

Abstract
SHOX in the short arm pseudoautosomal region (PAR1) of sex chromosomes is one of the major growth genes in humans. SHOX haploinsufficiency results in idiopathic short stature and Léri-Weill dyschondrosteosis and is associated with the short stature of patients with Turner syndrome. The SHOX protein likely controls chondrocyte apoptosis by regulating multiple target genes including BNP,Fgfr3, Agc1, and Ctgf. SHOX haploinsufficiency frequently results from deletions and duplications in PAR1 involving SHOX exons and/or the cis-acting enhancers, while exonic point mutations account for a small percentage of cases. The clinical severity of SHOX haploinsufficiency reflects hormonal conditions rather than mutation types. Growth hormone treatment seems to be beneficial for cases with SHOX haploinsufficiency, although the long-term outcomes of this therapy require confirmation. Future challenges in SHOX research include elucidating its precise function in the developing limbs, identifying additional cis-acting enhancers, and determining optimal therapeutic strategies for patients.
Key Words: Bone, Léri-Weill syndrome, Mutation, Pseudoautosomal region, Short stature, Skeletal deformity, Turner syndrome
In 1997, SHOX was reported as the causative gene for short stature in Turner syndrome [Rao et al., 1997]. Subsequently, heterozygous mutations of SHOX have been identified in patients with nonsyndromic short stature (idiopathic short stature, ISS) and Léri-Weill dyschondrosteosis (LWD) [Rao et al., 1997; Belin et al., 1998; Shears et al., 1998; Rappold et al., 2002]. Furthermore, SHOX abnormalities have been associated with various skeletal features of Turner syndrome such as scoliosis, high-arched palate, and micrognathia [Kosho et al., 1999; Binder, 2011]. Genetic defects leading to SHOX haploinsufficiency include intragenic mutations and deletions as well as copy number variations (CNVs) in the gene-flanking regions that possibly affect cis-regulatory machinery. This article provides an overview of the current understanding of SHOX.
The SHOX Gene
SHOX is located in the short arm pseudoautosomal region (PAR1) of the X and Y chromosomes (fig. 1). SHOX consists of exons 1-5 and alternatively spliced exons 6a and b (fig. 2) [Rao et al., 1997]. Like other genes in PAR1, SHOX escapes X-inactivation and therefore is present in 2 active forms in both males and females [Rao et al., 1997]. SHOX is expressed in the developing limbs and pharyngeal arches in human embryos and likely regulates differentiation and proliferation of chondrocytes [Clement-Jones et al., 2000]. Loss-of-function mutations of SHOX affect skeletal growth in a dose-dependent manner.
Fig. 1.

Genomic structure of SHOX and its putative enhancer regions. SHOX (red box) is located in PAR1 of sex chromosomes. Previous studies identified 7 highly evolutionarily conserved noncoding DNA elements (CNEs) with cis-regulatory activity (blue boxes). These elements were designated as CNE-5, −3, −2, 4, 5, and 9 [Chen et al., 2009; Durand et al., 2010]; evolutionarily conserved region (ECR) 1 [Benito-Sanz et al., 2012b]; and evolutionarily conserved sequence (ECS) 4 [Fukami et al., 2006]. The horizontal line indicates the physical distance from the Xp/Yp telomere (hg 19, build 37).
Fig. 2.

A, B Structural comparison among SHOXa, SHOX2a, and SHOXb. All of these proteins contain a homeodomain, while an OAR domain is present only in SHOXa and SHOX2a.
SHOX haploinsufficiency underlies the short stature of Turner syndrome patients and is associated with ISS and LWD. SHOX haploinsufficiency is estimated to account for 2-3% of ISS cases and ∼70% of LWD cases. Although the frequency of SHOX mutations and deletions in previously reported ISS and LWD cases varied from 1.5 to 16.9% and from 33.9 to 100%, respectively (table 1), this may reflect the differences in methods of mutation screening and inclusion criteria of participants. SHOX nullizygosity leads to Langer mesomelic dysplasia, an extremely rare condition characterized by severe short stature and skeletal deformity [Shears et al., 2002; Zinn et al., 2002]. Thus,SHOX is one of the major growth genes in humans. Although SHOX is located in the sex chromosomes, SHOX haploinsufficiency follows an autosomal dominant inheritance pattern. This phenomenon is defined as pseudoautosomal dominant inheritance [Shears et al., 2002]. Kant et al. [2011] demonstrated that heterozygous SHOX mutations can be transferred from the Y chromosome to the X chromosome and vice versa (‘the jumping SHOX gene’). Overdosage of SHOX has been implicated in the tall stature of individuals with 47,XXY (Klinefelter syndrome) or 47,XXX karyotypes (triple-X syndrome). Furthermore, trisomy of PAR1 involving SHOX was observed in a female with tall stature [Ogata et al., 2001a, 2002]. However, microduplications involving only small genomic intervals around SHOX have been identified in a few patients with ISS and LWD [Iughetti et al., 2010; Benito-Sanz et al., 2011; Fukami et al., 2015]. The mechanism of such diverse effects of SHOX overdosage on stature is discussed below.
Table 1.
Frequency of SHOX abnormalities in patients with idiopathic short stature, bilateral Madelung deformity, or LWD
| Report | Methods for mutation screening | Ethnic origin | Patient | Frequency of SHOX abnormality |
|---|---|---|---|---|
| Binder et al. [2000] | SSCP and microsatellite genotyping | not described | ISS | 1/68 (1.5) |
| Rappold et al. [2002] | SSCP | Japanese, German, | ISS | 9/750 (1.2) |
| FISH | Greek | 3/150 (2.0) | ||
| Flanagan et al. [2002] | FISH and direct sequencing | not described | BMD | 12/18 (66.7) |
| Schneider et al. [2005] | SNP/microsatellite genotyping and FISH | international | LWD | 40/118 (33.9) |
| Huber et al. [2006] | SNP/microsatellite marker genotyping and direct | French | LWD | 42/56 (75.0) |
| sequencing | ISS | 12/84 (14.3) | ||
| Benito-Sanz et al. [2006] | SNP/microsatellite genotyping and MLPA | Spanish | LWD | 16/26 (61.5) |
| Gatta et al. [2007] | MLPA and direct sequencing | not described | LWD | 7/15 (46.7) |
| Jorge et al. [2007] | Southern blotting, microsatellite marker | not described | LWD | 8/9 (88.9) |
| genotyping, FISH, and direct sequencing | ISSa | 2/63 (3.2) | ||
| DSS | 2/9 (22.2) | |||
| Fukami et al. [2008] | MLPA and direct sequencing | Japanese | LWD | 26/29 (89.7) |
| Chen et al. [2009] | SNP/microsatellite marker genotyping, FISH and | international | LWD | 29/58 (50.0) |
| MLPA | ISS | 31/735 (4.2) | ||
| Funari et al. [2010] | MLPA | Brazilian | LWD | 8/8 (100) |
| DSS | 4/36 (11.1) | |||
| Benito-Sanz et al. [2011] | MLPA | not described | LWD | 9/122 (7.4)b |
| ISS | 6/613 (0.9)b | |||
| Benito-Sanz et al., [2012b] | MLPA | international | LWD | 19/124 (15.3)c |
| ISS | 11/576 (1.9)c | |||
| Rosilio et al. [2012] | SNP genotyping and direct sequencing | French | LWD | 87/178 (48.9) |
| ISSa | 49/290 (16.9) | |||
| DSS | 13/69 (18.8) | |||
| Hirschfeldova et al. [2012] | MLPA and direct sequencing | not described | LWD | 11/16 (68.7) |
| ISS | 6/51 (11.8) | |||
| DSS | 6/6 (100) | |||
| Bunyan et al. [2013] | MLPA and direct sequencing | not described | LWD | 88/132 (66.7) |
| Sandoval et al., [2014] | MLPA | Colombian | ISS | 5/62 (8.1) |
| Poggi et al. [2015] | MLPA | Chilean | LWD | 18/27 (66.7) |
| ISS | 4/18 (22.2) |
BMD = Bilateral Madelung deformity; DSS = disproportionate short stature; ISS = idiopathic short stature; SSCP = single-strand conformation polymorphism. Percentages are given in parentheses.
This patient cohort included both proportionate and disproportionate short stature.
This study focused on copy number gain of SHOX.
This study focused on SHOX downstream deletion.
A paralog of SHOX, SHOX2, is located in chromosome 3 [Blaschke et al., 1998]. SHOX2 and SHOX protein structures share significant similarity (fig. 2). Although the precise function of SHOX2 remains to be clarified, previous studies have suggested that SHOX2 might play a role in the development of cardiac and neurological systems [Espinoza-Lewis et al., 2009; Rosin et al., 2015]. Thus, SHOX and SHOX2 likely have distinct functions, although both proteins may be involved in the skeletal growth [Blaschke and Rappold, 2006; Bobick and Cobb, 2012].
The SHOX Protein
The SHOX protein contains a homeodomain, a structure frequently seen in transcription factors involved in body patterning [Izpisúa-Belmonte and Duboule, 1992]. SHOX likely controls chondrocyte development by transactivating multiple target genes. In vitro assays demonstrated that SHOX induces oxidative stress in osteosarcoma cells and causes lysosomal membrane rupture to release active cathepsin B to the cytosol [Hristov et al., 2014]. Thus, SHOX appears to regulate the cell death process of chondrocytes in the growth plate. Since there is no SHOX ortholog in rodents, a knockout mouse approach cannot be applied to study the function of SHOX. Previous studies have utilized in vitro analysis and in vivo assays using chick micromass culture to identify putative target genes of SHOX [Tiecke et al., 2006; Aza-Carmona et al., 2011; Decker et al., 2011; Durand et al., 2012]. These studies suggested that SHOX exerts positive and negative effects on the expression of BNP and Fgfr3, respectively [Marchini et al., 2007; Decker et al., 2011]. In addition, SHOX interacts with the SOX trio, i.e. SOX5, SOX6, and SOX9, which function as the major chondrogenic factors, and thereby activates the enhancer of Agc1, a gene encoding the major component of cartilage [Aza-Carmona et al., 2011]. Moreover, HOXA9 has been reported as a regulator of SHOX [Durand et al., 2012]. Recently, Beiser et al. [2014] generated transgenic mice in which human SHOX is expressed under the control of the murine Col2a1 promoter and enhancer. The transgenic mice manifested no remarkable phenotypes, possibly because of low expression levels of SHOX in skeletal tissues. Nevertheless, detailed molecular analysis of the mice suggested that SHOX controls the expression of extracellular matrix genes including Ctgf in the developing limbs.
Molecular Basis of SHOX Haploinsufficiency
Previously reported SHOX abnormalities included various missense and nonsense mutations as well as nucleotide insertions or deletions in the coding exons 2-6a [Niesler et al., 2007; Binder, 2011]. These nucleotide alterations are listed in the SHOX mutation database (http://grenada.lumc.nl/LOVD2/MR/home.php?select_db=SHOX) [Niesler et al., 2007]. Known pathogenic SHOX mutations are widely distributed in exons 2-6a without hotspots [Binder, 2011]. Although a few nucleotide changes in exon 6b have been submitted to the database, the clinical significance of these substitutions is unclear. Indeed, SHOX isoform b encoded by exons 1-6b lacks a functionally important OAR domain and is therefore likely to be a nonfunctioning protein [Rao et al., 1997]. However, it is possible that the SHOXb isoform affects skeletal growth by regulating mRNA levels of the major SHOXa isoform [Durand et al., 2011]. Alternatively, SHOXb may compete with SHOXa cofactors such as SOX5 or SOX6, as suggested for SHOX2a and b [Aza-Carmona et al., 2014].
SHOX haploinsufficiency is more frequently caused by CNVs than point mutations [Benito-Sanz et al., 2005,2006,2011,2012a,b; Fukami et al., 2008; Chen et al., 2009; Rosilio et al., 2012]. Various submicroscopic microdeletions in PAR1 involving SHOX exons and/or its flanking regions have been identified in ISS and LWD patients. In particular, microdeletions in the SHOX downstream region have been reported as the most common genetic defects in LWD patients of Spanish origin [Benito-Sanz et al., 2006]. Microdeletions in PAR1 leading to LWD and ISS are predicted to affect exons and/or cis-acting enhancer elements of SHOX. Since monoallelic SHOX expression was confirmed in the skeletal tissues of a patient with a downstream deletion [Flanagan et al., 2002], elimination of SHOX enhancers seems to be sufficient to abolish gene expression. Although the actual positions of the upstream and downstream enhancers of SHOX have yet to be determined, they are likely located within highly conserved noncoding DNA elements (CNEs) around the gene because cis-acting enhancers are usually conserved among species [Pennacchio et al., 2006]. To date, 7 CNEs with in vitro or in vivo cis-regulatory activity have been identified in PAR1: 3 in the SHOX upstream region and 4 in the downstream region (fig. 1) [Fukami et al., 2006; Chen et al., 2009; Durand et al., 2010; Benito-Sanz et al., 2012b]. These CNEs are predicted to contain SHOX enhancers. Indeed, physical interaction between the CNEs and SHOX has been indicated by in vitro 3C assays [Benito-Sanz et al., 2012b; Verdin et al., 2015] and by in vivo assays using zebrafish [Kenyon et al., 2011]. Furthermore, since microdeletions in the far downstream region of known CNEs have recently been identified in patients with LWD-compatible skeletal features and/or short stature [Bunyan et al., 2014; Sandoval et al., 2014; Fukami et al., 2015], there may be a hitherto unidentified cis-acting element(s) of SHOX. In vitro assays have suggested that SHOX transcription could be regulated by multiple cis-acting elements distributed in a >1-Mb region in PAR1 [Verdin et al., 2015].
Recently, a few submicroscopic PAR1 microduplications were identified in patients with ISS and LWD [Iughetti et al., 2010; Benito-Sanz et al., 2011; Fukami et al., 2015]. This finding argues against the previous notion that SHOX overdosage underlies tall stature in individuals with Klinefelter syndrome and 47,XXX females. These apparently conflicting results can be reconciled by assuming that relatively large duplications involving all SHOX exons and cis-acting enhancers result in SHOX overexpression, while small duplications encompassing only some of these components may reduce SHOX expression levels by disrupting the cis-regulatory machinery [Fukami et al., 2015]. However, since microduplications around SHOX have also been identified in several individuals with normal stature [Benito-Sanz et al., 2011; Fukami et al., 2015], the pathogenicity of these CNVs needs to be confirmed in future studies. It is possible that the clinical consequence of each PAR1-linked duplication is determined by its genomic position and structure.
Characterization of the breakpoints of some PAR1-linked deletions suggested that nonallelic homologous recombination and nonhomologous end-joining play a role in the development of these CNVs [Fukami et al., 2006; Benito-Sanz et al., 2012b]. Likewise, the breakpoints of one duplication have been characterized, showing that this CNV was a tandem duplication mediated by Alu repeats [Fukami et al., 2015]. Notably, PAR1 is enriched with Alu repeats [Blaschke and Rappold, 2006], which may underlie the high frequency of CNVs in this region. Moreover, the high recombination rate of PAR1 during spermatogenesis may also be associated with the high frequency of PAR1-linked CNVs. Indeed, the average of the recombination rate in PAR1 during male meiosis is ∼17 times higher than the average of the genome [Hinch et al., 2014]. While there were no breakpoint hotspots, a 47.5-kb deletion in the SHOX downstream region was repeatedly identified in English and Scandinavian patients and ascribed to a founder effect [Bunyan et al., 2013].
SHOX abnormalities were absent in about 20% of LWD patients (table 1), and the genetic defects of these cases remained unknown until recently. Hisado-Oliva et al. [2015] demonstrated that heterozygous mutations in NPR2, a causative gene for Maroteaux-type acromesomelic dysplasia, result in LWD-like phenotypes. These findings provide the first indication of the locus heterogeneity for LWD. Thus, mutation analysis of NPR2 should be considered for ISS/LWD patients without SHOX abnormalities.
Clinical Manifestations of Patients with SHOX Haploinsufficiency
Patients with SHOX haploinsufficiency usually present with mesomelic short stature. In most cases, head circumferences and sitting height are within the normal range, while arm span is decreased and sitting height/height ratio is increased [Rappold et al., 2007; Binder, 2011; Malaquias et al., 2013]. Although apparent mesomelia can be absent in patients with SHOX haploinsufficiency, axiological examinations detect body disproportion in most patients [Rappold et al., 2007; Binder, 2011]. Longitudinal follow-up studies of female patients with SHOX haploinsufficiency showed that body disproportion often deteriorates during puberty [Fukami et al., 2003, 2004].
Growth failure in patients with SHOX haploinsufficiency usually occurs from the first years of age [Binder et al., 2004]. The mean adult height of ISS patients with SHOX haploinsufficiency and normal karyotype is around −2.2 SD, although growth failure can be more severe in patients with LWD phenotypes [Binder, 2011]. Thus, the mean growth deficit of SHOX haploinsufficiency is estimated to be ∼12 cm. This suggests that SHOX haploinsufficiency does not necessarily cause clinically discernible short stature. Consistent with this,SHOX haploinsufficiency has been identified in several individuals with normal stature. Since the mean adult height of females with Turner syndrome is about −3.2 SD, it appears that SHOX deficiency accounts for most but not all of the short stature in Turner patients.
The most characteristic clinical feature of SHOX haploinsufficiency is Madelung deformity (fig. 3). Madelung deformity is a combination of anatomical changes in the wrist consisting of bowing and shortening of the radius, prominence of the ulnar head, and palmar and ulnar deviation of the carpal bones (fig. 3) [Binder et al., 2001; Schmidt-Rohlfing et al., 2001]. Madelung deformity can be radiologically diagnosed by the absence or narrowing of the ulnar portion of the distal radial physis, anterior bowing of the radial shaft, and dorsal subluxation of the ulnar head (fig. 3). Histopathological analysis showed a disturbed columnar arrangement of chondrocytes in the growth plate, where tandem stacking of chondrocytes was replaced by a side-by-side arrangement [Munns et al., 2001]. Furthermore, abnormal enchondral ossification was indicated by hypertrophic osteoid in the radial metaphysis. The primary lesion of Madelung deformity appears to be the premature fusion of the distal radial epiphysis, which possibly results from an aberrant cell death process in the growth plate [Seki et al., 2014]. Furthermore, an aberrant ligament tethering the lunate to the distal portion of the radius was found in patients with Madelung deformity [Vickers and Nielsen, 1992; Harley et al., 2006; Steinman et al., 2013; Seki et al., 2014]. This ‘Vickers ligament’ likely compresses the distal epiphysis of the radius and further disturbs its linear growth. This ligament is predicted to develop under an aberrant mechanical force due to asymmetrical growth of the radius and ulna. Notably, although Madelung deformity is a characteristic feature of LWD, it can also occur in association with other disorders such as multiple exostoses syndrome, multiple epiphyseal dysplasia, mucopolysaccharidosis, pseudohypoparathyroidism type 1b, and injury.
Fig. 3.
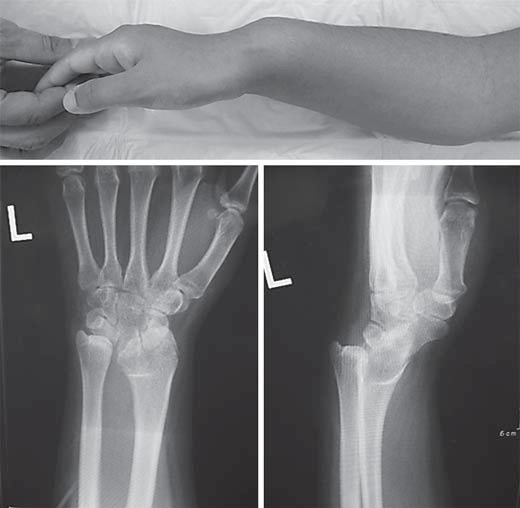
Madelung deformity of LWD. Bowing and shortening of the radius, prominence of the ulnar head, palmar and ulnar deviation of the carpal bones, and dorsal subluxation of the ulnar head are shown.
The severity of skeletal changes of SHOX haploinsufficiency is variable among patients and tends to be more severe in females than in males [Kosho et al., 1999; Binder, 2011]. While adult female patients often present with LWD, adult male patients and children usually exhibit ISS or only mild Madelung deformity. Relatively severe manifestations in adult females can be explained by the effect of gonadal estrogens. Since estrogens are known to enhance the fusion of growth plates in healthy males and females, they may accelerate premature epiphyseal fusion in individuals with SHOX haploinsufficiency. A relatively low frequency of LWD in Turner patients despite SHOX deficiency is consistent with attenuated estrogen production in these individuals.
Soucek et al. [2013] investigated bone mineral density and bone geometry in prepubertal patients with SHOX haploinsufficiency. They found a significantly increased total bone area, decreased relative cortical bone area, and a thin cortex. A possible interpretation of these findings is that the total bone area was increased to maintain the bone strength under the presence of mechanical loading. Similar findings were observed in prepubertal Turner patients, suggesting that SHOX plays a major role in bone geometrical changes of Turner syndrome.
SHOX haploinsufficiency is associated with additional clinical features [Rappold et al., 2007; Binder, 2011]. Of these, muscular hypertrophy in the lower limbs is of clinical importance because it has been reported in about one-third of SHOX-deficient patients [Rappold et al., 2007]. In addition, skeletal features of Turner syndrome, such as scoliosis, high-arched palate, short metacarpals, and micrognathia, were shared by a certain percentage of patients with SHOX haploinsufficiency and a normal karyotype [Rappold et al., 2007; Binder, 2011; Rosilio et al., 2012].
Genotype-Phenotype Correlation
The phenotypic severity of individuals with SHOX haploinsufficiency does not reflect the mutation types [Binder et al., 2004]. In fact, identical SHOX abnormalities have been detected in patients with ISS and LWD and in individuals with normal stature. Furthermore, no apparent phenotypic differences were reported between patients with missense mutations and those with nonsense or frameshift mutations [Binder et al., 2004]. Since the clinical manifestation of SHOX haploinsufficiency is usually more severe in adult female patients than in adult male and prepubertal patients, hormonal conditions rather than mutation types seem to determine the clinical consequences of SHOX haploinsufficiency. On the other hand, Rosilio et al. [2012] suggested that CNVs involving only the downstream enhancer regions lead to slightly milder phenotypes than mutations or deletions in the exons. SHOX downstream deletions may be associated with a broad clinical spectrum because Chen et al. [2009] documented a prominent phenotype in patients with such deletions. Furthermore, Donze et al. [2015] reported that patients with enhancer deletions were equally short as those with SHOX intragenic defects but were less disproportionate and showed better responses to growth hormone (GH) therapy. Benito-Sanz et al. [2011] suggested that SHOX duplications are often associated with relatively mild phenotypes. These findings suggest that there may be some phenotypic difference between SHOX exonic mutations/deletions and enhancer abnormalities.
Diagnosis of SHOX Abnormalities
SHOX haploinsufficiency can be diagnosed by the presence of Madelung deformity and mesomelic short stature, although these features are shared by only a certain percentage of the patients. A family history of autosomal dominant short stature supports the diagnosis of SHOX haploinsufficiency. Malaquias et al. [2013] demonstrated that body disproportion is a useful indicator of SHOX haploinsufficiency. Rappold et al. [2007] developed a scoring system for identification of the appropriate subjects for SHOX genetic testing. Score items of the system included arm span/height ratio, sitting height/height ratio, body mass index, cubitus valgus, short forearm, bowing of the forearm, muscular hypertrophy, and dislocation of the ulna.
Molecular analysis is useful to confirm the diagnosis of SHOX haploinsufficiency. Since SHOX haploinsufficiency is more frequently caused by submicroscopic CNVs than exonic point mutations, copy number analysis should be the first approach for molecular diagnosis. Multiplex ligation-dependent probe amplification (MLPA; MRC Holland, Amsterdam, The Netherlands) is frequently used for the initial screening of SHOX abnormalities (table 1) because it allows detection of copy number gains and losses of SHOX exons and the CNEs in a single assay. Array CGH is frequently used to confirm and characterize the CNVs identified by MLPA. Mutation analysis of the SHOX-coding region should be performed for patients without pathogenic CNVs. The SHOX mutation database is useful to assess the pathogenicity of missense mutations.
Treatment of Patients with SHOX Abnormalities
To date, management protocols for patients with SHOX haploinsufficiency have not been fully established. GH treatment has successfully improved growth velocity in several patients [Blum et al., 2013; Wit and Oostdijk, 2015]. The effects of GH on stature growth were comparable between patients with SHOX haploinsufficiency and those with Turner syndrome [Blum et al., 2013]. However, long-term outcomes of GH treatment need to be evaluated in future studies. Since Donze et al. [2015] reported that GH treatment was more effective in patients with enhancer deletions than in those with intragenic abnormalities, patients should be classified according to their mutation types. In addition, while Ogata et al. [2001b] suggested that gonadal suppression therapy may be useful to prevent the development of Madelung deformity in female patients, the outcome of this therapy remains to be investigated.
Surgical interventions have been carried out to reduce pain or improve wrist function in a few cases with severe Madelung deformity. In addition, previous studies suggested that surgical removal of the Vickers ligament in combination with dome osteotomy may benefit patients with Madelung deformity [Vickers and Nielsen, 1992; Harley et al., 2006; Steinman et al., 2013; Seki et al., 2014]. However, an optimal surgical procedure for Madelung deformity has yet to be determined.
Conclusions
SHOX is one of the major growth genes in humans, and its haploinsufficiency underlies syndromic and nonsyndromic short stature. SHOX haploinsufficiency represents a unique pseudoautosomal dominant disorder that mainly results from submicroscopic CNVs in PAR1. Future challenges in SHOX research include elucidation of its precise role in the developing limbs, identification of further cis-acting enhancers, and the development of optimal therapeutic strategies for patients.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
A.S. and T.O. have no conflicts of interest to disclose.
Acknowledgments
This work is supported by the Grants-in-Aid from the Japan Society for the Promotion of Science and by the Grants from the Japan Agency for Medical Research and Development, National Center for Child Health and Development, and Takeda Foundation. M.F. received a research grant from JCR Pharmaceuticals Co., Ltd.
References
- 1.Aza-Carmona M, Shears DJ, Yuste-Checa P, Barca-Tierno V, Hisado-Oliva A, et al. SHOX interacts with the chondrogenic transcription factors SOX5 and SOX6 to activate the aggrecan enhancer. Hum Mol Genet. 2011;20:1547–1559. doi: 10.1093/hmg/ddr032. [DOI] [PubMed] [Google Scholar]
- 2.Aza-Carmona M, Barca-Tierno V, Hisado-Oliva A, Belinchón A, Gorbenko-del Blanco D, et al. NPPB and ACAN, two novel SHOX2 transcription targets implicated in skeletal development. PLoS One. 2014;9:e83104. doi: 10.1371/journal.pone.0083104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beiser KU, Glaser A, Kleinschmidt K, Scholl I, Röth R, et al. Identification of novel SHOX target genes in the developing limb using a transgenic mouse model. PLoS One. 2014;9:e98543. doi: 10.1371/journal.pone.0098543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belin V, Cusin V, Viot G, Girlich D, Toutain A, et al. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet. 1998;19:67–69. doi: 10.1038/ng0198-67. [DOI] [PubMed] [Google Scholar]
- 5.Benito-Sanz S, Thomas NS, Huber C, Gorbenko del Blanco D, Aza-Carmona M, et al. A novel class of pseudoautosomal region 1 deletions downstream of SHOX is associated with Léri-Weill dyschondrosteosis. Am J Hum Genet. 2005;77:533–544. doi: 10.1086/449313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benito-Sanz S, del Blanco DG, Aza-Carmona M, Magano LF, Lapunzina P, et al. PAR1 deletions downstream of SHOX are the most frequent defect in a Spanish cohort of Léri-Weill dyschondrosteosis (LWD) probands. Hum Mutat. 2006;27:1062. doi: 10.1002/humu.9456. [DOI] [PubMed] [Google Scholar]
- 7.Benito-Sanz S, Barroso E, Heine-Suñer D, Hisado-Oliva A, Romanelli V, et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Léri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metab. 2011;96:E404–E412. doi: 10.1210/jc.2010-1689. [DOI] [PubMed] [Google Scholar]
- 8.Benito-Sanz S, Aza-Carmona M, Rodríguez-Estevez A, Rica-Etxebarria I, Gracia R, et al. Identification of the first PAR1 deletion encompassing upstream SHOX enhancers in a family with idiopathic short stature. Eur J Hum Genet. 2012a;20:125–127. doi: 10.1038/ejhg.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benito-Sanz S, Royo JL, Barroso E, Paumard-Hernández B, Barreda-Bonis AC, et al. Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J Med Genet. 2012b;49:442–450. doi: 10.1136/jmedgenet-2011-100678. [DOI] [PubMed] [Google Scholar]
- 10.Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011;75:81–89. doi: 10.1159/000324105. [DOI] [PubMed] [Google Scholar]
- 11.Binder G, Schwarze CP, Ranke MB. Identification of short stature caused by SHOX defects and therapeutic effect of recombinant human growth hormone. J Clin Endocrinol Metab. 2000;85:245–249. doi: 10.1210/jcem.85.1.6375. [DOI] [PubMed] [Google Scholar]
- 12.Binder G, Fritsch H, Schweizer R, Ranke MB. Radiological signs of Leri-Weill dyschondrosteosis in Turner syndrome. Horm Res. 2001;55:71–76. doi: 10.1159/000049973. [DOI] [PubMed] [Google Scholar]
- 13.Binder G, Renz A, Martinez A, Keselman A, Hesse V, et al. SHOX haploinsufficiency and Leri-Weill dyschondrosteosis: prevalence and growth failure in relation to mutation, sex, and degree of wrist deformity. J Clin Endocrinol Metab. 2004;89:4403–4408. doi: 10.1210/jc.2004-0591. [DOI] [PubMed] [Google Scholar]
- 14.Blaschke RJ, Rappold G. The pseudoautosomal regions, SHOX and disease. Curr Opin Genet Dev. 2006;16:233–239. doi: 10.1016/j.gde.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Blaschke RJ, Monaghan AP, Schiller S, Schechinger B, Rao E, et al. SHOT, a SHOX-related homeobox gene, is implicated in craniofacial, brain, heart, and limb development. Proc Natl Acad Sci USA. 1998;95:2406–2411. doi: 10.1073/pnas.95.5.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blum WF, Ross JL, Zimmermann AG, Quigley CA, Child CJ, et al. GH treatment to final height produces similar height gains in patients with SHOX deficiency and Turner syndrome: results of a multicenter trial. J Clin Endocrinol Metab. 2013;98:E1383–E1392. doi: 10.1210/jc.2013-1222. [DOI] [PubMed] [Google Scholar]
- 17.Bobick BE, Cobb J. Shox2 regulates progression through chondrogenesis in the mouse proximal limb. J Cell Sci. 2012;125:6071–6083. doi: 10.1242/jcs.111997. [DOI] [PubMed] [Google Scholar]
- 18.Bunyan DJ, Baker KR, Harvey JF, Thomas NS. Diagnostic screening identifies a wide range of mutations involving the SHOX gene, including a common 47.5 kb deletion 160 kb downstream with a variable phenotypic effect. Am J Med Genet A. 2013;161A:1329–1338. doi: 10.1002/ajmg.a.35919. [DOI] [PubMed] [Google Scholar]
- 19.Bunyan DJ, Taylor EJ, Maloney VK, Blyth M. Homozygosity for a novel deletion downstream of the SHOX gene provides evidence for an additional long range regulatory region with a mild phenotypic effect. Am J Med Genet A. 2014;164A:2764–2768. doi: 10.1002/ajmg.a.36724. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Wildhardt G, Zhong Z, Röth R, Weiss B, et al. Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory domain. J Med Genet. 2009;46:834–839. doi: 10.1136/jmg.2009.067785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet. 2000;9:695–702. doi: 10.1093/hmg/9.5.695. [DOI] [PubMed] [Google Scholar]
- 22.Decker E, Durand C, Bender S, Rödelsperger C, Glaser A, et al. FGFR3 is a target of the homeobox transcription factor SHOX in limb development. Hum Mol Genet. 2011;20:1524–1535. doi: 10.1093/hmg/ddr030. [DOI] [PubMed] [Google Scholar]
- 23.Donze SH, Meijer CR, Kant SG, Zandwijken GR, van der Hout AH, et al. The growth response to GH treatment is greater in patients with SHOX enhancer deletions compared to SHOX defects. Eur J Endocrinol. 2015;173:611–621. doi: 10.1530/EJE-15-0451. [DOI] [PubMed] [Google Scholar]
- 24.Durand C, Bangs F, Signolet J, Decker E, Tickle C, Rappold G. Enhancer elements upstream of the SHOX gene are active in the developing limb. Eur J Hum Genet. 2010;18:527–532. doi: 10.1038/ejhg.2009.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durand C, Roeth R, Dweep H, Vlatkovic I, Decker E, et al. Alternative splicing and nonsense-mediated RNA decay contribute to the regulation of SHOX expression. PLoS One. 2011;6:e18115. doi: 10.1371/journal.pone.0018115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durand C, Decker E, Roeth R, Schneider KU, Rappold G. The homeobox transcription factor HOXA9 is a regulator of SHOX in U2OS cells and chicken micromass cultures. PLoS One. 2012;7:e45369. doi: 10.1371/journal.pone.0045369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev Biol. 2009;327:376–385. doi: 10.1016/j.ydbio.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flanagan SF, Munns CF, Hayes M, Williams B, Berry M, et al. Prevalence of mutations in the short stature homeobox containing gene (SHOX) in Madelung deformity of childhood. J Med Genet. 2002;39:758–763. doi: 10.1136/jmg.39.10.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukami M, Matsuo N, Hasegawa T, Sato S, Ogata T. Longitudinal auxological study in a female with SHOX (short stature homeobox containing gene) haploinsufficiency and normal ovarian function. Eur J Endocrinol. 2003;149:337–341. doi: 10.1530/eje.0.1490337. [DOI] [PubMed] [Google Scholar]
- 30.Fukami M, Nishi Y, Hasegawa Y, Miyoshi Y, Okabe T, et al. Statural growth in 31 Japanese patients with SHOX haploinsufficiency: support for a disadvantageous effect of gonadal estrogens. Endocr J. 2004;51:197–200. doi: 10.1507/endocrj.51.197. [DOI] [PubMed] [Google Scholar]
- 31.Fukami M, Kato F, Tajima T, Yokoya S, Ogata T. Transactivation function of an approximately 800-bp evolutionarily conserved sequence at the SHOX 3′ region: implication for the downstream enhancer. Am J Hum Genet. 2006;78:167–170. doi: 10.1086/499254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukami M, Dateki S, Kato F, Hasegawa Y, Mochizuki H, et al. Identification and characterization of cryptic SHOX intragenic deletions in three Japanese patients with Léri-Weill dyschondrosteosis. J Hum Genet. 2008;53:454–459. doi: 10.1007/s10038-008-0269-z. [DOI] [PubMed] [Google Scholar]
- 33.Fukami M, Naiki Y, Muroya K, Hamajima T, Soneda S, et al. Rare pseudoautosomal copy-number variations involving SHOX and/or its flanking regions in individuals with and without short stature. J Hum Genet. 2015;60:553–556. doi: 10.1038/jhg.2015.53. [DOI] [PubMed] [Google Scholar]
- 34.Funari MF, Jorge AA, Souza SC, Billerbeck AE, Arnhold IJ, et al. Usefulness of MLPA in the detection of SHOX deletions. Eur J Med Genet. 2010;53:234–238. doi: 10.1016/j.ejmg.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Gatta V, Antonucci I, Morizio E, Palka C, Fischetto R, et al. Identification and characterization of different SHOX gene deletions in patients with Leri-Weill dyschondrosteosys by MLPA assay. J Hum Genet. 2007;52:21–27. doi: 10.1007/s10038-006-0074-5. [DOI] [PubMed] [Google Scholar]
- 36.Harley BJ, Brown C, Cummings K, Carter PR, Ezaki M. Volar ligament release and distal radius dome osteotomy for correction of Madelung's deformity. J Hand Surg Am. 2006;31:1499–1506. doi: 10.1016/j.jhsa.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 37.Hinch AG, Altemose N, Noor N, Donnelly P, Myers SR. Recombination in the human pseudoautosomal region PAR1. PLoS Genet. 2014;10:e1004503. doi: 10.1371/journal.pgen.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirschfeldova K, Solc R, Baxova A, Zapletalova J, Kebrdlova V, et al. SHOX gene defects and selected dysmorphic signs in patients of idiopathic short stature and Léri-Weill dyschondrosteosis. Gene. 2012;491:123–127. doi: 10.1016/j.gene.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 39.Hisado-Oliva A, Garre-Vázquez AI, Santaolalla-Caballero F, Belinchón A, Barreda-Bonis AC, et al. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Léri-Weill dyschondrosteosis. J Clin Endocrinol Metab. 2015;100:E1133–1142. doi: 10.1210/jc.2015-1612. [DOI] [PubMed] [Google Scholar]
- 40.Hristov G, Marttila T, Durand C, Niesler B, Rappold GA, Marchini A. SHOX triggers the lysosomal pathway of apoptosis via oxidative stress. Hum Mol Genet. 2014;23:1619–1630. doi: 10.1093/hmg/ddt552. [DOI] [PubMed] [Google Scholar]
- 41.Huber C, Rosilio M, Munnich A, Cormier-Daire V. French SHOX GeNeSIS Module: High incidence of SHOX anomalies in individuals with short stature. J Med Genet. 2006;43:735–739. doi: 10.1136/jmg.2006.040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iughetti L, Capone L, Elsedfy H, Bertorelli R, Predieri B, et al. Unexpected phenotype in a boy with trisomy of the SHOX gene. J Pediatr Endocrinol Metab. 2010;23:159–169. doi: 10.1515/jpem.2010.23.1-2.159. [DOI] [PubMed] [Google Scholar]
- 43.Izpisúa-Belmonte JC, Duboule D. Homeobox genes and pattern formation in the vertebrate limb. Dev Biol. 1992;152:26–36. doi: 10.1016/0012-1606(92)90153-8. [DOI] [PubMed] [Google Scholar]
- 44.Jorge AA, Souza SC, Nishi MY, Billerbeck AE, Libório DC, et al. SHOX mutations in idiopathic short stature and Leri-Weill dyschondrosteosis: frequency and phenotypic variability. Clin Endocrinol (Oxf) 2007;66:130–135. doi: 10.1111/j.1365-2265.2006.02698.x. [DOI] [PubMed] [Google Scholar]
- 45.Kant SG, van der Kamp HJ, Kriek M, Bakker E, Bakker B, et al. The jumping SHOX gene -crossover in the pseudoautosomal region resulting in unusual inheritance of Leri-Weill dyschondrosteosis. J Clin Endocrinol Metab. 2011;96:E356–E359. doi: 10.1210/jc.2010-1505. [DOI] [PubMed] [Google Scholar]
- 46.Kenyon EJ, McEwen GK, Callaway H, Elgar G. Functional analysis of conserved non-coding regions around the short stature hox gene (shox) in whole zebrafish embryos. PLoS One. 2011;6:e21498. doi: 10.1371/journal.pone.0021498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kosho T, Muroya K, Nagai T, Fujimoto M, Yokoya S, et al. Skeletal features and growth patterns in 14 patients with haploinsufficiency of SHOX: implications for the development of Turner syndrome. J Clin Endocrinol Metab. 1999;84:4613–4621. doi: 10.1210/jcem.84.12.6289. [DOI] [PubMed] [Google Scholar]
- 48.Malaquias AC, Scalco RC, Fontenele EG, Costalonga EF, Baldin AD, et al. The sitting height/height ratio for age in healthy and short individuals and its potential role in selecting short children for SHOX analysis. Horm Res Paediatr. 2013;80:449–456. doi: 10.1159/000355411. [DOI] [PubMed] [Google Scholar]
- 49.Marchini A, Häcker B, Marttila T, Hesse V, Emons J, et al. BNP is a transcriptional target of the short stature homeobox gene SHOX. Hum Mol Genet. 2007;16:3081–3087. doi: 10.1093/hmg/ddm266. [DOI] [PubMed] [Google Scholar]
- 50.Munns CF, Glass IA, LaBrom R, Hayes M, Flanagan S, et al. Histopathological analysis of Leri-Weill dyschondrosteosis: disordered growth plate. Hand Surg. 2001;6:13–23. doi: 10.1142/s0218810401000424. [DOI] [PubMed] [Google Scholar]
- 51.Niesler B, Röth R, Wilke S, Fujimura F, Fischer C, Rappold G. The novel human SHOX allelic variant database. Hum Mutat. 2007;28:933–938. doi: 10.1002/humu.20542. [DOI] [PubMed] [Google Scholar]
- 52.Ogata T, Matsuo N, Nishimura G. SHOX haploinsufficiency and overdosage: impact of gonadal function status. J Med Genet. 2001a;38:1–6. doi: 10.1136/jmg.38.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogata T, Onigata K, Hotsubo T, Matsuo N, Rappold G. Growth hormone and gonadotropin-releasing hormone analog therapy in haploinsufficiency of SHOX. Endocr J. 2001b;48:317–322. doi: 10.1507/endocrj.48.317. [DOI] [PubMed] [Google Scholar]
- 54.Ogata T, Inokuchi M, Ogawa M. Growth pattern and body proportion in a female with short stature homeobox-containing gene overdosage and gonadal estrogen deficiency. Eur J Endocrinol. 2002;147:249–254. doi: 10.1530/eje.0.1470249. [DOI] [PubMed] [Google Scholar]
- 55.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 56.Poggi H, Vera A, Avalos C, Lagos M, Mellado C, et al. A deletion of more than 800 kb is the most recurrent mutation in Chilean patients with SHOX gene defects. Horm Res Paediatr. 2015;84:254–257. doi: 10.1159/000439109. [DOI] [PubMed] [Google Scholar]
- 57.Rao E, Weiss B, Fukami M, Rump A, Niesler B, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure idiopathic short stature and Turner syndrome. Nat Genet. 1997;16:54–63. doi: 10.1038/ng0597-54. [DOI] [PubMed] [Google Scholar]
- 58.Rappold GA, Fukami M, Niesler B, Schiller S, Zumkeller W, et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab. 2002;87:1402–1406. doi: 10.1210/jcem.87.3.8328. [DOI] [PubMed] [Google Scholar]
- 59.Rappold G, Blum WF, Shavrikova EP, Crowe BJ, Roeth R, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet. 2007;44:306–313. doi: 10.1136/jmg.2006.046581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosilio M, Huber-Lequesne C, Sapin H, Carel JC, Blum WF, Cormier-Daire V. Genotypes and phenotypes of children with SHOX deficiency in France. J Clin Endocrinol Metab. 2012;97:E1257–E1265. doi: 10.1210/jc.2011-3460. [DOI] [PubMed] [Google Scholar]
- 61.Rosin JM, McAllister BB, Dyck RH, Percival CJ, Kurrasch DM, Cobb J. Mice lacking the transcription factor SHOX2 display impaired cerebellar development and deficits in motor coordination. Dev Biol. 2015;399:54–67. doi: 10.1016/j.ydbio.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 62.Sandoval GT, Jaimes GC, Barrios MC, Cespedes C, Velasco HM. SHOX gene and conserved noncoding element deletions/duplications in Colombian patients with idiopathic short stature. Mol Genet Genomic Med. 2014;2:95–102. doi: 10.1002/mgg3.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmidt-Rohlfing B, Schwöbel B, Pauschert R, Niethard FU. Madelung deformity: clinical features, therapy and results. J Pediatr Orthop B. 2001;10:344–348. [PubMed] [Google Scholar]
- 64.Schneider KU, Sabherwal N, Jantz K, Röth R, Muncke N, et al. Identification of a major recombination hotspot in patients with short stature and SHOX deficiency. Am J Hum Genet. 2005;77:89–96. doi: 10.1086/431655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seki A, Jinno T, Suzuki E, Takayama S, Ogata T, Fukami M. Skeletal deformity associated with SHOX deficiency. Clin Pediatr Endocrinol. 2014;23:65–72. doi: 10.1297/cpe.23.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shears DJ, Vassal HJ, Goodman FR, Palmer RW, Reardon W, et al. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat Genet. 1998;19:70–73. doi: 10.1038/ng0198-70. [DOI] [PubMed] [Google Scholar]
- 67.Shears DJ, Guillen-Navarro E, Sempere-Miralles M, Domingo-Jimenez R, Scambler PJ, Winter RM. Pseudodominant inheritance of Langer mesomelic dysplasia caused by a SHOX homeobox missense mutation. Am J Med Genet. 2002;110:153–157. doi: 10.1002/ajmg.10421. [DOI] [PubMed] [Google Scholar]
- 68.Soucek O, Zapletalova J, Zemkova D, Snajderova M, Novotna D, et al. Prepubertal girls with Turner syndrome and children with isolated SHOX deficiency have similar bone geometry at the radius. J Clin Endocrinol Metab. 2013;98:E1241–E1247. doi: 10.1210/jc.2013-1113. [DOI] [PubMed] [Google Scholar]
- 69.Steinman S, Oishi S, Mills J, Bush P, Wheeler L, Ezaki M. Volar ligament release and distal radial dome osteotomy for the correction of Madelung deformity: long-term follow-up. J Bone Joint Surg Am. 2013;95:1198–1204. doi: 10.2106/JBJS.L.00714. [DOI] [PubMed] [Google Scholar]
- 70.Tiecke E, Bangs F, Blaschke R, Farrell ER, Rappold G, Tickle C. Expression of the short stature homeobox gene Shox is restricted by proximal and distal signals in chick limb buds and affects the length of skeletal elements. Dev Biol. 2006;298:585–596. doi: 10.1016/j.ydbio.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 71.Verdin H, Fernández-Miñán A, Benito-Sanz S, Janssens S, Callewaert B, et al. Profiling of conserved non-coding elements upstream of SHOX and functional characterisation of the SHOX cis-regulatory landscape. Sci Rep. 2015;5:17667. doi: 10.1038/srep17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vickers D, Nielsen G. Madelung deformity: surgical prophylaxis (physiolysis) during the late growth period by resection of the dyschondrosteosis lesion. J Hand Surg Br. 1992;17:401–407. doi: 10.1016/s0266-7681(05)80262-1. [DOI] [PubMed] [Google Scholar]
- 73.Wit JM, Oostdijk W. Novel approaches to short stature therapy. Best Pract Res Clin Endocrinol Metab. 2015;29:353–366. doi: 10.1016/j.beem.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 74.Zinn AR, Wei F, Zhang L, Elder FF, Scott CI, et al. Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet. 2002;110:158–163. doi: 10.1002/ajmg.10422. [DOI] [PubMed] [Google Scholar]