

ABSTRACT
Keratinocytes are known to synthesize cortisol through activation of the enzyme 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1). To confirm the function of 11β-HSD1 in keratinocytes during inflammation in vivo, we created keratinocyte-specific-11β-HSD1 knockout mice (K5-Hsd11b1-KO mice) and analyzed the response to narrow-band ultraviolet B (NB-UVB) irradiation. Firstly, we measured the mRNA and protein levels of 11β-HSD1 following NB-UVB irradiation and found that the expression of 11β-HSD1 in keratinocytes of mouse ear skin was enhanced at 3 and 24 hours after 250 mJ/cm2, 500 mJ/cm2, 1 J/cm2, and 2 J/cm2 NB-UVB irradiation. Next, we determined that 24 hours after exposure to 1 J/cm2 NB-UVB irradiation, the numbers of F4/80-, CD45-, and Gr-1-positive cells were increased in K5-Hsd11b1-KO mice compared to wild type (WT) mice. Furthermore, the expression of the chemokine (C-X-C-motif) ligand 1 (CXCL1) and interleukin (IL)-6 was also significantly enhanced in NB-UVB-irradiated K5-Hsd11b1-KO mice compared with WT mice. In addition, activation of nuclear factor-kappa B (NF-κB) after NB-UVB irradiation was enhanced in K5-Hsd11b1-KO mice compared to that in WT mice. Thus, NB-UVB-induced inflammation is augmented in K5-Hsd11b1-KO mice compared with WT mice. These results indicate that 11β-HSD1 may suppress NB-UVB-induced inflammation via inhibition of NF-κB activation.
KEYWORDS: corticosterone, inflammation, keratinocytes, narrow-band UVB, 11β-hydroxysteroid dehydrogenase 1
Introduction
Since the discovery of cortisone in the 1950s, glucocorticoid (GC) has been used as one of the most effective anti-inflammatory drugs for the treatment of inflammatory and autoimmune diseases and is also widely used as a topical applying for inflammatory dermatitis. In keratinocytes, GCs also affect proliferation, differentiation, and metabolism of cells in addition to exerting anti-inflammatory effects.1
The anti-inflammatory effects of GCs are achieved at concentrations that are much higher than the physiological levels of GCs. At physiological doses, GCs have the potential to enhance or suppress the immune response depending on the concentration and duration of exposure.2,3 In keratinocytes, interleukin-1β-induced interleukin-6 (IL-6) production is stimulated by low dose (10−10 M) cortisol but is suppressed with a high dose (10−5 M).4 The endogenous GCs in humans and rodents are cortisol and corticosterone, respectively. Cortisol in skin cells was first demonstrated in human melanoma cells.5 Thereafter, cortisol in human melanocytes and fibroblasts was reported.6,7 Corticosterone was first reported in rodent skin.8 The stressors such as inflammation, viral infection, and trauma stimulate the hypothalamo-pituitary-adrenal (HPA) axis, which triggers the production of systemic cortisol. It is reported that HPA axis also exists in the skin and synthesizes cortisol.6,7,9-13 The skin is reported to have neuroendocrine capabilities that express elements of the HPA axis, key enzyme of corticosteroid glucocorticoids.14,15 In addition to cortisol production from the HPA axis, extra-adrenal cortisol production in various tissues, including colon, heart, and lung, has also been reported.16-20
Skin cells are also known to synthesize cortisol21-24 or corticosterone25 through a de novo pathway involving an activating enzyme. The enzyme that catalyzes the intracellular conversion of hormonally-inactive cortisone into active cortisol is 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1).4 Two iso-enzymes exist: 11β-HSD1 activates cortisol from cortisone, and 11β-HSD2 inactivates cortisol to cortisone.26,27 11β-HSD1is expressed in many tissues, with the highest levels found in the liver, lung, adipose tissue, ovaries, and central nervous system.26 In the skin, 11β-HSD1 is expressed in epidermal keratinocytes, dermal fibroblasts, and root sheath cells of the outer hair follicle.28-31
Our and other group previously reported a role for 11β-HSD1 in the skin and demonstrated that the expression of 11β-HSD1 increased with age in mouse skin and negatively regulated cutaneous wound healing by modulating proliferation of keratinocytes and fibroblasts.28,32,33 The expression of 11β-HSD1 was decreased in benign and malignant tumors of the skin, such as squamous cell carcinoma, basal cell carcinoma, and seborrhoeic keratosis, indicating the association of 11β-HSD1 with cell proliferation.34 Skobowiat et al. reported that UVB and UVC, but not UVA irradiation, increased the expression of 11β-HSD1 in human keratinocytes. Conversely, irradiation with UVA, but not UVB and UVC, induced the expression of 11β-HSD2.35 Alteration of cortisol by UV irradiation may alter the maintenance of homeostasis in keratinocytes.
In this study, we investigated the importance of 11β-HSD1 in keratinocytes during narrow-band (NB-UVB)-induced inflammation in vivo using keratinocyte-specific 11β-HSD1 knockout (K5-Hsd11b1-KO) mice. We previously tried to investigate the effects of 11β-HSD1 in skin using systemic 11β-HSD1 knockout mice; however, analysis of these mice was complicated, as these animals showed increased serum corticosterone levels probably due to some compensatory mechanism.32,36 Thus, in the current study, we generated K5-Hsd11b1-KO mouse to investigate the function of 11β-HSD1 in keratinocytes and to eliminate the increased serum corticosterone levels observed in systemic11β-HSD1 knockout mice.
Materials and methods
Cell culture
The isolation and culture of mouse keratinocytes was carried out as follows. Full-thickness skin harvested from 2-month-old mice was treated with 4 mg/ml of dispase (Gibco; Invitrogen, Paisley, UK) for 1 hour at 37°C. Next, the epidermis was peeled from the dermis, trypsinised to prepare single cells, and incubated in EpiLife Medium with 60 μM of calcium (Invitrogen) in EpiLife Human Keratinocyte Growth Supplement (Invitrogen) Medium for 24 hours at 37°C under an atmosphere of 5% CO2. Non-adherent cells were removed by 2 washes with phosphate-buffered saline (PBS), and then cells were cultured for 2 d in EpiLife Medium including Keratinocyte Growth Supplement. Non-adherent cells were again removed with 2 washes with PBS, and then cells were cultured for one day in EpiLife medium without bovine pituitary extract and hydrocortisone prior to use in the experiments.
Mice
Animal care was in strict accordance with the institutional guidelines of Osaka University. All of the animal experiments were carried out with the approval of the Animal Experiments Committee of Osaka University (#20-003-0).
Generation of K5-Hsd11b1-KO mice is that Hsd11b1tm1a/+ ES cells were purchased from the Knockout Mouse Project (KOMP) Repository (Davis, CA, USA). The ES cells were injected into blastocysts collected from superovulated BALB/c female mice. The treated blastocysts were then transferred into the uterus of pseudopregnant ICR female mice (SLC Japan) to obtain chimeric mice. Male chimeras were mated with female C57BL/6 mice, resulting in germline transmission of the Hsd11b1 tm1a allele. Hsd11b1tm1a/tm1a mice were obtained by intercrossing Hsd11b1tm1a/+ mice. To create Hsd11b1flox/flox mice, Hsd11b1tm1a/tm1a mice were crossed with B6.Cg-Tg (ACTFLPe) 9205Dym/J purchased from The Jackson Laboratory (Bar Harbor, ME, USA) to remove poly (A) signal cassette. Generation of K5-Cre transgenic mice was previously described.37 K5-Cre transgenic mice were bred with Hsd11b1flox/flox mice to generate mice carrying the K5-Cre transgene and a floxed Hsd11b1 allele (K5Cre/0; Hsd11b1flox/+). These mice were then mated with each other. Offsprings carrying K5Cre/0; Hsd11b1flox/flox and K5Cre/0; Hsd11b1+/+ were used for further analyses as K5-Hsd11b1-KO and wild type (WT), respectively.
UVB irradiation
The source of UV irradiation was M-DER-320 (Tokodenki, Tokyo, Japan). The lamps of UV irradiation were NB-UVB fluorescent lamp (TL-20 W/ 01; Philips, Rosenthal, Holland). 85% of the UVB emission was at 311 ± 2 nm. The dose of UVB was quantified using a UV radiometer (Topcon Corporation, Tokyo, Japan), and murine skin was irradiated with a single dose of 40 mJ/cm2 (in vitro) or 250 mJ /cm2, 500 mJ /cm2, 1 J/cm2, or 2 J /cm2 (in vivo) NB-UVB.
Histopathological analysis
The samples were fixed in 10% formaldehyde for 24 hours, followed by embedding in paraffin and microtome sectioning. The sections (4 μm thick) were then stained with haematoxylin and eosin (H&E). For immunohistochemical analysis, the sections were hydrated via passage through xylene and graded ethanol. Following antigen retrieval for 10 minutes at 110°C in citric buffer (pH 6.0), the slides were blocked with serum-free protein block (Dako-Cytomation, Carpinteria, CA, USA) for 10 minutes, then incubated with the primary antibody overnight at 4°C (rabbit anti-11β-HSD1 antibody 1:100 dilution, Abcam, Cambridge, UK). After washing with Tris-buffered saline (TBS) containing 0.05% Triton-X100, the slides were mounted using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) followed by counterstaining with haematoxylin. For immunofluorescence analysis, the sections were hydrated as described above and incubated with the following primary antibodies: rat anti-F4/80 antibody diluted at 1:100 (AbD Serotec, Oxford, UK), rat anti-CD45 antibody diluted at 1:100 (BD Biosciences, San Jose, CA, USA), rat anti-Gr-1 antibody diluted at 1:100 (eBioscience, San Diego, CA, USA), and rabbit anti-nuclear factor kappaB (NF-κB) antibody diluted at 1:100 (Cell Signaling Technology, Beverly, MA, USA). Sections were then incubated with the secondary antibody (anti-rat Alexa Fluor 555 or anti-rabbit Alexa Fluor 555, Invitrogen). Rabbit IgG and Rat IgG (DAKO-Cytomation, Carpinteria, CA) were used as an isotype control, respectively.
Myeloperoxidase (MPO) activity assessment
The MPO activity in tissue lysates derived from the skin samples was determined using the MPO Assay Kit (Bio Vision, Mountain View, CA, USA) according to the manufacturer's instructions. Briefly, samples of mouse skin were cut into small pieces with scissors and crushed by sonication. Samples were then subjected to centrifugation at 1,500 × g for 15 minutes, and the supernatants of the mouse skin tissue lysates were collected and diluted 5-fold. The MPO activity of each sample was subsequently determined and normalized to its respective protein concentration.
Western blotting
The skin samples were crushed in liquid nitrogen and solubilized at 4°C in lysis buffer (0.5% sodium deoxycholate, 1% Nonidet P40, 0.1% sodium dodecyl sulfate, 100 μg/ml phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, and protease inhibitor cocktail). Proteins (30 μg) were separated on SDS-polyacrylamide gels and transferred onto polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA). Non-specific protein binding was blocked by incubating the membranes in 5% w/v non-fat milk powder in TBS-T (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 0.1% v/v Tween-20), and then the membranes were incubated with rabbit anti-phospho-NF-κB p65 antibodies or rabbit anti-NF-κB p65 antibodies (Cell Signaling Technology) diluted 1:1,000 overnight at 4°C or with mouse anti-β-actin antibody (Sigma-Aldrich, St. Louis, MO, USA) diluted 1:10,000 for 30 minutes at room temperature. The membranes were then washed 3 times in TBS-T for 5 minutes each and finally incubated with either HRP-conjugated anti-mouse or anti-rabbit antibodies at a dilution of 1:10,000 for 60 minutes at room temperature. Protein bands were detected using the ECL Plus kit (GE Healthcare, Buckinghamshire, UK), and the intensity of the bands was quantified using the NIH Image J software program.
RNA isolation and quantitative real-time polymerase chain reaction (RT-PCR)
Total RNA was isolated from the cells using the SV Total RNA Isolation System (Promega, Madison, WI, USA). The product was reverse transcribed into first-strand cDNA (cDNA), and the expression levels of Hsd11b1,chemokine (C-X-C-motif) ligand1 (CXCL1), and IL-6 were measured using the Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's protocol. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize the mRNA, and sequence-specific primers were designed as follows: Hsd11b1, sense 5′-aaaattacctcctcccgatcct-3′, antisense 5′-ggcagcgagacactaccttc-3′; CXCL1, sense 5′-gcctatcgccaatgagctg-3′, antisense 5′-tctccgttacttggggacac-3′; IL-6, sense 5′-tgggaaatcgtggaaatgaga-3′, antisense 5′-aagtgcatcatcgttgttcataca-3′; and GAPDH, sense 5′- tgtcatcatacttggcaggtttct-3′, antisense 5′-catggccttccgtgttccta-3′. RT-PCR (40 cycles of denaturation at 92°C for 15 seconds followed by annealing at 60°C for 60 seconds) was run on an ABI 7000 Prism system (Applied Biosystems).
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed on the tissue lysates derived from mouse skin samples using Quantikine Immunoassay (R&D Systems, Minneapolis, MN, USA). The assay was performed according to the manufacturer's instructions. Samples of mouse skin were cut into small pieces with scissors and then crushed by sonication. Following centrifugation at 1,500× g for 15 minutes, the supernatants of the mouse skin tissue lysates were collected. The concentrations of CXCL1 and IL-6 were normalized to the respective protein concentration.
Statistical analysis
The data are expressed as the mean ± standard deviation (SD). The Student's t-test was used to determine the level of significance of the differences between the 2 groups. An analysis of variance (ANOVA) was performed followed by the Bonferroni-Dunn test for multiple comparisons to allow for pairwise testing of significant differences between the groups. Statistical significance was defined with a P-value of <0.05.
Results
Generation of K5-Hsd11b1-KO mice
To investigate the function of 11β-HSD1 in keratinocytes, we generated K5-Hsd11b1-KO mice by breeding K5-Cre transgenic mice with Hsd11b1flox/flox. The expression of 11β-HSD1 was significantly decreased in epidermis of K5-Hsd11b1-KO mice (Fig. 1A). The mRNA expression level of 11β-HSD1 was also decreased in keratinocytes derived from K5-Hsd11b1-KO mice (Fig. 1B).
Figure 1.
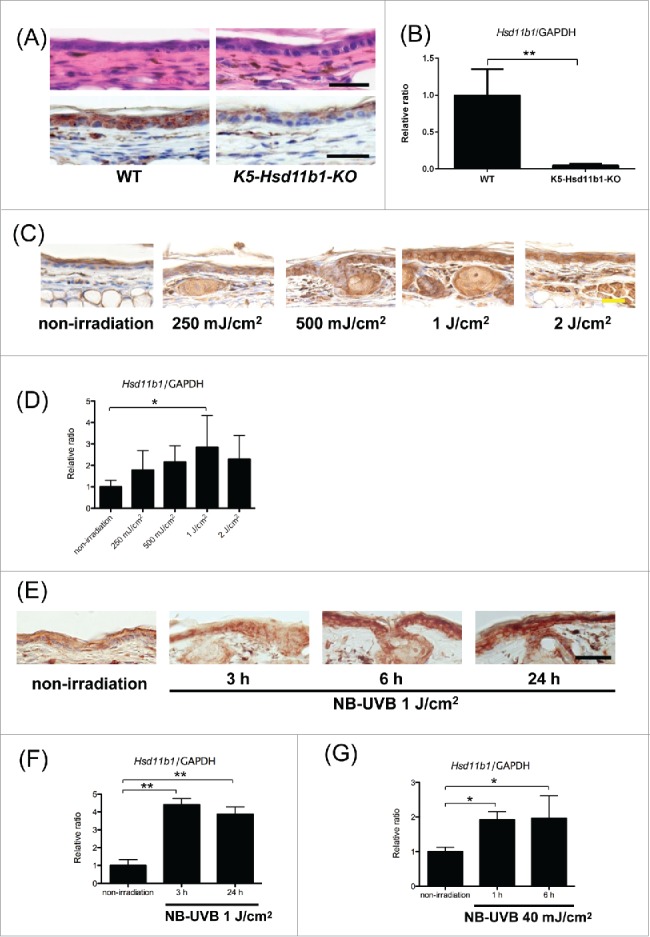
(For figure legend, see page 6.) Figure 1 (see previous page). Generation of K5-Hsd11b1-KO mice. The expression of 11β-HSD1 in the skin was enhanced following NB-UVB irradiation in WT mice. (A) Representative H&E staining (upper panel) and 11β-HSD1 staining (lower panel) of 2-month-old WT and K5-Hsd11b1-KO mouse skin Bar = 50 μm. (B) The relative expression levels of 11β-HSD1 in keratinocytes derived from WT and K5-Hsd11b1-KO mice were assessed using RT-PCR. GAPDH was used as an internal control. The bars indicate the mean ± SD (N = 5; *P < 0.05, Student's t-test). (C) Expression of 11β-HSD1 in 2-month-old WT mice irradiated with 250 mJ/cm2, 500 mJ/cm2, 1 J/cm2, and 2 J/cm2 NB-UVB. Skin specimens were harvested 24 hours after NB-UVB irradiation. Rabbit IgG was used as isotype control. Bar = 20 μm (N = 6 per group). (D) The relative expression levels of 11β-HSD1 in WT mice irradiated with 250 mJ/cm2, 500 mJ/cm2, 1 J/cm2, and 2 J/cm2 NB-UVB were assessed using RT-PCR. Skin samples were collected 24 hours after NB-UVB irradiation. GAPDH was used as an internal control. The bars indicate the mean ± SD (N = 6; **P<0.01, one-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons). (E) Expression of 11β-HSD1 in 2-month-old WT mice irradiated with 1 J/cm2 of NB-UVB. Skin specimens were harvested 3, 6, and 24 hours after NB-UVB irradiation. Rabbit IgG was used as isotype control. Bar = 50 μm (N = 6 per group). (F) The relative expression levels of 11β-HSD1 in WT mice irradiated with 1 J/cm2 NB-UVB were assessed using RT-PCR. Skin samples were collected at the indicated time points for unirradiated mice and 3 and 24 hours after NB-UVB irradiation. GAPDH was used as an internal control. The bars indicate the mean ± SD (N = 6; **P < 0.01, one-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons). (G) The relative expression levels of 11β-HSD1 in WT mouse-derived keratinocytes after NB-UVB irradiation with 40 mJ/cm2 NB-UVB were assessed using RT-PCR. Keratinocytes were collected at the indicated time points for unirradiated mice and 1 and 6 hours after NB-UVB irradiation. GAPDH was used as an internal control. The bars indicate the mean ± SD (N = 6; *P < 0.05, one-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons).
NB-UVB irradiation increased the expression of 11β-HSD1 in mouse skin
To examine the dynamics of 11β-HSD1 expression following NB-UVB irradiation, we first irradiated mouse ear skin with various doses of NB-UVB. The expression of 11β-HSD1 in epidermis increased following exposure to 250 mJ/cm2, 500 mJ/cm2, 1 J/cm2, and 2 J/cm2 NB-UVB irradiation (Fig. 1C). The mRNA expression of 11β-HSD1 also increased in a dose-dependent manner 24 hours after NB-UVB irradiation, and peak mRNA levels were observed following 1 J/cm2 NB-UVB irradiation (Fig. 1D).
Thus, we irradiated mouse ear skin with 1 J/cm2 of NB-UVB in subsequent experiments. The protein expression of 11β-HSD1 began to increase 3 hours after NB-UVB irradiation and continued to increase until 24 hours after NB-UVB irradiation (Fig. 1E). The mRNA expression of 11β-HSD1 also increased in skin irradiated with NB-UVB (Fig. 1F). In addition, increased expression of 11β-HSD1 was observed in NB-UVB-irradiated keratinocytes derived from ear and tail in vitro (Fig. 1G).
Inflammatory cell infiltration after NB-UVB irradiation was augmented in K5-Hsd11b1-KO mouse skin
Following NB-UVB irradiation of WT and K5-Hsd11b1-KO mice, the function of increased 11β-HSD1 in keratinocytes was evaluated. NB-UVB irradiation significantly increased the number of infiltrated inflammatory cells in the dermis (Fig. 2A). Specifically, the numbers of F4/80-, CD45-, and Gr-1-positive cells were significantly higher in irradiated K5-Hsd11b1-KO mice than in irradiated WT mice (Fig. 2A-D). Accordingly, myeloperoxidase (MPO) activity was significantly increased in the NB-UVB-irradiated K5-Hsd11b1-KO mice compared to that in the irradiated WT mice (Fig. 2E).
Figure 2.

NB-UVB-induced infiltration of inflammatory cells was increased in the K5-Hsd11b1-KO mouse skin. (A) H&E staining (upper panel) and immunofluorescence staining of F4/80, CD45, and Gr-1 (red) and nuclei (Hoechst 33342, blue) (lower panel) in the 2-month-old WT and K5-Hsd11b1-KO mouse skin 24 hours after NB-UVB irradiation at 1 J/cm2 (N = 8 per group). Rat IgG was used as isotype control. Bar = 100 μm. (B-D) Numbers of F4/80- (B), CD45- (C), Gr-1-positive (D) cells per each high power field in the 2-month-old WT (white bar) and K5-Hsd11b1-KO (black bar) mouse skin after NB-UVB irradiation at 1 J/cm2. Three sections from each mouse were evaluated. The bars indicate the mean ± SD (N = 8; **P < 0.01, two-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons). (E) Myeloperoxidase (MPO) activity in skin from 2-month-old mice 24 hours after NB-UVB irradiation at 1 J/cm2. The bars indicate the mean ± SD (N = 6; **P < 0.01, 2-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons).
NB-UVB-induced pro-inflammatory cytokines were augmented in the K5-Hsd11b1-KO mice
Next, we investigated the expression of pro-inflammatory cytokines in WT and K5-Hsd11b1-KO mice following NB-UVB irradiation. The mRNA and protein levels of CXCL1 and IL-6 were significantly higher in NB-UVB-irradiated K5-Hsd11b1-KO mouse skin than in irradiated WT mouse skin (Fig. 3A-D). Furthermore, we examined the effect of various doses of cortisol on the inflammatory response of keratinocytes in culture. CXCL1 mRNA levels were significantly decreased in keratinocytes derived from WT mice exposed to higher doses of cortisol (1×10−7 M and 1 × 10−5 M) compared with WT keratinocytes cultured in cortisol-free media (Fig. 3E). Similarly, CXCL1 concentrations in culture media tended to decrease following exposure to higher doses of cortisol (Fig. 3F).
Figure 3.
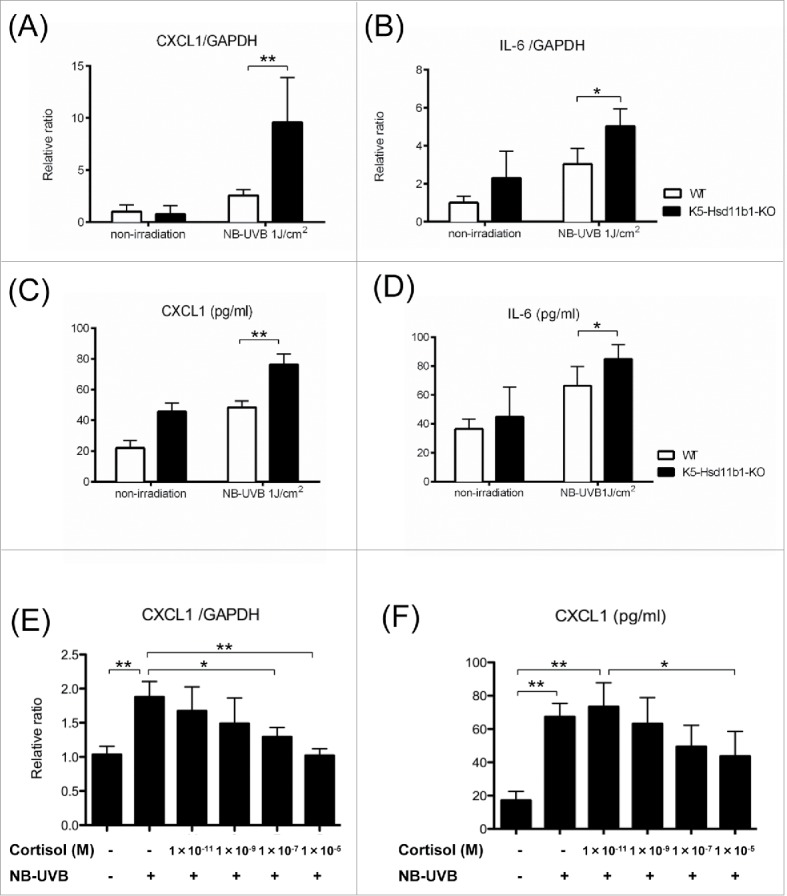
Inflammatory response to NB-UVB irradiation was enhanced in the K5-Hsd11b1-KO mouse skin. (A, B) Relative expression levels of CXCL1 (A) and IL-6 (B) in skin from 2-month-old mice 3 hours after NB-UVB irradiation at 1 J/cm2. GAPDH was used as an internal control. The bars indicate the mean ± SD (N = 6; **P < 0.01, *P < 0.05, two-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons). (C, D) The levels of CXCL1 (C) and IL-6 (D) in the skin of 2-month-old WT mice 10 hours after NB-UVB irradiation at 1 J/cm2 were measured using ELISA. The bars indicate the mean ± SD (N = 6; **P < 0.01, *P < 0.05, two-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons). (E) The indicated dose of cortisol was added to the cultured media of the WT mice-derived keratinocytes for 24 hours, and then cells were treated with or without 40 mJ/cm2 NB-UVB irradiation. Cells were harvested after 1 hour, and CXCL1 expression was investigated by rtPCR. GAPDH was used as an internal control. The bars indicate the mean ± SD (N = 6; **P < 0.01,*P < 0.05, one-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons). (F) The indicated dose of cortisol was added to the cultured media of WT mice-derived keratinocytes for 24 hours, and then cells were treated with or without 40 mJ/cm2 NB-UVB irradiation. Culture media was collected 10 h later, and concentrations of CXCL1 were measured by ELISA. The bars indicate the mean ± SD (N = 6; **P < 0.01, *P < 0.05, one-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons).
NB-UVB-induced NF-κB activation was augmented in the K5-Hsd11b1-KO mice
Subsequently, the activation of NF-κB by NB-UVB irradiation was evaluated in K5-Hsd11b1-KO mice. Immunohistochemical analysis revealed nuclear translocation of NF-κB 3 hours after NB-UVB irradiation of keratinocytes. The number of keratinocytes with activated NF-κB was higher in the irradiated K5-Hsd11b1-KO mouse skin than in the irradiated WT mouse skin (Fig. 4A). In addition, phosphorylation of NF-κB was enhanced one hour after NB-UVB irradiation in the irradiated K5-Hsd11b1-KO mouse skin as evaluated by western blotting, and the amount of phosphorylated NF-κB was significantly higher in K5-Hsd11b1-KO mouse skin extract than in WT mouse skin extract (Fig. 4B).
Figure 4.
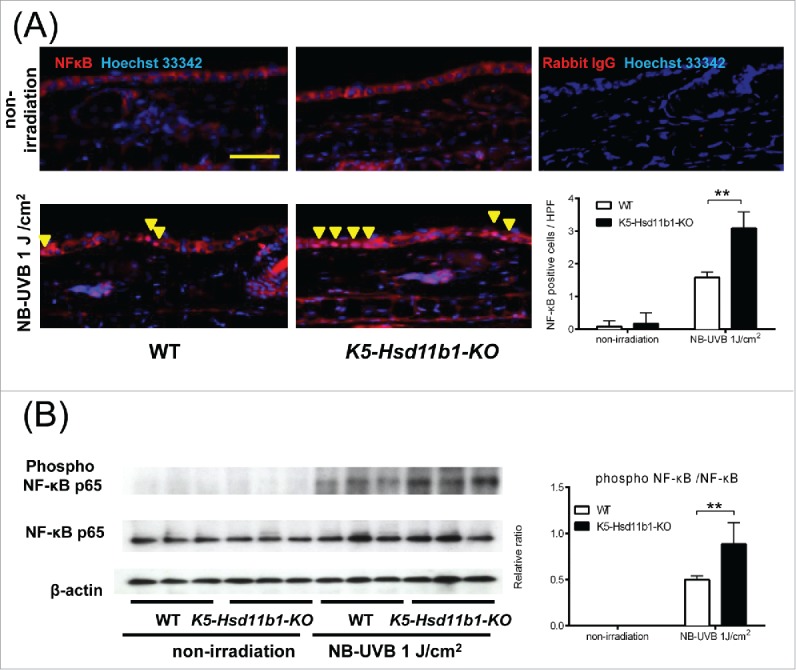
NB-UVB-induced activation of NF-κB in the K5-Hsd11b1-KO mouse skin was enhanced. (A) Immunofluorescence staining of NF-κB p65 (red) and nuclei (Hoechst 33342, blue) in skin of 2-month-old WT and K5-Hsd11b1-KO mice 3 hours after NB-UVB irradiation at 1 J/cm2. Rabbit IgG was used as isotype control. Bar = 100 μm. Three sections from each mouse were evaluated. The bars indicate the number of NF-κB p-65-positive cells translocated into the nucleus after NB-UVB irradiation (mean ± SD; N = 6; **P < 0.01, two-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons). (B) Western blot analysis of the phospho-NF-κB p65 expression in skin from 2-month-old WT and K5-Hsd11b1-KO mice 1 hour after NB-UVB irradiation at 1 J/cm2. The intensity of the bands was quantified using the NIH Image J software program. The phospho-NF-κB p65/NF-κB p65 was calculated. The bars indicate the mean ± SD (N = 3; *P < 0.05, 2-way ANOVA followed by the Bonferroni-Dunn test for multiple comparisons).
Discussion
In this study, we analyzed the inflammatory response to NB-UVB irradiation in K5-Hsd11b1-KO mice. The response to NB-UVB irradiation was enhanced in K5-Hsd11b1-KO mice compared with WT mice possibly through enhanced NF-κB activation. Therefore, these results suggest that local cortisol activation in keratinocytes through 11β-HSD1 is important for regulating NB-UVB-induced inflammation.
In WT mouse-derived keratinocytes in vitro, a higher dose of cortisol suppressed the immune response. We previously reported that 1x10−5 M cortisol attenuated IL-1β-induced IL-6 production in normal human epidermal keratinocytes.4 Additionally, 10−7 M and 10−5 M concentrations of corticosterone have been shown to suppress the expression of pro-inflammatory cytokines in mouse peritoneal macrophages,38 and these results are consistent with our findings in mouse-derived keratinocytes.
Association of 11β-HSD1 in various inflammatory diseases such as inflammatory bowel diseases, and rheumatoid arthritis has been reported.20,39-42 In Crohn's disease and ulcerative colitis in humans and in dextran sodium sulfate-induced colitis in rats, expression of 11β-HSD1 is upregulated whereas, the expression of 11β-HSD2 was downregulated, indicating GC activation is strongly increased in inflammatory bowel diseases.40,41 Local GC activation in synovial fluids in patients with rheumatoid arthritis has also been reported, and systemic11β-HSD1 activity was found to be higher in untreated rheumatoid arthritis patients.39 These results raise the possibilities that local GC activation was increased in these inflammatory diseases to either attenuate or to augment the local inflammation. A study using 11β-HSD1-deficient mice in a murine model of arthritis and peritonitis may provide an answer to the question of whether GC activation attenuates or augments local inflammation.43 In this study, the 11β-HSD1-deficient mice but not the 11β-HSD2-deficient mice exhibited earlier onset and slower resolution of inflammation than the WT mice in the K/BxN serum transfer model of arthritis, suggesting that increased 11β-HSD1 activity is involved in resolving inflammation. Similarly, in thioglycollate-induced sterile peritonitis, 11β-HSD1-deficient mice had more inflammatory cells in the peritoneum than 11β-HSD2-deficient mice.43 The results obtained in these arthritis and peritonitis models are in agreement with our finding that the response to NB-UVB irradiation was enhanced in K5-Hsd11b1-KO mice. In addition, our results demonstrated the importance of local corticosterone activation in keratinocytes during skin inflammation in K5-Hsd11b1-KO mice. As keratinocytes are located on the superficial surface of the skin and are exposed daily to various mechanical and chemical agents, this local cortisol activation mechanism may play a pivotal role to combat daily skin inflammation.
Recently, a mouse model of 11β-HSD1 deficiency in liver and adipose tissue has been reported.44,45 Adipose-specific-11β-HSD1-deficient mice were protected from hepatic steatosis and circulating fatty acid excess induced by excess corticosterone44; however, whether an increased circulating GC excess occurred in liver-specific 11β-HSD1-deficient mice was unclear from 2 studies.44,45 One study demonstrated that cortisone and 11-DHC-induced hepatic steatosis were not different between liver-specific-11β-HSD1-deficient mice and WT mice,44 whereas another study showed that liver-specific 11β-HSD1-deficient mice were protected from 11-DHC-induced metabolic effects.45 This difference may stem from the difference in the dose of 11-DHC given to mice, as the anti-inflammatory effect of GCs depends on its dose.
We lastly investigated the suppression of NB-UVB-induced inflammatory response in keratinocytes by 11β-HSD1. We focused on NF-κB, as this transcription factor regulates a variety of genes encoding proinflammatory cytokines. UVB irradiation phosphorylated NF-κB, which translocated from the cytosol to the nucleus. GCs are known to suppress the expression of inflammatory gene transcription factors, such as NF-κB.46,47 In our study, the activation of NF-κB after NB-UVB irradiation was enhanced in the K5-Hsd11b1-KO mice. Furthermore, previous research has also demonstrated that 11β-HSD1 regulates NF-κB activation by modulating the coordinated action of GC receptors and mineralocorticoid receptor.48 Therefore, we conclude that corticosterone activation by 11β-HSD1 in keratinocytes represses local inflammation induced by NB-UVB via suppression of NF-κB activation. In summary, we demonstrated that NB-UVB-induced inflammation was augmented in K5-Hsd11b1-KO mice compared with WT mice. Activation of corticosterme in keratinocytes is considered important for the suppression of local inflammation via the suppression of NF-κB activation. These findings indicate the importance of corticosterone production in keratinocytes as a local first-line defense to protect the skin from external stimuli.
Abbreviations
- 11β-HSD1
11β-hydroxysteroid dehydrogenase 1
- cDNA
complementary DNA
- CXCL1
chemokine (C-X-C-motif) ligand1
- ELISA
enzyme-linked immunosorbent assay
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GC
glucocorticoid
- HPA
hypothalamo-pituitary-adrenal
- IL-6
interleukin-6
- K5-Hsd11b1-KO
keratinocyte-specific 11β-HSD1 knockout
- MPO
myeloperoxidase
- NB-UVB
narrow-band UVB
- NF-κB
nuclear factor-kappa B
- PBS
phosphate-buffered saline
- SD
standard deviation
- TBS
Tris-buffered saline
- WT
wildtype
Disclosure of potential conflics of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Mr. Kenju Nishida, Ms. Sayaka Matsumura, Ms. Eriko Nobuyoshi, and Ms. Yumiko Fujii for research assistance.
Funding
This work was supported in part by a Grant-in-Aid for Research Activity Start-up (No. 15H06375) and the LYDIA O'LEARY Memorial Foundation.
References
- [1]. Stojadinovic O, Lee B, Vouthounis C, Vukelic S, Pastar I, Blumenberg M, Brem H, Tomic-Canic M. Novel genomic effects of glucocorticoids in epidermal keratinocytes: inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem 2007; 282:4021-34; PMID:17095510; http://dx.doi.org/10.1074/jbc.M606262200 [DOI] [PubMed] [Google Scholar]
- [2]. Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 2004; 125:697-706; PMID:15541765; http://dx.doi.org/10.1016/j.mad.2004.06.010 [DOI] [PubMed] [Google Scholar]
- [3]. Yeager MP, Guyre PM, Munck AU. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol Scand 2004; 48:799-813; PMID:15242423; http://dx.doi.org/10.1111/j.1399-6576.2004.00434.x [DOI] [PubMed] [Google Scholar]
- [4]. Itoi S, Terao M, Murota H, Katayama I. 11beta-Hydroxysteroid dehydrogenase 1 contributes to the pro-inflammatory response of keratinocytes. Biochem Biophys Res Commun 2013; 440:265-70; PMID:24055708; http://dx.doi.org/10.1016/j.bbrc.2013.09.065 [DOI] [PubMed] [Google Scholar]
- [5]. Slominski A, Gomez-Sanchez CE, Foecking MF, Wortsman J. Metabolism of progesterone to DOC, corticosterone and 18OHDOC in cultured human melanoma cells. FEBS Lett 1999; 455:364-6; PMID:10437805; http://dx.doi.org/10.1016/S0014-5793(99)00889-3 [DOI] [PubMed] [Google Scholar]
- [6]. Slominski A, Zbytek B, Szczesniewski A, Semak I, Kaminski J, Sweatman T, Wortsman J. CRH stimulation of corticosteroids production in melanocytes is mediated by ACTH. Am J Physiol Endocrinol Metab 2005; 288:E701-6; PMID:15572653; http://dx.doi.org/10.1152/ajpendo.00519.2004 [DOI] [PubMed] [Google Scholar]
- [7]. Slominski A, Zbytek B, Semak I, Sweatman T, Wortsman J. CRH stimulates POMC activity and corticosterone production in dermal fibroblasts. J Neuroimmunol 2005; 162:97-102; PMID:15833364; http://dx.doi.org/10.1016/j.jneuroim.2005.01.014 [DOI] [PubMed] [Google Scholar]
- [8]. Slominski A, Gomez-Sanchez CE, Foecking MF, Wortsman J. Active steroidogenesis in the normal rat skin. Biochimica et biophysica acta 2000; 1474:1-4; PMID:10699483; http://dx.doi.org/10.1016/S0304-4165(99)00215-9 [DOI] [PubMed] [Google Scholar]
- [9]. Slominski A, Ermak G, Mihm M. ACTH receptor, CYP11A1, CYP17 and CYP21A2 genes are expressed in skin. J Clin Endocrinol Metabol 1996; 81:2746-9; PMID:8675607 [DOI] [PubMed] [Google Scholar]
- [10]. Slominski A, Mihm MC. Potential mechanism of skin response to stress. Int J Dermatol 1996; 35:849-51; PMID:8970839; http://dx.doi.org/10.1111/j.1365-4362.1996.tb05049.x [DOI] [PubMed] [Google Scholar]
- [11]. Ito N, Ito T, Kromminga A, Bettermann A, Takigawa M, Kees F, Straub RH, Paus R. Human hair follicles display a functional equivalent of the hypothalamic-pituitary-adrenal axis and synthesize cortisol. FASEB J 2005; 19:1332-4; PMID:15946990 [DOI] [PubMed] [Google Scholar]
- [12]. Slominski A, Wortsman J, Tuckey RC, Paus R. Differential expression of HPA axis homolog in the skin. Mol Cell Endocrinol 2007; 265-266:143-9; PMID:17197073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Slominski AT, Zmijewski MA, Zbytek B, Tobin DJ, Theoharides TC, Rivier J. Key role of CRF in the skin stress response system. Endocr Rev 2013; 34:827-84; PMID:23939821; http://dx.doi.org/10.1210/er.2012-1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Skobowiat C, Nejati R, Lu L, Williams RW, Slominski AT. Genetic variation of the cutaneous HPA axis: an analysis of UVB-induced differential responses. Gene 2013; 530:1-7; PMID:23962689; http://dx.doi.org/10.1016/j.gene.2013.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Skobowiat C, Slominski AT. UVB activates hypothalamic-pituitary-adrenal axis in C57BL/6 mice. J Investigat Dermatol 2015; 135:1638-48; PMID:25317845; http://dx.doi.org/10.1038/jid.2014.450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Noti M, Corazza N, Mueller C, Berger B, Brunner T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 2010; 207:1057-66; PMID:20439544; http://dx.doi.org/10.1084/jem.20090849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Mueller M, Cima I, Noti M, Fuhrer A, Jakob S, Dubuquoy L, Schoonjans K, Brunner T. The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J Exp Med 2006; 203:2057-62; PMID:16923850; http://dx.doi.org/10.1084/jem.20060357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Young MJ, Clyne CD, Cole TJ, Funder JW. Cardiac steroidogenesis in the normal and failing heart. J Clin Endocrinol Metabol 2001; 86:5121-6; PMID:11701663; http://dx.doi.org/10.1210/jcem.86.11.7925 [DOI] [PubMed] [Google Scholar]
- [19]. Kayes-Wandover KM, White PC. Steroidogenic enzyme gene expression in the human heart. J Clin Endocrinol Metab 2000; 85:2519-25; PMID:10902803 [DOI] [PubMed] [Google Scholar]
- [20]. Hostettler N, Bianchi P, Gennari-Moser C, Kassahn D, Schoonjans K, Corazza N, Brunner T. Local glucocorticoid production in the mouse lung is induced by immune cell stimulation. Allergy 2012; 67:227-34; PMID:22111694; http://dx.doi.org/10.1111/j.1398-9995.2011.02749.x [DOI] [PubMed] [Google Scholar]
- [21]. Slominski A, Zbytek B, Szczesniewski A, Wortsman J. Cultured human dermal fibroblasts do produce cortisol. J Investigat Dermatol 2006; 126:1177-8; PMID:16484985; http://dx.doi.org/10.1038/sj.jid.5700204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Slominski A, Zjawiony J, Wortsman J, Semak I, Stewart J, Pisarchik A, Sweatman T, Marcos J, Dunbar C, CT R. A novel pathway for sequential transformation of 7-dehydrocholesterol and expression of the P450scc system in mammalian skin. Eur J Biochem 2004; 271:4178-88; PMID:15511223; http://dx.doi.org/10.1111/j.1432-1033.2004.04356.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23]. Slominski AT, Li W, Kim TK, Semak I, Wang J, Zjawiony JK, Tuckey RC. Novel activities of CYP11A1 and their potential physiological significance. J Steroid Biochem Mol Biol 2015; 151:25-37; PMID:25448732; http://dx.doi.org/10.1016/j.jsbmb.2014.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Slominski AT, Zmijewski MA, Skobowiat C, Zbytek B, Slominski RM, Steketee JD. Sensing the environment: regulation of local and global homeostasis by the skin's neuroendocrine system. Adv Anat Embryol Cell Biol 2012; 212:v, vii, 1-115; PMID:NOT_FOUND; http://dx.doi.org/10.1007/978-3-642-19683-6_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Vukelic S, Stojadinovic O, Pastar I, Rabach M, Krzyzanowska A, Lebrun E, Davis SC, Resnik S, Brem H, Tomic-Canic M. Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J Biol Chem 2011; 286:10265-75; PMID:21239489; http://dx.doi.org/10.1074/jbc.M110.188268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 2004; 25:831-66; PMID:15466942; http://dx.doi.org/10.1210/er.2003-0031 [DOI] [PubMed] [Google Scholar]
- [27]. Seckl JR. 11beta-hydroxysteroid dehydrogenases: changing glucocorticoid action. Curr Opin Pharmacol 2004; 4:597-602; PMID:15525550; http://dx.doi.org/10.1016/j.coph.2004.09.001 [DOI] [PubMed] [Google Scholar]
- [28]. Terao M, Murota H, Kimura A, Kato A, Ishikawa A, Igawa K, Miyoshi E, Katayama I. 11beta-Hydroxysteroid dehydrogenase-1 is a novel regulator of skin homeostasis and a candidate target for promoting tissue repair. PLoS One 2011; 6:e25039; PMID:21949844; http://dx.doi.org/10.1371/journal.pone.0025039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Tiganescu A, Walker EA, Hardy RS, Mayes AE, Stewart PM. Localization, age- and site-dependent expression, and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in skin. J Investigat Dermatol 2011; 131:30-6; PMID:20739946; http://dx.doi.org/10.1038/jid.2010.257 [DOI] [PubMed] [Google Scholar]
- [30]. Hennebert O, Chalbot S, Alran S, Morfin R. Dehydroepiandrosterone 7alpha-hydroxylation in human tissues: possible interference with type 1 11beta-hydroxysteroid dehydrogenase-mediated processes. J Steroid Biochem Mol Biol 2007; 104:326-33; PMID:17467270; http://dx.doi.org/10.1016/j.jsbmb.2007.03.026 [DOI] [PubMed] [Google Scholar]
- [31]. Cirillo N, Prime SS. Keratinocytes synthesize and activate cortisol: first characterisation of a novel epidermal glucocorticoid system. J Cell Biochem 2011; PMID:21344493 [DOI] [PubMed] [Google Scholar]
- [32]. Terao M, Tani M, Itoi S, Yoshimura T, Hamasaki T, Murota H, Katayama I. 11beta-hydroxysteroid dehydrogenase 1 specific inhibitor increased dermal collagen content and promotes fibroblast proliferation. PLoS One 2014; 9:e93051; PMID:24667799; http://dx.doi.org/10.1371/journal.pone.0093051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Tiganescu A, Tahrani AA, Morgan SA, Otranto M, Desmouliere A, Abrahams L, Hassan-Smith Z, Walker EA, Rabbitt EH, Cooper MS, et al. 11beta-Hydroxysteroid dehydrogenase blockade prevents age-induced skin structure and function defects. J Clin Invest 2013; 123:3051-60; PMID:23722901; http://dx.doi.org/10.1172/JCI64162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]. Terao M, Itoi S, Murota H, Katayama I. Expression profiles of cortisol-inactivating enzyme, 11beta-hydroxysteroid dehydrogenase-2, in human epidermal tumors and its role in keratinocyte proliferation. Exp Dermatol 2013; 22:98-101; PMID:23362866; http://dx.doi.org/10.1111/exd.12075 [DOI] [PubMed] [Google Scholar]
- [35]. Skobowiat C, Sayre RM, Dowdy JC, Slominski AT. Ultraviolet radiation regulates cortisol activity in a waveband-dependent manner in human skin ex vivo. Br J Dermatol 2013; 168:595-601; PMID:23363016; http://dx.doi.org/10.1111/bjd.12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Holmes MC, Kotelevtsev Y, Mullins JJ, Seckl JR. Phenotypic analysis of mice bearing targeted deletions of 11beta-hydroxysteroid dehydrogenases 1 and 2 genes. Mol Cell Endocrinol 2001; 171:15-20; PMID:11165006; http://dx.doi.org/10.1016/S0303-7207(00)00386-5 [DOI] [PubMed] [Google Scholar]
- [37]. Tarutani M, Itami S, Okabe M, Ikawa M, Tezuka T, Yoshikawa K, Kinoshita T, Takeda J. Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc Natl Acad Sci U S A 1997; 94:7400-5; PMID:9207103; http://dx.doi.org/10.1073/pnas.94.14.7400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Lim HY, Muller N, Herold MJ, van den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 2007; 122:47-53; PMID:17451463; http://dx.doi.org/10.1111/j.1365-2567.2007.02611.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. Ergang P, Vytackova K, Svec J, Bryndova J, Miksik I, Pacha J. Upregulation of 11beta-hydroxysteroid dehydrogenase 1 in lymphoid organs during inflammation in the rat. J Steroid Biochem Mol Biol 2011; 126:19-25; PMID:21513799; http://dx.doi.org/10.1016/j.jsbmb.2011.04.002 [DOI] [PubMed] [Google Scholar]
- [40]. Zbankova S, Bryndova J, Leden P, Kment M, Svec A, Pacha J. 11beta-hydroxysteroid dehydrogenase 1 and 2 expression in colon from patients with ulcerative colitis. J Gastroenterol Hepatol 2007; 22:1019-23; PMID:17608848; http://dx.doi.org/10.1111/j.1440-1746.2006.04529.x [DOI] [PubMed] [Google Scholar]
- [41]. Stegk JP, Ebert B, Martin HJ, Maser E. Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Mol Cell Endocrinol 2009; 301:104-8; PMID:19022342; http://dx.doi.org/10.1016/j.mce.2008.10.030 [DOI] [PubMed] [Google Scholar]
- [42]. Hardy R, Rabbitt EH, Filer A, Emery P, Hewison M, Stewart PM, Gittoes NJ, Buckley CD, Raza K, Cooper MS. Local and systemic glucocorticoid metabolism in inflammatory arthritis. Ann Rheum Dis 2008; 67:1204-10; PMID:18420938; http://dx.doi.org/10.1136/ard.2008.090662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43]. Coutinho AE, Gray M, Brownstein DG, Salter DM, Sawatzky DA, Clay S, Gilmour JS, Seckl JR, Savill JS, Chapman KE. 11beta-Hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology 2012; 153:234-40; PMID:22067318; http://dx.doi.org/10.1210/en.2011-1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. Morgan SA, McCabe EL, Gathercole LL, Hassan-Smith ZK, Larner DP, Bujalska IJ, Stewart PM, Tomlinson JW, Lavery GG. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci U S A 2014; 111:E2482-91; PMID:24889609; http://dx.doi.org/10.1073/pnas.1323681111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45]. Harno E, Cottrell EC, Keevil BG, DeSchoolmeester J, Bohlooly YM, Andersen H, Turnbull AV, Leighton B, White A. 11-Dehydrocorticosterone causes metabolic syndrome, which is prevented when 11beta-HSD1 is knocked out in livers of male mice. Endocrinology 2013; 154:3599-609; PMID:23832962; http://dx.doi.org/10.1210/en.2013-1362 [DOI] [PubMed] [Google Scholar]
- [46]. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 2000; 21:55-89; PMID:10696570 [DOI] [PubMed] [Google Scholar]
- [47]. Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol 2001; 79:376-84; PMID:11488985; http://dx.doi.org/10.1046/j.1440-1711.2001.01025.x [DOI] [PubMed] [Google Scholar]
- [48]. Chantong B, Kratschmar DV, Nashev LG, Balazs Z, Odermatt A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J Neuroinflammation 2012; 9:260; PMID:23190711; http://dx.doi.org/10.1186/1742-2094-9-260 [DOI] [PMC free article] [PubMed] [Google Scholar]