
Experimental studies using an animal model of IA have clarified the crucial role of persistent inflammation, presumably triggered by high wall shear stress (WSS) loaded on intracranial arterial walls at bifurcation sites and maintained/amplified by the formation of a positive feedback loop that includes the cyclooxygenase (COX)-2-prostaglandin (PG) E2-EP2-NF-κB cascade in endothelial cells (ECs) [4–6]. Computational fluid dynamic (CFD) analyses of human intracranial artery or IA lesions have supported this notion because high WSS can be detected at the prospective site of IA formation or the neck portion of IAs [7, 8]. Because the pharmacological inhibition of pro-inflammatory factors or genetic deletion of pro-inflammatory genes significantly suppresses not only the incidence but also the enlargement of IAs [4, 5, 9–11], long-lasting inflammation in intracranial arteries plays a role in both these steps. In terms of inflammation, therefore, the initiation and enlargement/progression of IAs share the same machinery. However, the hemodynamic status during these two steps is completely opposite. In the dome of growing IAs, the region of low WSS significantly overlaps with that of the enlarging portion [12, 13], suggesting the role of low WSS in the enlargement of IAs. Importantly, at the rupture point of IAs, the presence of low WSS and concomitant turbulent flow have also been demonstrated [14, 15], although there is a controversy regarding WSS status relating with the rupture of IAs [16]. However, the contribution of low WSS with or without concomitant turbulent flow loaded on the ECs of IA walls to enlargement and rupture remains to be elucidated. Considered with the fact that the target of treatment is of course pre-existing IAs and that a recent cohort study in Japan clearly demonstrated the positive correlation of the size of IAs with the annual risk of rupture [17], factors induced in ECs under low WSS condition may be good candidates for development of therapeutic drugs to prevent enlargement and presumably rupture of pre-existing IAs.
In the present study, we loaded WSS calculated from human cases by three-dimensional (3D) computational simulation on primary culture of ECs and examined the change in gene expression profiles and the temporal sequence of their expression in lesions.
Materials and methods
Data acquisition
Usage of 3D-computational tomography angiography (3D-CTA) data in the present study was approved by the review committee of the National Hospital Organization, Kyoto Medical Center in Kyoto, Japan.
Three patients with unruptured aneurysm were enrolled in the present study. In order to calculate WSS acting on the dome of human IAs and parent arteries, we employed three patient-specific, anatomically realistic arterial geometries of unruptured IAs. The geometries were segmented precisely from the volume data set of 3D-CTA using the Vascular Modeling Toolkit (VMTK) [18]. To apply CFD technique to these geometries, we generated high-quality computational meshes, including wall boundary-fitted layers, which were necessary to capture the steep velocity profiles near the wall boundary and to calculate WSS accurately, using the Mixed-Element Grid Generator in 3 Dimensions (MEGG3D) [19, 20]. A well-validated CFD solver, OpenFOAM, was used to perform blood flow analyses. In the CFD analyses, blood was treated as a Newtonian fluid and the wall was assumed to be rigid. We used patient-specific flow velocities measured with the ultrasound Doppler technique as the inlet boundary condition of the CFD analyses.
Primary culture of endothelial cells from human carotid artery
The primary culture of ECs from human carotid artery was purchased from Cell Applications (San Diego, CA, USA). The characteristics of these cells were confirmed to be compatible with those of ECs in some methods, including immunohistochemistry [4].
Loading shear stress
The primary culture of ECs, cultured on gelatin-coated glass slides, were loaded with a shear stress at 0.05 or 3.0 Pa according to the result from CFD analyses with a custom-made apparatus, as previously described [21]. After 24 h of shear stress loading, the cells were harvested. In addition, turbulent flow was loaded on ECs with a custom-made apparatus, as previously reported [22].
RNA extraction and cap analysis of gene expression analysis
Using an RNeasy Plus Mini Kit (QIAGEN, Hilden, Germany), total RNA was prepared from ECs loaded on shear stress or kept in a static condition as a control; the quality was checked by an RNA analyzer. RNA sequence-based cap analysis of gene expression (CAGE) analysis, which was conducted by the Genome Network Analysis Support Facility (GeNAS) at RIKEN (http://www.osc.riken.jp/genas/), was employed to obtain the gene expression profile [23, 24]. Pathway and gene ontology (GO) analyses were performed with the Platform for Drug Discovery housed at the Data Analysis Center of the National Institute of Genetics (http://cell-innovation.nig.ac.jp/wiki2/tiki-index.php?page=5.+CAGE) using peak data obtained by the CAGE analysis.
Quantitative real time PCR
Total RNA was prepared as described in the previous section and reverse-transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Life Technologies Corporation, Carlsbad, CA). Then, quantitative real time PCR (RT-PCR) was performed with the SYBR Premix Ex Taq II (Takara, Shiga, Japan) and Real Time System CFX96 (Bio-Rad Laboratories, Irvine, CA). β-actin was used as an internal control. For quantification, the second derivative maximum method was used for crossing point determination. Primer sets used are listed in Table 1.
Table 1.
List of primer sets used in the present study
| Gene name | Reference Sequence | forward primer (5′ → 3′) | reverse primer (5′ → 3′) |
|---|---|---|---|
| ACTB | NM_001101.3 | CAT ACT CCT GCT TGC TGA TCC | GAT GCA GAA GGA GAT CAC TGC |
| CCL2 | NM_002982.3 | AGC TTC TTT GGG ACA CTT GC | ATA GCA GCC ACC TTC ATT CC |
| CCNB | NM_031966.3 | GGA TCA GCT CCA TCT TCT GC | TTT GGT TGA TAC TGC CTC TCC |
| CDCA7 | NM_031942.3 | GAA ACT AAG CGG TTG GAG AGG | TTC TGG AGC ATC ACA GAA GG |
| CDC20 | NM_001255.2 | CAG AGC ACA CAT TCC AGA TGC | GTT CCT CTG CAG ACA TTC ACC |
| CDK1 | NM_001786.4 | CTG GCC ACA CTT CAT TAT TGG | GCA CCA TAT TTG CTG AAC TAG C |
| CXCL6 | NM_002993.3 | TAG TGG TCA AGA GAG GGT TCG | GAG GGA TGA ATG CAG ATA AAG G |
| CX3CL1 | NM_002996.3 | CGT CAA AGG GAA CCT CTA ACC | CTT GAC CAT TCT CCA CCT TCC |
| E2F1 | NM_005225.2 | CGT TGG TGA TGT CAT AGA TGC | CAC TGA ATC TGA CCA CCA AGC |
| FANCI | NM_001113378.1 | TTT AAC AAG GTG TCC ACA CAG C | CTC TTC TCA GGC AAC CCT ACC |
| HIST1H1D | NM_005320.2 | CTC CTT AGA AGC TGC CAC TGC | TCC ACT TGC TCC TAC CAT TCC |
| HIST1H1E | NM_005321.2 | GCC TTC TTG TTG AGT TTG AAG G | GAC GTG GAG AAG AAC AAC AGC |
| HIST1H2AH | NM_080596.2 | CTG GAT ATT GGG CAA GAC ACC | ACA AGA AGA CCC GTA TCA TCC |
| HIST1H2BO | NM_003527.4 | GAG CTG GTG TAC TTG GTG ACG | CTG GCG CAT TAC AAC AAG C |
| HIST1H2I | NM_003525.1 | ACT TGG AGC TGG TGT ACT TGG | ACT TCC AGG GAG ATC CAA AGC |
| KIF11 | NM_004523.3 | CTG ATC AAG GAG ATG TTC ACG | TGG AAC AGG ATC TGA AAC TGG |
| KIF15 | NM_020242.1 | TGA AGA AAG CTC CTT GTC AGC | GAC CAA ACA GCA GGA AGA GC |
| MYBL2 | NM_002466.3 | GAG GCT GGA AGA GTT TGA AGG | CTC TGG CTC TTG ACA TTG TGG |
| NOS3 | NM_000603.4 | GTC CAG GAA GAA GGT GAG AGC | CAG TAG AGC AGC TGG AGA AGG |
| NUF21 | NM_145697.2 | CAC TTC CAA CTG ACA TGA AGG | TGA AAG ATA CGG TCC AGA AGC |
| SELE | NM_000450.2 | GTC TTG GTC TCT TCA CCT TTG C | AAA GAC TCA GTG TTC CCT TTC C |
| VCAM1 | NM_001078.3 | ACT TTG ACT TCT TGC TCA CAG C | CTG TGC AAA TCC TTG ATA CTG C |
Rodent IA models, histological analysis, and immunohistochemistry of induced IAs
All of the following experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Kyoto University Graduate School of Medicine.
Sprague-Dawley rats were purchased from Japan SLC (Shizuoka, Japan). To induce IA, male 7-week-old rats were subjected to ligation of the left carotid artery and ligation of the left renal artery under general anesthesia by intraperitoneal injection of pentobarbital sodium (50 mg/kg). Animals were fed a special chow containing 8 % sodium chloride and, in some experiments, 0.12 % 3-aminopropionitrile (Tokyo Chemical Industry, Tokyo, Japan), an inhibitor of lysyl oxidase that catalyzes cross-linking of collagen and elastin. The procedure to induce IAs (defined as “aneurysm induction”) was designed to increase the local hemodynamic stress on the right bifurcation site of the anterior cerebral artery and olfactory artery a contralateral side of carotid ligation [4, 25, 26]. For histological analyses, animals were deeply anesthetized by intraperitoneal injection of a lethal dose of pentobarbital sodium and transcardinally perfused with a fixative, 4 % paraformaldehyde. Then, the circle of Willis including IA lesion was dissected out and serial frozen sections of 5-μm thickness were prepared. For immunohistochemistry, after blocking with 3 % donkey serum (Jackson ImmunoResearch, Baltimore, MD), the sections were incubated with primary antibodies followed by incubation with secondary antibodies conjugated with fluorescent dye (Jackson ImmunoResearch). Finally, immunofluorescence images were acquired with a confocal fluorescence microscope system (Lsm710; Carl Zeiss Microscopy GmBH, Gottingen, Germany). The following primary antibodies were used: mouse monoclonal anti-smooth muscle alpha actin antibody (#MS113; Thermo Scientific, Waltham, MA), goat polyclonal anti-MCP-1 antibody (#sc-1785; Santa Cruz Biotechnology, Dallas, TX), goat polyclonal anti-CX3CL1 antibody (#AF537; R&D Systems, Minneapolis, MN).
Detection of proliferative cells in rat intracranial arteries
To detect proliferative cells in vivo, rats were given an intraperitoneal injection of 5-ethynyl-2′-deoxyuridine (EdU, 80 mg/kg) and, after 24 h, a specimen of intracranial arteries was prepared as described above. EdU intercalated in genome was detected and visualized by click reaction using Alexa488-conjugated azide according to the manufacturer’s instruction (Click-iT EdU Imaging Kits, Life Technologies).
Statistics
Data are shown in mean ± SEM. Statistical comparisons between more than two groups were conducted using the Kruskal-Wallis test followed by post-hoc Dunn’s test. P less than 0.05 was considered statistically significant.
Results
Calculation of wall shear stress
Three patients with unruptured IAs were enrolled in the present study. WSS loaded on the dome of each IA and the corresponding parent artery was calculated by CFD analysis (Fig. 1). Mean WSS calculated from the three cases were 0.017 Pa (±0.0027, n = 3) at the minimum value in the dome of IAs or 6.50 Pa (±0.66, n = 3) in the neck portion of IAs, respectively. Notably, WSS in the dome of IAs were strikingly lower compared with those in the parent artery (3.0 Pa), which is consistent with previous reports [12, 13].
Fig. 1.
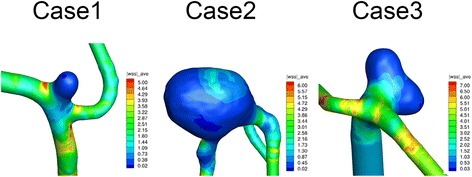
CFD analysis of intracranial aneurysm from three human patients. The value of wall shear stress (WSS) is indicated in the color phase
Change of gene expression profile
To verify the effect of each WSS on gene expression, a primary culture of ECs from human carotid artery was cultured and shear stress corresponding to the value (3.0 Pa, 0.05 Pa) obtained in CFD was loaded on these cells for 24 h. In addition, because some studies have demonstrated the presence of turbulent flow in addition to low WSS at the rupture point of IAs in human cases [14, 15], cultured ECs were loaded with the turbulent flow in combination with low WSS. Then, after the quality of purified RNA from the stimulated cells was confirmed by RNA analyzer (RIN > 9.50), the gene expression profile was obtained by RNA sequence–based CAGE analyses. The read count, which was successfully mapped to the reference genome using the MOIRAI workflow system [27], was around 10,000,000, enough for further analyses (Table 2). Then, profiling differentially expressing genes (peaks in RNA sequence mapped to the reference genome) between experimental groups was obtained by a pipeline, RECLU [28]. Here, the RECLU pipeline extracted two types of clusters, top peaks and bottom ones [28]. Briefly, top peaks or bottom ones represent the narrowest or broadest reproducible peaks in an RNA sequence [28]. Upregulated or downregulated peaks in a 0.05 Pa–loaded group compared with those in a 3.0 Pa–loaded group were identified; the total number was around 300 (Table 3). In another comparison, differentially expressed genes were also identified (Table 3). For the GO analyses, GO terms related with cell division/proliferation, such as nucleosome, mitotic cell cycle, and DNA replication, were highly overrepresented in the 0.05 Pa + turbulent flow–loaded group compared with the 3.0 Pa-loaded group (Table 4). Indeed, overexpressed genes in the 0.05 Pa + turbulent flow–loaded group included those related with cell division and proliferation (Table 5), which is consistent with a previous study that turbulent flow enhances proliferation of ECs [29]. Underrepresented terms in this comparison included IL-33 receptor activity and IL-1 receptor activity, but the p value was much less compared with that of the overrepresented terms (Table 4). In comparisons between the 0.05 Pa-loaded group and the 3.0 Pa-loaded group or 0.05 Pa + turbulent flow–loaded group and 0.05 Pa-loaded group, GO terms related with cell division/proliferation, such as mitosis, were flagged as overrepresented in the 0.05 Pa-loaded (Table 6) or 0.05 Pa + turbulent flow–loaded group (Table 7), respectively, suggesting that cell division/proliferation was enhanced in a low WSS condition and further amplified by turbulent flow. In pathway analyses, biological processes consistently related with cell division/proliferation, such as telomere maintenance, mitotic cell cycle, and DNA replication, were overrepresented in a set of genes from the low WSS-loaded group compared with that of the 3.0 Pa-loaded group (Table 8).
Table 2.
Read count obtained in RNA-sequence-based CAGE analysis
| sample No. | Mapped Read Count | rRNA read count | |
|---|---|---|---|
| 0.05Pa | 1 | 11,216,634 | 2,265,251 |
| 2 | 8,936,578 | 2,108,157 | |
| 3.0Pa | 1 | 11,293,958 | 1,776,765 |
| 2 | 9,160,492 | 2,264,215 | |
| 0.05Pa + TF | 1 | 6,765,681 | 1,171,547 |
| 2 | 9,547,702 | 1,795,395 |
TF turbulent flow
Table 3.
Number of differentially expressed peaks between each comparison
| up-regulated peaks | down-regulated peaks | |||
|---|---|---|---|---|
| Top peaks | Bottom peaks | Top peaks | Bottom peaks | |
| 3.0Pa vs. 0.05Pa | 314 | 331 | 261 | 385 |
| 3.0Pa vs. 0.05Pa + TF | 604 | 562 | 496 | 395 |
| 0.05Pa vs. 0.05Pa + TF | 78 | 214 | 74 | 148 |
TF turbulent flow
Table 4.
GO term analysis of differentially expressing genes between 3.0Pa-loaded and 0.05Pa + turbulent flow-loaded group
| Over-presented term in 0.05Pa + turbulent flow-loaded group | |||
| Accession | Term | P-value | FDR |
| GO:0000786 | nucleosome | 7.7E-54 | 3.0E-51 |
| GO:0006334 | nucleosome assembly | 6.4E-53 | 1.3E-50 |
| GO:0000278 | mitotic cell cycle | 3.8E-26 | 5.1E-24 |
| GO:0007067 | mitosis | 9.7E-26 | 9.6E-24 |
| GO:0046982 | protein heterodimerization activity | 9.7E-24 | 7.7E-22 |
| GO:0005654 | nucleoplasm | 5.6E-19 | 3.7E-17 |
| GO:0060968 | regulation of gene silencing | 8.7E-15 | 4.7E-13 |
| GO:0003677 | DNA binding | 9.5E-15 | 4.7E-13 |
| GO:0005819 | spindle | 8.5E-14 | 3.7E-12 |
| GO:0000775 | chromosome, centromeric region | 3.5E-13 | 1.4E-11 |
| GO:0005634 | nucleus | 4.8E-13 | 1.7E-11 |
| GO:0000777 | condensed chromosome kinetochore | 9.2E-13 | 3.1E-11 |
| GO:0034080 | CENP-A containing nucleosome assembly at centromere | 5.9E-12 | 1.8E-10 |
| GO:0007059 | chromosome segregation | 1.6E-11 | 4.5E-10 |
| GO:0006260 | DNA replication | 1.1E-10 | 2.9E-09 |
| GO:0030496 | midbody | 1.6E-10 | 3.9E-09 |
| GO:0031536 | positive regulation of exit from mitosis | 2.1E-10 | 4.8E-09 |
| GO:0000922 | spindle pole | 3.6E-10 | 7.8E-09 |
| GO:0005876 | spindle microtubule | 1.9E-09 | 4.0E-08 |
| GO:0006281 | DNA repair | 2.5E-09 | 5.0E-08 |
| Under-presented term in 0.05Pa + turbulent flow-loaded group | |||
| Accession | Term | P-value | FDR |
| GO:0002113 | interleukin-33 binding | 2.9E-07 | 5.9E-05 |
| GO:0002114 | interleukin-33 receptor activity | 2.9E-07 | 5.9E-05 |
| GO:0043032 | positive regulation of macrophage activation | 7.9E-07 | 1.1E-04 |
| GO:0090197 | positive regulation of chemokine secretion | 1.0E-06 | 1.1E-04 |
| GO:0004908 | interleukin-1 receptor activity | 1.5E-06 | 1.2E-04 |
| GO:0002826 | negative regulation of T-helper 1 type immune response | 1.7E-06 | 1.2E-04 |
| GO:0032754 | positive regulation of interleukin-5 production | 3.6E-05 | 2.1E-03 |
| GO:0070698 | type I activin receptor binding | 5.9E-05 | 3.0E-03 |
| GO:0071310 | cellular response to organic substance | 1.1E-04 | 4.9E-03 |
| GO:0030617 | inhibitory cytoplasmic mediator activity | 1.2E-04 | 4.9E-03 |
FDR false discovery rate
Table 5.
List of over-expressed genes in 0.05Pa + turbulent flow-loaded group compared with 3.0Pa-loaded group
| Gene Symbol | Gene Accession | Log Fold Change | P-value | Peaks |
|---|---|---|---|---|
| PRND | ENST00000305817 | 5.780 | 6.90E-14 | BOTTOM |
| LTB | ENST00000429299 | 6.170 | 3.80E-10 | TOP |
| KIF15 | ENST00000438321 | 6.110 | 8.80E-10 | TOP |
| KIF20A | ENST00000508792 | 8.290 | 1.40E-09 | TOP |
| CENPA | ENST00000233505 | 6.020 | 1.60E-09 | TOP |
| NCAPH | ENST00000240423 | 7.770 | 1.80E-07 | TOP |
| KCNAB1 | ENST00000389634 | 7.430 | 4.60E-06 | TOP |
| LRRC17 | ENST00000498487 | 7.270 | 6.00E-06 | TOP |
| ASPM | ENST00000294732 | 7.040 | 4.00E-05 | TOP |
| CDC45 | ENST00000404724 | 6.890 | 0.00011 | TOP |
| RP11-322E11.5 | ENST00000593122 | 6.770 | 0.00021 | TOP |
| HIST1H2AH | ENST00000377459 | 6.710 | 0.00026 | TOP |
| SPC24 | ENST00000423327 | 6.830 | 0.0004 | TOP |
| FAM64A | ENST00000572447 | 6.510 | 0.0018 | TOP |
| MCM7 | ENST00000489841 | 6.360 | 0.0029 | TOP |
| HIST1H1B | ENST00000331442 | 6.100 | 0.0047 | BOTTOM |
| DAB2IP | ENST00000436835 | 6.100 | 0.0047 | BOTTOM |
| RRM2 | ENST00000459969 | 6.190 | 0.0052 | TOP |
| DEPDC1B | ENST00000453022 | 6.100 | 0.0082 | TOP |
| EXO1 | ENST00000423131 | 6.100 | 0.009 | TOP |
| PDLIM3 | ENST00000284770 | 6.000 | 0.01 | TOP |
| CENPM | ENST00000402420 | 6.000 | 0.015 | TOP |
| PAK1 | ENST00000528592 | 6.000 | 0.015 | TOP |
| ME3 | ENST00000526834 | 5.900 | 0.015 | TOP |
| HIST1H2BI | ENST00000377733 | 5.780 | 0.017 | BOTTOM |
| TTK | ENST00000509313 | 5.900 | 0.019 | TOP |
| ITM2B | ENST00000463839 | 6.100 | 0.02 | TOP |
| ERCC6L | ENST00000373657 | 5.790 | 0.024 | TOP |
| KIT | ENST00000514582 | 5.790 | 0.024 | TOP |
| CENPA | ENST00000419525 | 5.900 | 0.025 | TOP |
Table 6.
Over-presented term in 0.05Pa -loaded group compared with 3.0Pa-loaded group in GO term analysis
| Accession | Term | P-value | FDR |
|---|---|---|---|
| GO:0000786 | nucleosome | 3.5E-27 | 4.9E-25 |
| GO:0006334 | nucleosome assembly | 1.5E-24 | 1.1E-22 |
| GO:0046982 | protein heterodimerization activity | 1.3E-14 | 6.0E-13 |
| GO:0060968 | regulation of gene silencing | 4.9E-10 | 1.7E-08 |
| GO:0000278 | mitotic cell cycle | 3.4E-09 | 9.6E-08 |
| GO:0003677 | DNA binding | 4.7E-09 | 1.1E-07 |
| GO:0005654 | nucleoplasm | 7.3E-09 | 1.5E-07 |
| GO:0031145 | anaphase-promoting complex-dependent proteasomal ubiquitin-dependent protein catabolic process | 1.9E-06 | 3.3E-05 |
| GO:0006260 | DNA replication | 1.3E-05 | 1.7E-04 |
| GO:0010994 | free ubiquitin chain polymerization | 1.3E-05 | 1.7E-04 |
| GO:0000083 | regulation of transcription involved in G1/S transition of mitotic cell cycle | 1.4E-05 | 1.7E-04 |
| GO:0051437 | positive regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle | 2.8E-05 | 3.3E-04 |
| GO:0051439 | regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle | 3.3E-05 | 3.4E-04 |
| GO:0043565 | sequence-specific DNA binding | 3.4E-05 | 3.4E-04 |
| GO:0031536 | positive regulation of exit from mitosis | 4.6E-05 | 4.3E-04 |
| GO:0007067 | mitosis | 5.4E-05 | 4.7E-04 |
| GO:0008054 | cyclin catabolic process | 6.6E-05 | 5.4E-04 |
| GO:0000281 | mitotic cytokinesis | 7.2E-05 | 5.6E-04 |
| GO:0005634 | nucleus | 8.0E-05 | 5.9E-04 |
| GO:0034501 | protein localization to kinetochore | 1.0E-04 | 7.1E-04 |
FDR false discovery rate
Table 7.
Over-presented term in 0.05Pa + turbulent flow-loaded group compared with 0.05Pa-loaded group in GO term analysis
| Accession | Term | P-value | FDR |
|---|---|---|---|
| GO:0000775 | chromosome, centromeric region | 9.3E-07 | 3.6E-05 |
| GO:0007067 | mitosis | 9.9E-07 | 3.6E-05 |
| GO:0000786 | nucleosome | 1.9E-06 | 4.7E-05 |
| GO:0000278 | mitotic cell cycle | 8.4E-06 | 1.5E-04 |
| GO:0031536 | positive regulation of exit from mitosis | 1.2E-05 | 1.7E-04 |
| GO:0030496 | midbody | 1.8E-05 | 2.1E-04 |
| GO:0006334 | nucleosome assembly | 2.3E-05 | 2.4E-04 |
| GO:0000079 | regulation of cyclin-dependent protein kinase activity | 6.5E-05 | 5.9E-04 |
| GO:0007059 | chromosome segregation | 1.4E-04 | 1.2E-03 |
| GO:0051382 | kinetochore assembly | 2.0E-04 | 1.5E-03 |
| GO:0000070 | mitotic sister chromatid segregation | 2.8E-04 | 1.9E-03 |
| GO:0000922 | spindle pole | 3.2E-04 | 1.9E-03 |
| GO:0051301 | cell division | 3.7E-04 | 2.1E-03 |
| GO:0000086 | G2/M transition of mitotic cell cycle | 8.6E-04 | 4.5E-03 |
| GO:0007094 | mitotic spindle assembly checkpoint | 9.3E-04 | 4.5E-03 |
| GO:0005164 | tumor necrosis factor receptor binding | 1.3E-03 | 6.0E-03 |
| GO:0000777 | condensed chromosome kinetochore | 2.9E-03 | 1.2E-02 |
| GO:0007165 | signal transduction | 3.8E-03 | 1.5E-02 |
| GO:0046982 | protein heterodimerization activity | 3.9E-03 | 1.5E-02 |
| GO:0000910 | cytokinesis | 4.1E-03 | 1.5E-02 |
FDR false discovery rate
Table 8.
Over-represented pathways in 0.05Pa-loaded or 0.05Pa + turbulent flow-loaded group compared with 3.0Pa-loaded group
| Comparison | Peaks | REACTOME ID | Pathway | P-value | FDR |
|---|---|---|---|---|---|
| 3.0Pa < 0.05Pa + TF | Top peaks | REACT_7970 | Telomere Maintenance | 7.53E-12 | 1.36E-10 |
| REACT_8017 | APC-Cdc20 mediated degradation of Nek2A | 3.74E-03 | 2.32E-02 | ||
| REACT_152 | Cell Cycle, Mitotic | 3.86E-03 | 2.32E-02 | ||
| Bottom peaks | REACT_7970 | Telomere Maintenance | 2.67E-14 | 4.01E-13 | |
| REACT_152 | Cell Cycle, Mitotic | 8.97E-03 | 6.73E-02 | ||
| 3.0Pa < 0.05Pa | Top peaks | REACT_7970 | Telomere Maintenance | 5.21E-08 | 5.21E-07 |
| REACT_383 | DNA Replication | 2.24E-02 | 1.12E-01 | ||
| REACT_1538 | Cell Cycle Checkpoints | 8.86E-02 | 2.48E-01 | ||
| REACT_6850 | Cdc20:Phospho-APC/C mediated degradation of Cyclin A | 1.20E-01 | 2.48E-01 | ||
| REACT_152 | Cell Cycle, Mitotic | 1.35E-01 | 2.48E-01 | ||
| REACT_8017 | APC-Cdc20 mediated degradation of Nek2A | 1.49E-01 | 2.48E-01 | ||
| Bottom peaks | REACT_7970 | Telomere Maintenance | 1.16E-06 | 6.96E-06 | |
| REACT_152 | Cell Cycle, Mitotic | 1.03E-02 | 3.09E-02 | ||
| REACT_383 | DNA Replication | 3.15E-02 | 6.30E-02 |
FDR false discovery rate
In addition to genes located at the head of the list, which included many genes related with cell division or proliferation (Table 5), upregulated genes in the 0.05 Pa or 0.05 Pa + turbulent flow–loaded group included chemoattractants or adhesion molecules for inflammatory cells. Next, we focused on these chemoattractants and adhesion molecules because inflammatory cells, especially macrophages, play a crucial role in IA formation/progression [10, 11, 30]. In the low WSS condition (0.05 Pa), expression of CX3CL1, a chemoattractant for monocytes, and VCAM-1 were induced compared with the 3.0 Pa-loaded group (Table 9). Presence of turbulent flow concomitant with low WSS further augmented the expression of chemoattractants and adhesion molecules such as CCL2 (gene for MCP-1), a chemoattractant for macrophages, CXCL6, a chemoattractant for granulocytes, and SELE (gene for E-selectin) (Table 9). These results suggest that turbulent flow with concomitant low WSS induces chemoattractants and adhesion molecules for inflammatory cells in ECs that in turn recruit cells in lesions to evoke inflammation.
Table 9.
Up-regulation of chemoattractants and adhesion molecules for inflammatory cells in 0.05Pa or 0.05Pa + turbulent flow-loaded group
| 3.0Pa < 0.05Pa | ||||||
| Symbol | Gene Accession | Start | End | Log Fold Change | P-value | Peaks |
| CX3CL1 | ENST00000006053 | 57406348 | 57406458 | 3.20 | 2.9E-03 | BOTTOM |
| CX3CL1 | ENST00000563383 | 57406400 | 57406402 | 3.69 | 2.1E-02 | TOP |
| VCAM1 | ENST00000370119 | 101185296 | 101185298 | 4.89 | 1.7E-03 | TOP |
| VCAM1 | ENST00000370119 | 101185284 | 101185300 | 4.37 | 6.9E-06 | BOTTOM |
| 0.05Pa < 0.05Pa + turbulent flow | ||||||
| Symbol | Gene Accession | Start | End | Log Fold Change | P-value | Peaks |
| CCL2 | ENST00000225831 | 32582302 | 32582304 | 2.27 | 2.4E-04 | TOP |
| CCL2 | ENST00000225831 | 32582222 | 32582386 | 2.06 | 1.2E-06 | BOTTOM |
| CXCL1 | ENST00000509101 | 74735046 | 74735111 | 1.45 | 1.2E-02 | BOTTOM |
| CXCL6 | ENST00000515050 | 74702302 | 74702396 | 1.87 | 2.6E-02 | BOTTOM |
| ICAM1 | ENST00000423829 | 10381756 | 10381818 | 1.39 | 7.5E-03 | BOTTOM |
| ICAM2 | ENST00000581417 | 62084077 | 62084249 | 1.38 | 5.5E-03 | BOTTOM |
| SELE | ENST00000367779 | 169703219 | 169703222 | 2.95 | 7.0E-03 | BOTTOM |
| VCAM1 | ENST00000370119 | 101185275 | 101185302 | 1.33 | 2.6E-02 | BOTTOM |
| 3.0Pa < 0.05Pa + turbulent flow | ||||||
| Symbol | Gene Accession | Start | End | Log Fold Change | P-value | Peaks |
| CCL2 | ENST00000225831 | 32582302 | 32582304 | 2.08 | 2.6E-03 | TOP |
| CCL2 | ENST00000225831 | 32582222 | 32582386 | 1.94 | 4.4E-04 | BOTTOM |
| CCL2 | ENST00000225831 | 32582222 | 32582224 | 2.15 | 4.5E-02 | TOP |
| CX3CL1 | ENST00000563383 | 57406400 | 57406402 | 2.19 | 4.7E-04 | TOP |
| CX3CL1 | ENST00000006053 | 57406348 | 57406402 | 2.03 | 1.7E-04 | BOTTOM |
| CXCL11 | ENST00000306621 | 76957183 | 76957237 | 2.89 | 1.4E-02 | TOP |
| CXCL6 | ENST00000515050 | 74702302 | 74702396 | 2.11 | 1.6E-02 | BOTTOM |
| SELE | ENST00000367779 | 169703220 | 169703222 | 3.69 | 1.5E-02 | TOP |
| SELE | ENST00000367779 | 169703219 | 169703222 | 2.94 | 2.8E-03 | BOTTOM |
| VCAM1 | ENST00000370119 | 101185275 | 101185302 | 1.33 | 2.6E-02 | BOTTOM |
RT-PCR analyses of genes differentially expressed under each WSS condition
To confirm results of the gene expression profile obtained by the RNA sequence–based CAGE analysis, we examined the expression of selected genes by RT-PCR and found that the results could be reproduced (Fig. 2). For example, genes related to cell proliferation/division, such as histone subunit genes (HIST1HID, etc.), cyclin or cyclin-dependent kinase (CDK1, CCNB), transcription factor regulating cell cycle (E2F1), genes regulating cell division (CDC20, CDCA7), the nuclear protein promoting cell cycle (MYBL2), gene regulating chromatin segregation (NUF2), kinesins that transport materials in cytoplasm (KIF11, KIF15), or gene encoding protein stabilizing DNA (FANCI) were induced in ECs loaded on low WSS (Fig. 2). Interestingly, NOS3 (eNOS) expression was downregulated under low WSS conditions in RT-PCR (Fig. 2), suggesting that there is a dysfunction of ECs in this WSS condition, as demonstrated by previous studies in which there was a loss of gap junction during IA formation [31] or a decrease of eNOS staining in the dome of IAs [32]. We also confirmed that the genes for chemoattractants and adhesion molecules, VCAM1, CX3CL1, SELE, CCL2, CXCL6, were induced under low WSS or low WSS with concomitant turbulent flow condition (Fig. 2) during the RNA sequence analysis (Table 9). Again, we noted the remarkable contribution of turbulent flow in addition to concomitant low WSS to induction of these genes.
Fig. 2.
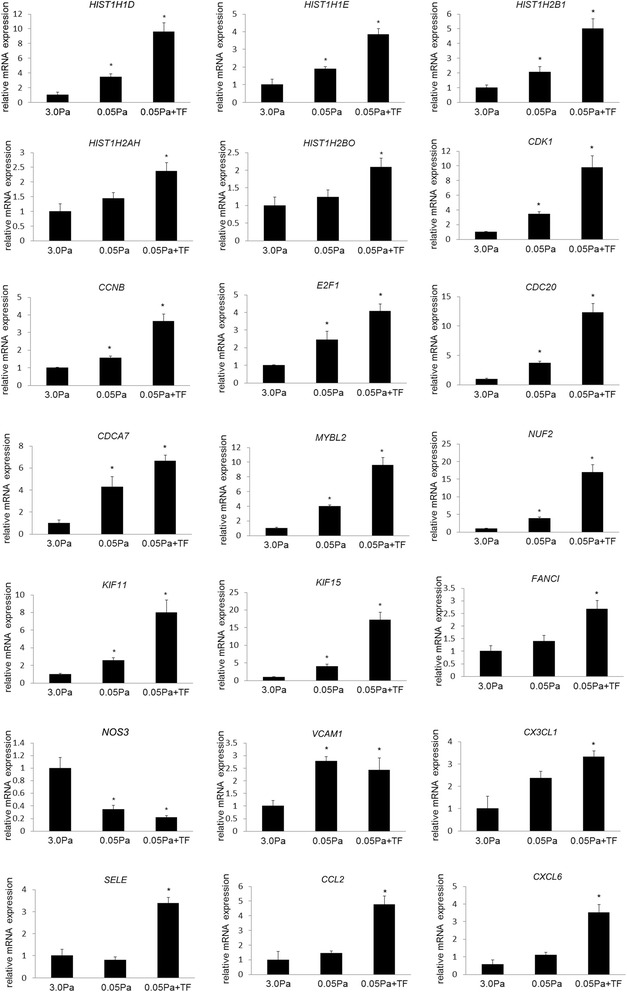
Induction of genes in cultured endothelial cells (ECs) loaded on low WSS and turbulent flow. RNA was purified from cultured ECs loaded on 3.0 Pa, 0.05 Pa, or 0.05 Pa with turbulent flow (TF), and gene expressions were analyzed by RT-PCR analysis. All bars indicate mean ± SEM (n = 3). * p < 0.05 compared with 3.0 Pa-loaded group
Proliferative status of intracranial arterial walls during IA formation
In response to the results of an in vitro study that showed pathways related with proliferation of ECs are overrepresented under low WSS and concomitant turbulent flow, we examined the proliferation of cells in intracranial arterial walls in vivo. In this experiment, we used Edu, which was intercalated in the genome during replication of proliferative cells as an alternative of thymidine, to detect proliferative cells in rat tissue. In the small intestine, which was used as a positive control, proliferative cells incorporated with EdU were detected as Alexa488-positive cells in all rats that were examined (Fig. 3a), confirming the proper procedure to detect proliferative cells in vivo. In intracranial arterial walls before and after IA induction, a small number of Alexa488-positive proliferative cells were identified, but no signaling was detectable in the endothelial cell layer (Fig. 3b and c), suggesting that ECs in intracranial arteries do not actively proliferate.
Fig. 3.
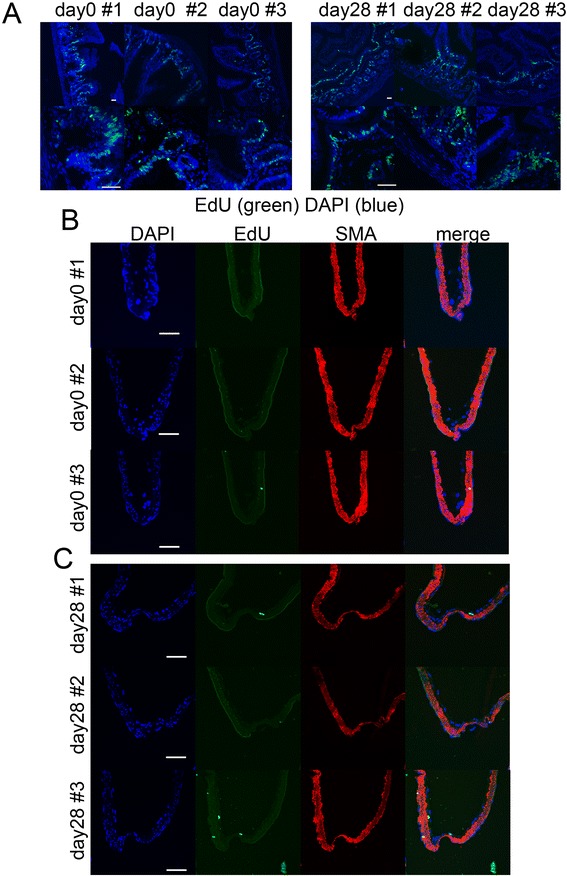
Detection of proliferative cells in intracranial aneurysm lesions of rat. a, Proliferative cells in the small intestine of rats after intraperitoneal injection of EdU (80mg/kg) were detected by click reaction (green color). Merged images with nuclear staining by DAPI are shown. Magnified images are shown in the lower panels. Scale bar = 50μm. b and c, Proliferative cells in intracranial arterial walls of rats before (day 0, b) or after (day 28, c) aneurysm induction were labeled after intraperitoneal injection of EdU. Immunostaining for α-smooth muscle actin (SMA) is shown to visualize the arterial walls. Scale bar = 50μm
Temporal sequence of MCP-1 and CXCL3 expression in IA lesions in vivo
To verify the in vivo relevance of our in vitro study, we examined the temporal sequence of MCP-1 and CX3CL1 expression in the rat IA model. We selected these two molecules because they could recruit macrophages, which serve as major inflammatory cells in lesions both in human and animal models [33, 34]. In addition, MCP-1–mediated recruitment of macrophages plays a crucial role in IA formation/progression via the secretion of a variety of pro-inflammatory factors because genetic deletion or inhibition of MCP-1 has been shown to almost completely suppress IA formation in animal models [11, 30].
MCP-1 expression was only weakly detected in ECs of intracranial arteries before induction (Fig. 4a and b). Its intensity in immunostaining increased and spread to adventitia of arterial walls after IA induction, which was consistent with our previous study [30]. Importantly, expression of MCP-1 in ECs of IA walls was sustained, not decreased, during IA formation (Fig. 4a and b), suggesting the in vivo relevance of in vitro study.
Fig. 4.
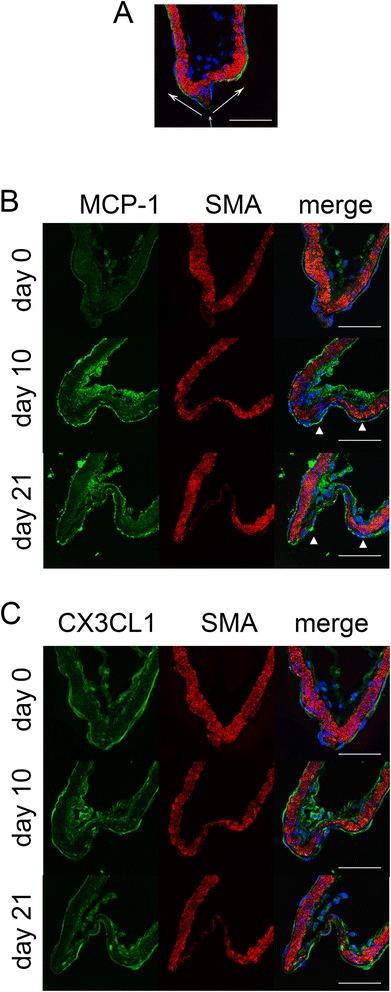
Induction of MCP-1 in the endothelial cell layer during intracranial aneurysm formation in rats. a, Representative image to show the anatomical structure of the bifurcation site of the intracranial artery. A merged image of immunohistochemistry for smooth muscle marker, α-smooth muscle actin (SMA, red), an endothelial cell marker, CD31 (green), and nuclear staining by DAPI (blue) is shown. White arrows indicate the direction of blood flow. Scale bar = 50μm. b and c, Expression of MCP-1 (b) and CX3CL1 (c) in the endothelial cell layer of arterial walls during IA formation. Immunohistochemistry for MCP-1 (green in b), CX3CL1 (green in c), SMA (red), and for the merged images with DAPI (blue) are shown. Scale bar = 50μm
In the case of CX3CL1, expression was persistently detectable in intracranial arteries, including endothelial cell layer, from day 0 to day 21 of IA induction (Fig. 4c). However, the signal intensity did not change during IA formation (Fig. 4c), which was consistent with our previous study of the gene expression profile of ECs from IA lesions [35], indicating the independence of CX3CL1 in IA formation.
Discussion
Recent studies have provided experimental evidence that long-lasting inflammatory responses in intracranial arteries play a crucial role in IA formation [10]. During this process, MCP-1-mediated macrophage recruitment and macrophage-evoked inflammation are critical for IA formation because of the genetic deletion of MCP-1, inhibition of MCP-1 by a dominant negative form (7-ND), or pharmacological depletion of macrophages that remarkably suppress IA formation [11, 30]. MCP-1 is induced in ECs in intracranial arteries under a high WSS condition loaded at an early stage of IA formation [30]. Although shear stress dramatically changes during IA progression, from high WSS at an early stage [7, 8] to low WSS, sometimes with turbulent flow, at a later stage [12–14], MCP-1 expression is sustained in ECs of IA lesions induced in a rat model both by immunostaining [30] and RT-PCR analysis [35]. Consistently, MCP-1 expression is also detected in ECs of human IA walls [30] in which low WSS is present. The current experiment addressed this issue and provided experimental evidence linking low WSS with concomitant turbulent flow and MCP-1 expression in ECs of IA walls.
MCP-1 expression under a low WSS stress condition is presumably critical for macrophage infiltration in pathological conditions, as observed in IA walls, to exacerbate inflammatory responses leading to progression of IAs. This is due to the fact that a low WSS condition facilitates adhesion of macrophages to ECs expressing MCP-1, but a high WSS condition interferes with adhesion. Furthermore, the present study again highlights the importance of MCP-1 over the entire period of IA formation/progression because induction of another chemoattractant for macrophages, CX3CL1, is not obvious during IA formation. Among genes induced under a low WSS condition in ECs, VCAM-1 [5] and E-selectin [36] induction in IA lesions, including ECs in the dome of a rat model, have been reported, suggesting the in vivo relevance of the present study. Consistent with our RNA-sequence and RT-PCR analyses that VCAM-1 can be induced under a low WSS condition, VCAM-1 expression in ECs in the dome of IAs induced in a rat model has been demonstrated [5]. However, the contribution of this molecule to IA formation and progression is not clear. E-selectin can also be induced in ECs in the dome of IAs during IA formation [36]. However, because inhibition of E-selectin expression by cimetidine failed to suppress infiltration of macrophages in lesion and IA formation [36], the contribution of E-selectin to IA formation could be interpreted as negative. Intriguingly, the present study clearly demonstrated the importance of concomitant turbulent flow with low WSS in the exacerbation of macrophage infiltration via MCP-1 induction. Since the media of IA walls becomes gradually thinner and smooth muscle cells in media are shed during IA progression, these histopathological changes of IA walls and MCP-1 induction under a low WSS condition remarkably facilitates macrophage infiltration and the resultant exacerbation of inflammation and tissue destruction that presumably leads to rupture of IAs. Indeed, massive macrophage infiltration of lesions has been reported in ruptured human IAs, but not in unruptured ones, suggesting the role of macrophages in the rupture of IAs. However, the rupture of IAs and resultant inflammatory response itself may greatly increase macrophage infiltration [37–39]. This hypothesis goes hand-in-hand with the observation that turbulent flow with low WSS can be detected at the rupture point of IAs [14, 15], although contribution of abnormally high WSS to the rupture of IAs is also presumed [16].
The present study includes several limitations. First, there are potential limitations related to CFD. Specifically, blood was treated as a Newtonian fluid and the wall was assumed to be rigid in our CFD simulations. These simplified properties might lead to a different state compared with the in vivo state. However, the effect of a non-Newtonian property on large artery hemodynamics is believed to be small [40, 41]. In addition, it has been reported that the overall feature of WSS distribution does not considerably change when incorporating wall deformation [42]. Thus, our CFD simulations provide a reasonable WSS approximation of the in vivo state. Another limitation is that our sample size was small (3 cases of IA). We infer that the present low-WSS characteristics in the dome are consistent among clinical cases, as demonstrated by previous studies [12, 13, 43]. However, future studies will have to allow for more realistic estimation of WSS, considering the wide range of geometries that intracranial aneurysms can have. The IA rat model used in the present study also has some intrinsic limitations. IAs induced in a rat model are not saccular with a narrow neck, but rather have a mountain-like shape with a wide neck. Further, this model does not allow for assessment of enlargement in the same animal due to the small size of the lesions nor rupture because of low incidence of spontaneous rupture [25]. Therefore, we exclusively analyzed the enlargement of IAs in animal models, and from this reference point we have provided evidence geared toward developing therapeutic drugs to prevent the enlargement of IAs. Here, because statins suppressed enlargement of IAs in a rat model [44–46] and usage of this class of drugs reduced the relative risk of subarachnoid hemorrhage in a Japanese case-control study [47], some drugs preventing progression of IAs in animal models should be considered for human IAs to prevent rupture. However, because approximately one third of human IAs rupture but the remaining never rupture [48], there may be different pathological processes at work. In addition, we used primary culture of ECs from the carotid artery, which is an extra-cranial artery, because we could not obtain ECs from human intracranial artery. Differences in the origins of ECs may influence results and, therefore, we need to pay careful attention when analyzing data. One final major limitation of the present study is that we could not reconstitute whole arterial walls, especially the physical or chemical interaction with adjacent cells or structures such as medial smooth muscle cells or internal elastic lamina. Indeed, turbulent flow concomitant with low WSS greatly activates proliferation of ECs in vitro, but not in IA walls in vivo. To examine the detailed interaction of hemodynamic forces with arterial walls, including ECs leading to pathological situations, organ culture using whole arterial walls may be necessary.
Conclusions
In the present study, we used primary culture of ECs loaded on shear stress, RNA sequencing of these cells, bioinformatic analysis of RNA-sequencing data, and verification of results from in vitro data using a rat model of IAs. These techniques showed that low wall shear stress with concomitant turbulent flow induced expressions of chemoattractant and adhesion molecules for macrophages such as MCP-1 in endothelial cells, which could be the mechanism behind sustained macrophage infiltration during IA progression and presumably rupture. In other words, we have clarified that MCP-1 expression is sustained during IA formation/progression independent of flow condition.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (#26861145, T.A.) and by Core Research for Evolutional Science and Technology (CREST) on Chronic Inflammation by the Japan Science and Technology Agency (S.N.).
We thank M. Kobori, K. Mizutani, and Y. Imai for technical assistance, and T. Arai and A. Washimi for secretarial assistance.
Abbreviations
- CAGE
cap analysis of gene expression
- CFD
computational fluid dynamic
- COX
cyclooxygenase
- EC
endothelial cell
- GO
gene ontology
- IA
intracranial aneurysm
- WSS
wall shear stress
Footnotes
Competing interests
T. A. and S. N. were supported by the Coordination Fund from Japan Science and Technology Agency and Astellas Pharma Inc. S.N. is a scientific advisor to Astellas Pharma. No potential conflicts of interest were disclosed by the other authors.
Authors’ contributions
TA conceived the study, carried out the molecular biological experiments, secured grants, and drafted the manuscript. KY and MF carried out the molecular biological experiments. YS carried out the 3-D computational simulations. SF corrected cases of aneurysms. SN participated in the design, secured grants, and assisted in coordinating the study. All authors read and approved the final manuscript.
References
- 1.van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet. 2007;369(9558):306–318. doi: 10.1016/S0140-6736(07)60153-6. [DOI] [PubMed] [Google Scholar]
- 2.Wiebers DO, Piepgras DG, Brown RD, Jr, Meissner I, Torner J, Kassell NF, Whisnant JP, Huston J, 3rd, Nichols DA. Unruptured aneurysms. J Neurosurg. 2002;96(1):50–51. doi: 10.3171/jns.2002.96.1.0050. [DOI] [PubMed] [Google Scholar]
- 3.Unruptured intracranial aneurysms--risk of rupture and risks of surgical intervention. International Study of Unruptured Intracranial Aneurysms Investigators (1998). N Engl J Med. 339 (24):1725-1733. doi:10.1056/NEJM199812103392401. [DOI] [PubMed]
- 4.Aoki T, Nishimura M, Matsuoka T, Yamamoto K, Furuyashiki T, Kataoka H, Kitaoka S, Ishibashi R, Ishibazawa A, Miyamoto S, Morishita R, Ando J, Hashimoto N, Nozaki K, Narumiya S. PGE2–EP2 signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-kappaB. Br J Pharmacol. 2011;163(6):1237–1249. doi: 10.1111/j.1476-5381.2011.01358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. NF-kappaB is a key mediator of cerebral aneurysm formation. Circulation. 2007;116(24):2830–2840. doi: 10.1161/CIRCULATIONAHA.107.728303. [DOI] [PubMed] [Google Scholar]
- 6.Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci. 2012;33(6):304–311. doi: 10.1016/j.tips.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Dolan JM, Kolega J, Meng H. High wall shear stress and spatial gradients in vascular pathology: a review. Ann Biomed Eng. 2013;41(7):1411–1427. doi: 10.1007/s10439-012-0695-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeuchi S, Karino T. Flow patterns and distributions of fluid velocity and wall shear stress in the human internal carotid and middle cerebral arteries. World Neurosurg. 2010;73(3):174–185. doi: 10.1016/j.surneu.2009.03.030. [DOI] [PubMed] [Google Scholar]
- 9.Jayaraman T, Berenstein V, Li X, Mayer J, Silane M, Shin YS, Niimi Y, Kilic T, Gunel M, Berenstein A. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery. 2005;57(3):558–564. doi: 10.1227/01.NEU.0000170439.89041.D6. [DOI] [PubMed] [Google Scholar]
- 10.Aoki T, Nishimura M. Targeting chronic inflammation in cerebral aneurysms: focusing on NF-kappaB as a putative target of medical therapy. Expert Opin Ther Targets. 2010;14(3):265–273. doi: 10.1517/14728221003586836. [DOI] [PubMed] [Google Scholar]
- 11.Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, Lawton MT, Young WL, Liang EI, Nuki Y, Hashimoto T. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke. 2011;42(1):173–178. doi: 10.1161/STROKEAHA.110.590976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boussel L, Rayz V, McCulloch C, Martin A, Acevedo-Bolton G, Lawton M, Higashida R, Smith WS, Young WL, Saloner D. Aneurysm growth occurs at region of low wall shear stress: patient-specific correlation of hemodynamics and growth in a longitudinal study. Stroke. 2008;39(11):2997–3002. doi: 10.1161/STROKEAHA.108.521617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanoue T, Tateshima S, Villablanca JP, Vinuela F, Tanishita K. Wall shear stress distribution inside growing cerebral aneurysm. AJNR Am J Neuroradiol. 2011;32(9):1732–1737. doi: 10.3174/ajnr.A2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Omodaka S, Sugiyama S, Inoue T, Funamoto K, Fujimura M, Shimizu H, Hayase T, Takahashi A, Tominaga T. Local hemodynamics at the rupture point of cerebral aneurysms determined by computational fluid dynamics analysis. Cerebrovasc Dis. 2012;34(2):121–129. doi: 10.1159/000339678. [DOI] [PubMed] [Google Scholar]
- 15.Fukazawa K, Ishida F, Umeda Y, Miura Y, Shimosaka S, Matsushima S, Taki W, Suzuki H. Using computational fluid dynamics analysis to characterize local hemodynamic features of middle cerebral artery aneurysm rupture points. World Neurosurg. 2013 doi: 10.1016/j.wneu.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 16.Cebral JR, Vazquez M, Sforza DM, Houzeaux G, Tateshima S, Scrivano E, Bleise C, Lylyk P, Putman CM. Analysis of hemodynamics and wall mechanics at sites of cerebral aneurysm rupture. J Neurointerv Surg. 2015;7(7):530–536. doi: 10.1136/neurintsurg-2014-011247. [DOI] [PubMed] [Google Scholar]
- 17.Morita A, Kirino T, Hashi K, Aoki N, Fukuhara S, Hashimoto N, Nakayama T, Sakai M, Teramoto A, Tominari S, Yoshimoto T. The natural course of unruptured cerebral aneurysms in a Japanese cohort. N Engl J Med. 2012;366(26):2474–2482. doi: 10.1056/NEJMoa1113260. [DOI] [PubMed] [Google Scholar]
- 18.Antiga L, Piccinelli M, Botti L, Ene-Iordache B, Remuzzi A, Steinman DA. An image-based modeling framework for patient-specific computational hemodynamics. Med Biol Eng Comput. 2008;46(11):1097–1112. doi: 10.1007/s11517-008-0420-1. [DOI] [PubMed] [Google Scholar]
- 19.Ito Y, Nakahashi K. Direct surface triangulation using stereolithography data. Aiaa J. 2002;40(3):490–496. doi: 10.2514/2.1672. [DOI] [Google Scholar]
- 20.Ito Y, Shih AM, Soni BK, Nakahashi K. Multiple marching direction approach to generate high-quality hybrid meshes. Aiaa J. 2007;45(1):162–167. doi: 10.2514/1.23260. [DOI] [Google Scholar]
- 21.Yamamoto K, Sokabe T, Matsumoto T, Yoshimura K, Shibata M, Ohura N, Fukuda T, Sato T, Sekine K, Kato S, Isshiki M, Fujita T, Kobayashi M, Kawamura K, Masuda H, Kamiya A, Ando J. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat Med. 2006;12(1):133–137. doi: 10.1038/nm1338. [DOI] [PubMed] [Google Scholar]
- 22.Sokabe T, Yamamoto K, Ohura N, Nakatsuka H, Qin K, Obi S, Kamiya A, Ando J. Differential regulation of urokinase-type plasminogen activator expression by fluid shear stress in human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287(5):H2027–2034. doi: 10.1152/ajpheart.00260.2004. [DOI] [PubMed] [Google Scholar]
- 23.Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, Fukuda S, Sasaki D, Podhajska A, Harbers M, Kawai J, Carninci P, Hayashizaki Y. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci U S A. 2003;100(26):15776–15781. doi: 10.1073/pnas.2136655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kodzius R, Kojima M, Nishiyori H, Nakamura M, Fukuda S, Tagami M, Sasaki D, Imamura K, Kai C, Harbers M, Hayashizaki Y, Carninci P. CAGE: cap analysis of gene expression. Nat Methods. 2006;3(3):211–222. doi: 10.1038/nmeth0306-211. [DOI] [PubMed] [Google Scholar]
- 25.Aoki T, Nishimura M. The development and the use of experimental animal models to study the underlying mechanisms of CA formation. J Biomed Biotechnol. 2011;2011:535921. doi: 10.1155/2011/535921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimoto N, Handa H, Hazama F. Experimentally induced cerebral aneurysms in rats. Surg Neurol. 1978;10(1):3–8. [PubMed] [Google Scholar]
- 27.Hasegawa A, Daub C, Carninci P, Hayashizaki Y, Lassmann T. MOIRAI: a compact workflow system for CAGE analysis. BMC Bioinformatics. 2014;15:144. doi: 10.1186/1471-2105-15-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohmiya H, Vitezic M, Frith MC, Itoh M, Carninci P, Forrest AR, Hayashizaki Y, Lassmann T. RECLU: a pipeline to discover reproducible transcriptional start sites and their alternative regulation using capped analysis of gene expression (CAGE) BMC Genomics. 2014;15:269. doi: 10.1186/1471-2164-15-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White CR, Haidekker M, Bao X, Frangos JA. Temporal gradients in shear, but not spatial gradients, stimulate endothelial cell proliferation. Circulation. 2001;103(20):2508–2513. doi: 10.1161/01.CIR.103.20.2508. [DOI] [PubMed] [Google Scholar]
- 30.Aoki T, Kataoka H, Ishibashi R, Nozaki K, Egashira K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke. 2009;40(3):942–951. doi: 10.1161/STROKEAHA.108.532556. [DOI] [PubMed] [Google Scholar]
- 31.Tada Y, Yagi K, Kitazato KT, Tamura T, Kinouchi T, Shimada K, Matsushita N, Nakajima N, Satomi J, Kageji T, Nagahiro S. Reduction of endothelial tight junction proteins is related to cerebral aneurysm formation in rats. J Hypertens. 2010;28(9):1883–1891. doi: 10.1097/HJH;0b013e32833c2273. [DOI] [PubMed] [Google Scholar]
- 32.Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Miyamoto S. Complementary inhibition of cerebral aneurysm formation by eNOS and nNOS. Lab Investig. 2011;91(4):619–626. doi: 10.1038/labinvest.2010.204. [DOI] [PubMed] [Google Scholar]
- 33.Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke. 2007;38(1):162–169. doi: 10.1161/01.STR.0000252129.18605.c8. [DOI] [PubMed] [Google Scholar]
- 34.Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery. 1999;45(5):1137–1146. doi: 10.1097/00006123-199911000-00024. [DOI] [PubMed] [Google Scholar]
- 35.Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Gene expression profile of the intima and media of experimentally induced cerebral aneurysms in rats by laser-microdissection and microarray techniques. Int J Mol Med. 2008;22(5):595–603. [PubMed] [Google Scholar]
- 36.Fukuda M, Aoki T, Manabe T, Maekawa A, Shirakawa T, Kataoka H, Takagi Y, Miyamoto S, Narumiya S. Exacerbation of intracranial aneurysm and aortic dissection in hypertensive rat treated with the prostaglandin F-receptor antagonist AS604872. J Pharmacol Sci. 2014;126(3):230–242. doi: 10.1254/jphs.14148FP. [DOI] [PubMed] [Google Scholar]
- 37.Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke. 1999;30(7):1396–1401. doi: 10.1161/01.STR.30.7.1396. [DOI] [PubMed] [Google Scholar]
- 38.Frosen J, Piippo A, Paetau A, Kangasniemi M, Niemela M, Hernesniemi J, Jaaskelainen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke. 2004;35(10):2287–2293. doi: 10.1161/01.STR.0000140636.30204.da. [DOI] [PubMed] [Google Scholar]
- 39.Tulamo R, Frosen J, Junnikkala S, Paetau A, Kangasniemi M, Pelaez J, Hernesniemi J, Niemela M, Meri S. Complement system becomes activated by the classical pathway in intracranial aneurysm walls. Lab Investig. 2010;90(2):168–179. doi: 10.1038/labinvest.2009.133. [DOI] [PubMed] [Google Scholar]
- 40.Perktold K, Peter R, Resch M. Pulsatile non-Newtonian blood flow simulation through a bifurcation with an aneurysm. Biorheology. 1989;26(6):1011–1030. doi: 10.3233/bir-1989-26605. [DOI] [PubMed] [Google Scholar]
- 41.Lee SW, Steinman DA. On the relative importance of rheology for image-based CFD models of the carotid bifurcation. J Biomech Eng. 2007;129(2):273–278. doi: 10.1115/1.2540836. [DOI] [PubMed] [Google Scholar]
- 42.Dempere-Marco L, Oubel E, Castro M, Putman C, Frangi A, Cebral J. CFD analysis incorporating the influence of wall motion: application to intracranial aneurysms. Medical image computing and computer-assisted intervention: MICCAI International Conference on Medical Image Computing and Computer-Assisted Intervention. 2006; 9 (Pt 2):438-445 [DOI] [PubMed]
- 43.Shojima M, Oshima M, Takagi K, Torii R, Hayakawa M, Katada K, Morita A, Kirino T. Magnitude and role of wall shear stress on cerebral aneurysm: computational fluid dynamic study of 20 middle cerebral artery aneurysms. Stroke. 2004;35(11):2500–2505. doi: 10.1161/01.STR.0000144648.89172.0f. [DOI] [PubMed] [Google Scholar]
- 44.Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Simvastatin suppresses the progression of experimentally induced cerebral aneurysms in rats. Stroke. 2008;39(4):1276–1285. doi: 10.1161/STROKEAHA.107.503086. [DOI] [PubMed] [Google Scholar]
- 45.Aoki T, Kataoka H, Ishibashi R, Nakagami H, Nozaki K, Morishita R, Hashimoto N. Pitavastatin suppresses formation and progression of cerebral aneurysms through inhibition of the nuclear factor kappaB pathway. Neurosurgery. 2009;64(2):357–365. doi: 10.1227/01.NEU.0000336764.92606.1D. [DOI] [PubMed] [Google Scholar]
- 46.Kimura N, Shimizu H, Eldawoody H, Nakayama T, Saito A, Tominaga T, Takahashi A. Effect of olmesartan and pravastatin on experimental cerebral aneurysms in rats. Brain Res. 2010;1322:144–152. doi: 10.1016/j.brainres.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 47.Yoshimura Y, Murakami Y, Saitoh M, Yokoi T, Aoki T, Miura K, Ueshima H, Nozaki K. Statin use and risk of cerebral aneurysm rupture: a hospital-based case-control study in Japan. J Stroke Cerebrovasc Dis. 2014;23(2):343–348. doi: 10.1016/j.jstrokecerebrovasdis.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 48.Korja M, Lehto H, Juvela S. Lifelong rupture risk of intracranial aneurysms depends on risk factors: a prospective Finnish cohort study. Stroke. 2014;45(7):1958–1963. doi: 10.1161/STROKEAHA.114.005318. [DOI] [PubMed] [Google Scholar]