

Abstract
Background:
Sensitizing effects of poly-ADP-ribose polymerase inhibitors have been studied in several preclinical models, but a clear understanding of predictive biomarkers is lacking. In this study, in vivo efficacy of veliparib combined with temozolomide (TMZ) was evaluated in a large panel of glioblastoma multiforme (GBM) patient-derived xenografts (PDX) and potential biomarkers were analyzed.
Methods:
The efficacy of TMZ alone vs TMZ/veliparib was compared in a panel of 28 GBM PDX lines grown as orthotopic xenografts (8–10 mice per group); all tests of statistical significance were two-sided. DNA damage was analyzed by γH2AX immunostaining and promoter methylation of DNA repair gene O6-methylguanine-DNA-methyltransferase (MGMT) by Clinical Laboratory Improvement Amendments–approved methylation-specific polymerase chain reaction.
Results:
The combination of TMZ/veliparib statistically significantly extended survival of GBM models (P < .05 by log-rank) compared with TMZ alone in five of 20 MGMT-hypermethylated lines (average extension in median survival = 87 days, range = 20–150 days), while the combination was ineffective in six MGMT-unmethylated lines. In the MGMT promoter–hypermethylated GBM12 line (median survival with TMZ+veliparib = 189 days, 95% confidence interval [CI] = 59 to 289 days, vs TMZ alone = 98 days, 95% CI = 49 to 210 days, P = .04), the profound TMZ-sensitizing effect of veliparib was lost when MGMT was overexpressed (median survival with TMZ+veliparib = 36 days, 95% CI = 28 to 38 days, vs TMZ alone = 35 days, 95% CI = 32 to 37 days, P = .87), and a similar association was observed in two nearly isogenic GBM28 sublines with an intact vs deleted MGMT locus. In comparing DNA damage signaling after dosing with veliparib/TMZ or TMZ alone, increased phosphorylation of damage-responsive proteins (KAP1, Chk1, Chk2, and H2AX) was observed only in MGMT promoter–hypermethylated lines.
Conclusion:
Veliparib statistically significantly enhances (P < .001) the efficacy of TMZ in tumors with MGMT promoter hypermethylation. Based on these data, MGMT promoter hypermethylation is being used as an eligibility criterion for A071102 (NCT02152982), the phase II/III clinical trial evaluating TMZ/veliparib combination in patients with GBM.
Temozolomide (TMZ) is a critical component of therapy for patients with glioblastoma (GBM), but the ultimate efficacy of TMZ is limited. Poly ADP ribose polymerase (PARP) enzymes play a critical role in repair of TMZ-induced DNA damage (1,2), and multiple preclinical studies have demonstrated excellent TMZ sensitizing effects of PARP inhibitors (3–6). While disruption of repair theoretically should sensitize essentially all tumors, the effects of PARP inhibitors are heterogeneous across tumor models (7,8). Moreover, for the PARP inhibitors in development, minimal brain penetration and/or excessive toxicity in combination with TMZ preclude use of some inhibitors in GBM (9,10). Based on initial promising results (6,11–13), the focus of this study was to define potential biomarkers associated with response to veliparib/TMZ in a panel of GBM patient-derived xenografts (PDXs).
PDX models provide a robust platform for evaluation of novel therapeutic strategies. By exclusive maintenance of tumors in mice, these models faithfully preserve the molecular, epigenetic, and genetic features of the original human specimens (14,15). The Mayo Clinic has developed a panel of GBM PDXs that are extensively characterized (16). The panel contains all major GBM expression subtypes (proneural, neural, classical, mesenchymal) (17), and molecular analyses demonstrate excellent genomic preservation between patient and xenograft tissues (unpublished results). The PDX models maintain promoter methylation of DNA repair gene O6-methylguanine-DNA-methyltransferase (MGMT), and similar to clinical experience MGMT promoter methylation in PDX models correlates with in vivo response to TMZ (18,19). These data suggest that the GBM PDX models are ideally suited for evaluation of TMZ sensitizing strategies.
In this study, extensive in vivo preclinical testing of TMZ/veliparib was used to guide the design of Alliance A071102 (NCT02152982), a randomized phase II/III clinical trial testing adjuvant TMZ combined with veliparib/placebo. Using a clinically relevant, cyclical dosing regimen, the efficacy of TMZ/veliparib was tested in orthotopic therapy studies in 28 GBM PDX models. In conjunction with studies in near-isogenic models differing in MGMT expression, the goal of this study was to delineate predictive biomarker strategy to enrich patients most likely to benefit from TMZ/veliparib combination.
Methods
Cell Culture, Drugs, and Antibodies
Short-term explant cultures of GBM12 were grown on laminin-coated flasks in neurobasal media (Life Technologies, Carlsbad, CA) (13). TMZ from the Mayo Clinic Pharmacy (Rochester, MN) was suspended in Ora-plus (Perrigo, Allegan, MN); veliparib from the Cancer Therapy Evaluation Program was diluted in saline. Antibodies used were phospho-S345-Chk1, phospho-T68-Chk2, γH2AX, Histone-H3, β-Actin, and PARP1 (Cell Signaling, Danvers, MA); Chk1, Chk2 (Millipore); phospho-S824-KAP1 (Abcam, Cambridge, MA); PAR (Trivigen, Gaithersburg, MD); KAP1 (Santa Cruz, Dallas, TX); MGMT (R&D, Minneapolis, MN). Western blotting was performed as described (13). Antibody dilutions and detailed methods for western blotting are provided in the Supplementary Methods (available online).
Genetic and Molecular Analyses
DNA extraction and polymerase chain reaction (PCR) were performed as described in Supplementary Methods (available online) (19–21). MGMT promoter methylation was analyzed using a Clinical Laboratory Improvement Amendments–validated quantitative real-time methylation-specific (MS-) PCR (22). Gene copy number was assessed using the Affymetrix 6.0 SNP array; the R package DNAcopy was used to detect abnormal copy number regions by circular binary segmentation (23).
Xenograft Studies
Studies were approved by the Mayo Clinic Institutional Animal Care and Use Committee, and all animal care procedures were followed. PDXs were maintained as previously described (16,18). Female athymic nude (Hsd:Athymic Nude-Foxn1nu, aged 6–7 weeks from Harlan, Indianapolis, IN) with established orthotopic tumors were randomized (8–10 mice per group) and treated with vehicle, TMZ, and/or veliparib by oral gavage, observed daily by staff blinded to treatment group, and euthanized upon reaching a moribund state. Pharmacokinetic assessment of veliparib in plasma and brain was as described previously and reported as mean ± standard deviation (13). For pharmacodynamic assessments, mice with established tumors were randomized (3 mice per group) and treated for five days and euthanized two or 72 hours after the last dose of TMZ. Immunostaining for γH2AX was as described (13,24).
Lentivirus Production and Cell Transduction
MGMT cDNA was cloned into pSIN-Luc-UbEm (25), lentivirus was packaged in HEK293T cells, and short-term explant cultures of GBM12 were transduced as previously described (13). Following transduction cells were FACS sorted and propagated as flank tumors.
Statistical Analyses
Survival was defined as time from tumor implantation to reach a moribund state. Median survival was estimated by the Kaplan-Meier method with corresponding 95% confidence interval (CI). Differences in survival across groups were assessed using the log-rank test. Survival ratios (fold change in median survival for TMZ or TMZ/veliparib treatment relative to placebo) were compared across treatment groups using the paired signed rank test. Differences in survival ratios between GBM lines with different molecular alterations or status were compared using two-sample Wilcoxon rank-sum test. Mead’s resource equation was used to determine the sample size for each experiment (26). Two-sided P values of less than .05 were considered statistically significant.
Results
Defining Optimal Dose and Schedule
The pharmacokinetics of TMZ is similar between mice and humans (27), and equivalent exposures of veliparib can be modeled with twice-daily dosing in mice (11). With this schedule, maximal plasma and brain veliparib concentrations were 370.1±76.4 and 173.3±21.5ng/mL, respectively (Figure 1A), which are similar to peak plasma exposure for veliparib in humans (28). Based on this and previous studies (6,12,29), veliparib was dosed at 12.5mg/kg twice daily, and all therapy evaluations were performed in orthotopic models.
Figure 1.

Pharmacokinetics and dosing schedule evaluation. A) Brain and plasma veliparib levels were measured up to six hours after the ninth dose (12.5mg/kg), administered twice daily; presented in the graphs are the averages from five observations, and vertical bars represent standard deviation. B-D) Evaluation of different dosing schedules for placebo, temozolomide (TMZ) alone, or combined with continuous veliparib were evaluated in a single experiment (n = 10 mice per group). Veliparib was dosed at 12.5mg/kg bid Monday through Friday (M-F) for six weeks combined with various TMZ regimens. B) Standard TMZ: 50mg/kg daily M-F x 1 week, (C) dose dense TMZ: TMZ 25mg/kg daily M-F x 3 weeks, and (D) metronomic TMZ: 15mg/kg M-F x 6 weeks. E) Standard TMZ (50mg/kg x 5 days) in combination with veliparib 12.5mg/kg bid x 5 or 12 days for two 28-day cycles (n = 10 mice per group, except TMZ group had n = 9 mice). P values by log-rank are reported for the comparison of TMZ vs TMZ + veliparib in all cases, except that the P value in E compares TMZ combined with veliparib x 5 days vs TMZ with veliparib x 12 days. All statistical tests were two-sided. AUC = area under the curve; TMZ = temozolomide.
An initial study was performed to evaluate three dosing schedules in the GBM12 model. Similar to previous results (12), a profound sensitizing effect was observed with conventional dosing of TMZ (50mg/kg, days 1–5) and veliparib (median survival = 113 days, 95% CI = 76 to 176) compared with TMZ alone (median survival = 60, 95% CI = 56 to 64, P < .001) (Figure 1B). In contrast, a dose-dense TMZ regimen (25mg/kg, days 1–5, 8–12, and 15–19) combined with veliparib resulted in increased toxicity-related deaths (5 of 10 mice) during therapy and did not statistically significantly extend survival compared with TMZ alone (Figure 1C). Metronomic TMZ (15mg/kg, days 1–5, weekly) with concurrent veliparib had inferior survival as compared with metronomic TMZ alone (Figure 1D). Similar results were seen in the GBM28B xenograft line, which lacks MGMT expression (Supplementary Figure 1, available online). Veliparib combined with standard TMZ statistically significantly extended median survival (189 days, 95% CI = 59 to 289) compared with TMZ alone vs (98, 95% CI = 49 to 210, P = .04), while veliparib combined with dose-dense TMZ was no different than monotherapy (Supplementary Figure 1, A and B, available online). In a subsequent GBM12 experiment, extending veliparib treatment beyond the end of TMZ therapy had no impact on efficacy (Figure 1E). Collectively, these data suggest that TMZ and veliparib dosed days 1–5 every 28 may provide superior efficacy and is better tolerated than alternative schedules.
Effect of MGMT Promoter Methylation Status on Treatment Efficacy
The efficacy of cyclical TMZ/veliparib was evaluated in 28 GBM PDXs; six models were MGMT promoter unmethylated, 20 hypermethylated, one indeterminate, and one MGMT deleted. As shown in Table 1, combined TMZ/veliparib provided a statistically significant but limited gain in median survival in only one unmethylated model (GBM6: 64 days, 95% CI = 57 to 69 vs 58, 95% CI = 50 to 58 with TMZ alone, P = .007). However, statistically significant increases (P < .05 by log-rank) in survival were observed in five of 20 MGMT promoter-hypermethylated GBM xenograft models, with an average increase in median survival of 87 days (range = 20–150 days) (Table-1; Supplementary Figures 2 and 3, available online). Despite an indeterminate MGMT status, GBM75 was highly sensitive to TMZ (placebo: median survival = 52, 95% CI = 36 to 73 vs 254; TMZ: 95% CI = 178 to 268, P < .001) and TMZ/veliparib was associated with a statistically nonsignificant (P = .08) but potentially meaningful 54-day (range = 26–71 days) median prolongation in survival as compared with TMZ alone (Table-1). Based on these results, the impact of MGMT status on response was analyzed by comparing the survival ratio (median survival for treatment relative to placebo) and the survival ratio difference (TMZ/veliparib - TMZ alone). As shown in Figure 2A, compared with the survival ratio difference in MGMT unmethylated lines (0.08, range = -1.53-0.24), the benefit in hypermethylated lines was statistically significantly greater (0.45, range = -0.48–8.64, P = .04). When stratified by methylation status, there was no statistically significant difference for unmethylated tumors in median survival ratio for TMZ/veliparib (1.7, range = 1.03–2.8) vs TMZ alone (1.5, range = 1.07–4.36), while for hypermethylated tumors there was a statistically significant increase in median survival ratio (TMZ/veliparib: 3.91, range = 1.2–12.6; TMZ alone: 3.1, range = 1.1–5.6, P < .001) (Figure 2, B and C). No statistically significant survival ratio difference (TMZ/veliparib vs TMZ alone) by PTEN (P = .22), p53 (P = .22), or EGFR (P = .37) status was observed (Supplementary Table 1 and Supplementary Figure 4, available online). In summary, meaningful and statistically significant prolongation in survival with TMZ/veliparib was limited to tumors with MGMT hypermethylation.
Table 1.
Response to treatment in orthotopic models of glioblastoma patient-derived xenografts*
| GBM lines | Treatment start, d |
Median survival (95% CI), d | Survival ratio† | |||||
|---|---|---|---|---|---|---|---|---|
| Placebo | Veliparib | TMZ | TMZ +veliparib |
TMZ | TMZ+ veliparib |
Δ-ratio‡ | ||
| MGMT unmethylated | ||||||||
| GBM6 | 12 | 41 (13 to 44) |
NA | 58 (50 to 58) |
64§ (57 to 69) |
1.41 | 1.56 | 0.15 |
| GBM14R | 6 | 27 (21 to 27) |
NA | 62 (27 to 69) |
69 (32 to 81) |
2.30 | 2.56 | 0.26 |
| GBM28A | 10 | 26 (26 to 31) |
NA | 33 (28 to 40) |
33 (26 to 62) |
1.27 | 1.27 | 0 |
| GBM43 | 7 | 14 (14 to 22) |
NA | 61 (53 to 65) |
39 (14 to 39) |
4.36 | 2.79 | -1.57 |
| GBM79 | 5 | 30 (29 to 31) |
NA | 32 (29 to 33) |
31 (29 to 31) |
1.07 | 1.03 | -0.04 |
| GBM122 | 63 | 80n=9
(66 to 96) |
82 (72 to 87) |
124 (90 to 149) |
140 (63 to 156) |
1.55 | 1.75 | 0.20 |
| MGMT hypermethylated | ||||||||
| GBM5 | 19 | 103n=9
(78 to 107) |
106n=9
(58 to 114) |
185 (19 to 267) |
268 (19 to 303) |
1.80 | 2.60 | 0.80 |
| GBM8 | 14 | 59 (49 to 61) |
52 (45 to 55) |
260 (160 to 296) |
231n=9
(146 to 314) |
4.41 | 3.91 | -0.50 |
| GBM12 | 4 | 15 (15 to 17) |
NA | 59 (3 to 62) |
189§ (75 to 257) |
3.93 | 12.60 | 8.67 |
| GBM15 | 34 | 71 (62 to 79) |
69 (61 to 72) |
249 (116 to 350) |
438 (171 to 452) |
3.51 | 6.17 | 2.66 |
| GBM22 | 7 | 20 (18 to 22) |
19 (17 to 32) |
58 (51 to 67) |
94§ (11 to 222) |
2.90 | 4.70 | 1.80 |
| GBM39 | 17 | 28n=8
(27 to 28) |
30 (26 to 31) |
138 (109 to 141) |
288§,n=7
(85 to 327) |
4.93 | 10.29 | 5.36 |
| GBM46R | 23 | 34 (25 to 45) |
38 (29 to 45) |
36 (23 to 43) |
49 (23 to 57) |
1.06 | 1.44 | 0.38 |
| GBM59 | 16 | 42n=20
(38 to 44) |
NA | 100n=20
(80 to 131) |
182n=20
(122 to 271) |
2.38 | 4.33 | 1.95 |
| GBM61 | 14 | 236 (152 to 278) |
315 (179 to 439) |
331 (125 to 465) |
435 (328 to 456) |
1.40 | 1.84 | 0.44 |
| GBM63 | 47 | 82n=9
(77 to 141) |
95 (76 to 117) |
263 (103 to 289) |
276 (224 to 294) |
3.21 | 3.37 | 0.16 |
| GBM76R | 17 | 77 (73 to 78) |
76 (73 to 78) |
216 (158 to 259) |
317§ (198 to 349) |
2.81 | 4.11 | 1.31 |
| GBM84 | 26 | 56 (46 to 67) |
58 (36 to 68) |
191 (159 to 234) |
219 (153 to 267) |
3.41 | 3.91 | 0.50 |
| GBM85 | 25 | 79n=9
(54 to 109) |
85 (64 to 101) |
233 (27 to 303) |
270 (211 to 325) |
2.95 | 3.42 | 0.47 |
| GBM102R | 25 | 71.5 (66 to 75) |
69 (65 to 69) |
160 (145 to 169) |
180§ (160 to 223) |
2.24 | 2.52 | 0.28 |
| GBM114 | 53 | 82 (74 to 93) |
95 (79 to 104) |
234 173 to 245 |
235 (84 to 245) |
2.85 | 2.87 | 0.02 |
| GBM115 | 23 | 140.5 (93 to 194) |
167 (134 to 210) |
173 (92 to 217) |
169 (116 to 187) |
1.23 | 1.20 | -0.03 |
| GBM116 | 25 | 61n=9
(42 to 219) |
59.5 (48 to 69) |
339 (140 to 366) |
343 (218 to 366) |
5.56 | 5.62 | 0.06 |
| GBM117 | 33 | 64.5 (60 to 91) |
62 (55 to 76) |
289 (199 to 328) |
279 (93 to 328) |
4.48 | 4.33 | -0.15 |
| GBM143R | 13 | 53.5 (38 to 71) |
56 (48 to 60) |
184 (21 to 202) |
183 (139 to 229) |
3.44 | 3.42 | -0.02 |
| GBM151 | 42 | 57 (54 to 63) |
58 (54 to 71) |
288 (103 to 423) |
317 (103 to 343) |
5.05 | 5.56 | 0.51 |
| MGMT methylation indeterminate | ||||||||
| GBM75 | 18 | 52n=9
(36 to 73) |
55 (35 to 59) |
254 (178 to 268) |
318 (235 to 326) |
4.88 | 6.11 | 1.23 |
| MGMT deleted | ||||||||
| GBM28B | 12 | 25 (23 to 25) |
26 (23 to 26) |
90 (35 to 91) |
124§ (96 to 268) |
3.60 | 4.96 | 1.36 |
* Each treatment group had 10 mice, with exceptions indicated by the superscript. CI = confidence interval; MGMT = O 6-methylguanine DNA methyltransferase; NA = not available; R = tumor line was established from a recurrent tumor; TMZ = temozolomide.
† Survival ratio was calculated by dividing the median survival in TMZ or TMZ+veliparib groups by median survival in placebo group.
‡ Δ-ratio was the difference in survival ratios between TMZ+velparib and TMZ groups.
§ Two-sided log-rank statistic P < .05 comparing median survival between TMZ vs TMZ+veliparib groups.
Figure 2.

Comparison of temozolomide (TMZ)/veliparib vs TMZ response in Mayo glioblastoma multiforme (GBM) patient-derived xenograft (PDX) models based on MGMT promoter methylation status. Ratio of median survival for treatment (TMZ/veliparib or TMZ alone) relative to placebo (survival ratio) and the difference of survival ratio for TMZ/veliparib minus survival ratio for TMZ alone (survival ratio difference) are presented as boxplots. A) Boxplots show the survival ratio difference based on O 6-methylguanine DNA methyltransferase (MGMT) methylation status for 26 PDX lines. B) Boxplots show the survival ratio for TMZ/veliparib or TMZ alone for six MGMT promoter-unmethylated PDX models and (C) 20 MGMT promoter-hypermethylated PDX GBM models. For each xenograft line, mice with established orthotopic xenografts were randomized to therapy with placebo, veliparib (12.5mg/kg bid x 5 days), TMZ alone (50mg/kg x 5 days), or the combination of TMZ/veliparib for three cycles. Mice were observed daily and killed upon reaching a moribund state. The P values denote a Wilcoxon rank-sum test for graph in (A) and a paired signed rank test for graphs in (B and C). All statistical tests were two-sided. Δ-MS ratio = survival ratio difference; MGMT = O 6-methylguanine DNA methyltransferase.
Impact of TMZ and Veliparib on DNA Damage Signaling
The potential association of MGMT promoter hypermethylation on pharmacodynamic effects of TMZ/veliparib was explored in two MGMT-hypermethylated (GBM12 and GBM39) and two MGMT-unmethylated (GBM6 and GBM43) models (Figure 3, A and B). Veliparib only treated tumors were harvested either two hours after (GBM12 and GBM39) or 72 hours after (GBM6 or GBM43) the last dose of drug, and as expected there was more profound PARP activity suppression in tumors harvested at the earlier time point. TMZ alone resulted in more robust DNA damage-induced phosphorylation of Chk1, Chk2, KAP1, and H2AX in hypermethylated compared with unmethylated tumors. Exclusively in the hypermethylated lines, the combination of TMZ/veliparib resulted in greater phosphorylation of KAP1 and H2AX as compared with TMZ alone (Figure 3, A and B). Interestingly, in the unmethylated GBM6, veliparib/TMZ resulted in a modest increase in Chk1 phosphorylation but no other signaling proteins. To validate the results from this flank tumor study, γH2AX foci staining was evaluated in GBM12 orthotopic tumors treated with TMZ or TMZ/veliparib. Consistent with the western blotting results, the fraction of cells with γH2AX staining was subtly higher following treatment with TMZ/veliparib vs TMZ alone (Figure 3C). Taken together, these results suggest that the addition of veliparib to TMZ treatment results in greater DNA damage in MGMT-hypermethylated lines.
Figure 3.
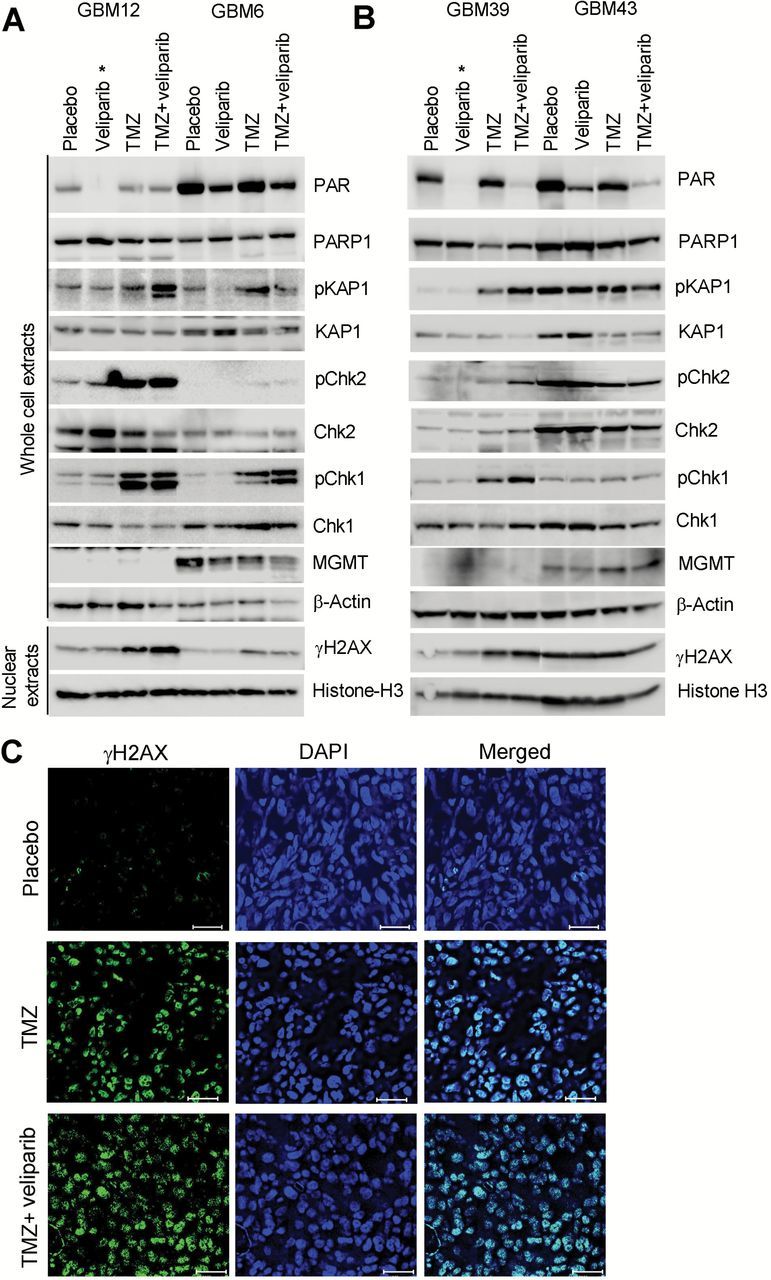
DNA damage signaling following temozolomide (TMZ)/veliparib treatment. Western blotting analysis of DNA damage response following treatment of flank tumor xenografts for five days with either placebo, veliparib, TMZ, or TMZ/veliparib (pooled samples from 3 mice per group) as in Figure 2 in (A) GBM12 vs GBM6 and (B) GBM39 vs GBM43. Tumor samples were harvested 72 hours after the last dose of TMZ, except veliparib-treated samples in GBM12 and GBM39 were harvested at two hours as denoted by an asterisk (*). C) Mice with orthotopic GBM12 tumors were treated as in (A) and processed for immunofluorescence for γH2AX (green) and DAPI (blue) 72 hours after the last dose of TMZ. Images captured with a 20X objective on a Leica AF6000 microscope; bar = 50 µm. Chk1 = checkpoint kinase 1; Chk2 = checkpoint kinase 2; DAPI = 4’,6-diamidino-2-phenylindole; KAP1 = KRAB-associated protein 1; MGMT = O 6-methylguanine DNA methyltransferase; PAR = poly ADP-ribose; PARP1 = poly ADP-ribose polymerase 1.
Impact of MGMT Expression on TMZ/Veliparib Efficacy
Promoter hypermethylation suppresses MGMT gene expression and is associated with enhanced TMZ sensitivity, while lack of methylation and increased MGMT expression is mechanistically linked to TMZ resistance. Our preceding results suggest MGMT expression may be an important determinant of veliparib sensitizing effects. Therefore, the impact of MGMT expression was evaluated in GBM12 using two different models: MGMT overexpression via lentiviral transduction (GBM12-MGMT) or acquired TMZ resistance associated with MGMT expression (GBM12TMZ#3080) (Figure 4A) (30). As expected, both MGMT-overexpressing models were highly resistant to TMZ alone (Figure 4, B and C). Moreover, MGMT expression was associated with prominent lack of efficacy for the combination of TMZ/veliparib compared with TMZ alone (GBM12-MGMT: median survival = 36 days, 95% CI = 28 to 38 days, vs 35 days, 95% CI = 32 to 37 days, respectively, P = .87; GBM12TMZ#3080: median survival = 17 days, 95% CI = 15 to 24 days, vs 15 days, 95% CI = 14 to 15 days, respectively, P = .001). Compared with the responsive parental GBM12 line, the lack of veliparib efficacy in these two MGMT-overexpressing models indicates that MGMT can reverse veliparib-mediated TMZ sensitization.
Figure 4.
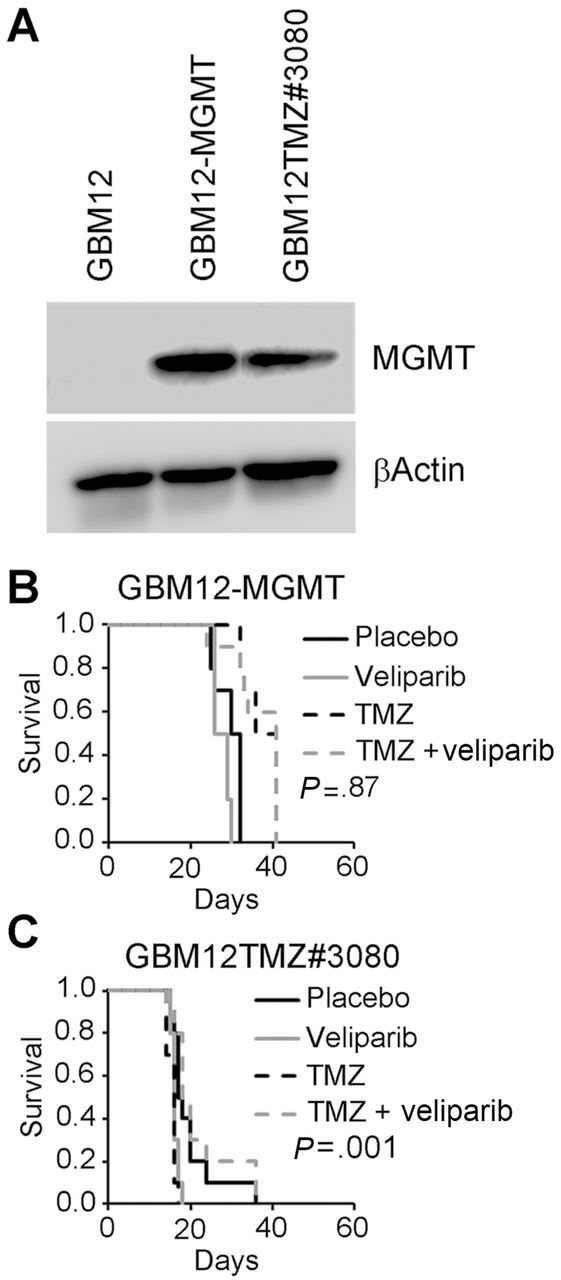
O 6-methylguanine DNA methyltransferase (MGMT) overexpression decreases sensitizing effects of veliparib. A) Western blot analysis of GBM12, GBM12-MGMT (GBM12 cells transduced with pSIN-MGMT-UbEm), or GBM12TMZ#3080 (temozolomide [TMZ]-resistant derivative of GBM12 with acquired MGMT expression). Assessment of efficacy of TMZ, veliparib, or the combination in orthotopic models (n = 10 mice per group) of (B) GBM12-MGMT and (C) GBM12TMZ#3080 using the dosing regimen in Figure 1B. The P values denote a log-rank test comparing survival in TMZ/veliparib vs TMZ alone. All statistical tests were two-sided. MGMT = O 6-methylguanine DNA methyltransferase.
The influence of MGMT expression on veliparib sensitization was further explored in GBM28. During routine passage, a GBM28 variant was identified, denoted as GBM28B, that harbored a bi-allelic deletion of MGMT as defined by genomic PCR (Figure 5A); for clarity, the parental line with intact MGMT is denoted as GBM28A. GBM28A has monosomy of chromosome 10, and comparing gene copy number across the genome the only the difference detected was a focal genetic deletion in GBM28B at chromosome 10 (band 10q26.3; base pair 130370868 to 131992367) (Figure 5B; Supplementary Figure 5, available online). This region encodes for three protein-encoding genes: MGMT, early B-cell factor 3 (EBF3), and glutaredoxin 3 (GLRX3). The PTEN gene on band 10q23.31 remained intact. As noted in Table 1 and Figure 5, C and D, GBM28A was highly resistant to TMZ alone or TMZ/veliparib (median survival = 33 days, 95% CI = 28 to 40 days, vs 33 days, 95% CI = 26 to 62 days, respectively, P = .24), while GBM28B was highly sensitive to TMZ compared with placebo (median survival = 90 days, 95% CI = 35 to 91 vs 25 days, 95% CI = 23 to 25 days, respectively, P < .001) (Figure 5D) and TMZ/veliparib was associated with a further prolongation of median survival (124 days, 95% CI = 96 to 268 days, P < .001). Collectively, these results support the concept that lack of MGMT expression is important for the sensitizing effects of veliparib combined with TMZ in vivo.
Figure 5.

Influence of O 6-methylguanine DNA methyltransferase (MGMT) status in GBM28 sublines. A) Polymerase chain reaction (PCR) amplification of MGMT and PTEN in GBM28 xenografts after indicated passage. Lanes representing passage 9 had both MGMT and PTEN intact and are denoted as GBM28A, while lanes representing late passage 13 with MGMT deleted but PTEN intact are denoted as GBM28B. B) aCGH analysis comparing GBM28A and GBM28B. Copy number variations on chromosome 10 are shown. C and D) The efficacy of TMZ, veliparib, or the combination using the dosing regimen in Figure 2 are shown in orthotopic models (n = 10 mice per group) of (C) GBM28A (D) GBM28B. The P values denote a log-rank test comparing survival in TMZ/veliparib vs TMZ alone. All statistical tests were two-sided. kBP = kilo basepairs of DNA; MGMT = O 6-methylguanine DNA methyltransferase; Pass# = passage number; PTEN = phosphatase and tensin homolog.
Discussion
PARP inhibitors enhance the efficacy of TMZ in multiple preclinical models and are a promising strategy for GBM (11,31–33). In vitro studies have demonstrated profound sensitization by PARP inhibitors in chemotherapy-resistant lines (3,34–36). In contrast, we recently reported that in vitro sensitizing effects of veliparib/TMZ in resistant lines are only possible at drug concentrations intolerable for mice (12,13). This is especially important in GBM as the blood brain barrier may further limit achievable drug levels (37,38). Notably, veliparib has moderate brain penetration with a brain to plasma ratio of approximately 50% (Figure 1A) (11). Previous preclinical efficacy studies have investigated the combination of veliparib and TMZ in several models using established cancer cell lines from a variety of tumors, including a rat glioma model and four established human GBM cell lines (11,39–41). Glioma genetically engineered mouse models (GEMMs) also have been used to investigate veliparib/TMZ (42), and the use of selected PDX models was reported previously by our group (12). Here, the efficacy of TMZ and veliparib was assessed in a much larger panel of 28 genetically diverse orthotopic GBM PDXs with a dosing regimen similar to that used clinically (29,43–45). These experiments demonstrate that a subset of PDX models profoundly benefit from the addition of veliparib to TMZ in vivo.
Clinically significant survival extension with veliparib/TMZ treatment was limited to models that were inherently sensitive to TMZ. Consistent with clinical experience, MGMT hypermethylation best defined sensitivity to TMZ in the GBM PDX models, and five of seven veliparib/TMZ-responsive models also were hypermethylated. The remaining two highly responsive models were MGMT deleted (GBM28B) or MGMT methylation indeterminate (analytical gray zone between methylated/unmethylated). While one unmethylated line had a six-day prolongation in survival, MGMT expression in isogenic PDX models (Figures 4–5) demonstrated that MGMT overexpression markedly suppresses the TMZ-sensitizing effects of veliparib. Although correlation of MGMT expression with in vivo response to TMZ/veliparib has not been explored previously, association between low MGMT expression and sensitizing effects of talazoparib in pediatric tumor PDX models was reported recently (46). MGMT expression is dynamically regulated in response to alkylation damage (19), which may explain why MGMT mRNA or protein levels are less robust predictors of TMZ efficacy in clinical studies (47–49). In contrast, promoter hypermethylation defines a closed chromatin state that limits MGMT upregulation following alkylator treatment, and the prognostic value of MGMT hypermethylation has been validated extensively in clinical trials (50–56). Based on these data, we identified MGMT promoter hypermethylation as a potential predictive biomarker that could enrich for patients most likely to benefit from TMZ/veliparib.
Utilization of large PDX panels for evaluation of novel therapies provides a platform to explore heterogeneity of response and potential predictive biomarkers. PDX models consistently preserve histopathological, genetic, and epigenetic profiles of the original tumors (18,30,57), while established tumor cell lines suffer from substantial genetic drift associated with long-term cell culture (58,59). GEMMs are powerful tools for dissecting genetic features associated with treatment response, but to date these models do not recapitulate the diverse epigenetic profiles of human cancers (60). Because response to TMZ is critically influenced by DNA methylation within the MGMT promoter, GEMMs may be less useful in dissecting the spectrum of response associated with TMZ-based therapies. Several studies from our group and others have correlated heterogeneous responses across PDX models with previously defined predictive biomarkers identified by analysis of large clinical patient datasets (18,61–64). These and other results have spurred tremendous interest in using PDX models to screen for novel therapies and identify corresponding predictive biomarkers to facilitate development of focused clinical trials. The current study represents the first example for any solid tumor in which a predictive biomarker was identified for a novel combination, exclusively based on an analysis of a large PDX panel, and subsequently used as an inclusion criteria for a definitive phase II/III clinical trial testing that combination.
The primary limitation of the current study is the paucity of MGMT-unmethylated tumor lines tested. Additionally, by testing 28 PDX lines and using P value threshold of .05, we would expect two of the 28 lines to have a statistically significant difference by chance. Therefore, P values were interpreted with caution and presented with effect sizes. However, the additional studies in GBM12 and GBM28 evaluating the impact of MGMT expression on treatment efficacy validate the primary conclusion that tumors expressing MGMT are unlikely to benefit from the combination. With only a quarter of MGMT-hypermethylated PDX lines benefiting from the TMZ/veliparib combination, additional mechanistic studies will be required to define a more precise predictive algorithm, and the availability of multiple sensitive and resistant models defined in this study will be instrumental for these studies. Ultimately, the utility of MGMT hypermethylation as an enrichment strategy will be defined in the ongoing A071102 clinical trial.
The definition of MGMT promoter hypermethylation as a predictive biomarker for response to adjuvant veliparib/TMZ in the current study was directly incorporated as an eligibility criterion for the Alliance A071102 (NCT02152982) randomized phase II/III clinical trial testing adjuvant TMZ combined with veliparib or placebo. Other ongoing or previous trials testing veliparib/TMZ in brain tumors—RTOG-0929 (65), ABTC-0801 (66), PBTC-033 (67), hepatocellular carcinoma (NCT01205828) (45), breast cancer (NCT01009788) (43), and colorectal cancer (NCT01051596) (44)—without a biomarker enrichment strategy may be underpowered because only a fraction of patients with TMZ-sensitive tumors may respond to the combination. Furthermore, trials in an unselected population of newly diagnosed GBM patients, in which 70% are MGMT promoter unmethylated, would require approximately 3400 patients to detect a survival benefit, as compared with 400 patients in the current design of A071102 restricted to patients with MGMT promoter hypermethylation. This enhancement in trial efficiency was only possible through a systematic evaluation of PARP inhibitor strategy in a panel of GBM PDXs. Beyond GBM, this same strategy may provide a paradigm for preclinical testing of novel therapeutics in other tumors types. Similar approaches are being pursued in industry and academic collaborations, such as the Pediatric Consortium (68,69), and the ultimate success of this paradigm will be judged based on the outcome of A071102 and other preclinically informed trials.
Funding
This work was supported by Mayo Clinic and funding from the National Institutes of Health (R01 CA176830, R01 NS77921, and P50 CA108961 to JNS). FBA is supported by the National Institutes of Health training grant T32-GM08685, and JMR by the P30 CA015083. SKG is the recipient of Eagles Cancer Fellowship from Mayo Clinic Cancer Center.
Supplementary Material
Funding agencies had no role in the designing of study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
JNS discloses research grant support from Merck, Basilea, Sanofi, Genentech Lilly, and Beigene. No potential conflicts of interest disclosed by other authors.
The authors thank Dr. Ikeda Yasuhiro, Mayo Clinic, for the pSIN-Luc-UbEm vector.
References
- 1. Ohba S, Mukherjee J, See WL, et al. Mutant IDH1-driven cellular transformation increases RAD51-mediated homologous recombination and temozolomide resistance. Cancer Res. 2014;74 (17):4836–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu X, Han EK, Anderson M, et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol Cancer Res. 2009;7 (10):1686–1692. [DOI] [PubMed] [Google Scholar]
- 3. Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96 (1):56–67. [DOI] [PubMed] [Google Scholar]
- 4. Thomas HD, Calabrese CR, Batey MA, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther. 2007;6 (3):945–956. [DOI] [PubMed] [Google Scholar]
- 5. Miknyoczki S, Chang H, Grobelny J, et al. The selective poly(ADP-ribose) polymerase-1(2) inhibitor, CEP-8983, increases the sensitivity of chemoresistant tumor cells to temozolomide and irinotecan but does not potentiate myelotoxicity. Mol Cancer Ther. 2007;6 (8):2290–2302. [DOI] [PubMed] [Google Scholar]
- 6. Liu X, Shi Y, Guan R, et al. Potentiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitor ABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks. Mol Cancer Res. 2008;6 (10):1621–1629. [DOI] [PubMed] [Google Scholar]
- 7. Fortini P, Pascucci B, Parlanti E, et al. The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie. 2003;85 (11):1053–1071. [DOI] [PubMed] [Google Scholar]
- 8. Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8 (3):193–204. [DOI] [PubMed] [Google Scholar]
- 9. Parrish KE, Cen L, Murray J, et al. Efficacy of PARP inhibitor rucaparib in orthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol Cancer Ther. 2015; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hopkins TA, Shi Y, Rodriguez LE, et al. Mechanistic Dissection of PARP1 Trapping and the Impact on in vivo Tolerability and Efficacy of PARP Inhibitors. Mol Cancer Res. 2015;13(11): 1465–1477. [DOI] [PubMed] [Google Scholar]
- 11. Donawho CK, Luo Y, Penning TD, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13 (9):2728–2737. [DOI] [PubMed] [Google Scholar]
- 12. Clarke MJ, Mulligan EA, Grogan PT, et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol Cancer Ther. 2009;8 (2):407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gupta SK, Mladek AC, Carlson BL, et al. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin Cancer Res. 2014;20 (14):3730–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Teicher BA. Tumor models for efficacy determination. Mol Cancer Ther. 2006;5 (10):2435–2443. [DOI] [PubMed] [Google Scholar]
- 15. Williams SA, Anderson WC, Santaguida MT, et al. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest. 2013;93 (9):970–982. [DOI] [PubMed] [Google Scholar]
- 16. Carlson BL, Pokorny JL, Schroeder MA, et al. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol. 2011;Chapter 14:Unit 14 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17 (1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carlson BL, Grogan PT, Mladek AC, et al. Radiosensitizing effects of temozolomide observed in vivo only in a subset of O6-methylguanine-DNA methyltransferase methylated glioblastoma multiforme xenografts. Int J Radiat Oncol Biol Phys. 2009;75 (1):212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kitange GJ, Carlson BL, Mladek AC, et al. Evaluation of MGMT promoter methylation status and correlation with temozolomide response in orthotopic glioblastoma xenograft model. J Neurooncol. 2009;92 (1):23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brat DJ, James CD, Jedlicka AE, et al. Molecular genetic alterations in radiation-induced astrocytomas. Am J Pathol. 1999;154 (5):1431–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kitange GJ, Carlson BL, Schroeder MA, et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-oncology. 2009;11 (3):281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vlassenbroeck I, Califice S, Diserens AC, et al. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn. 2008;10 (4):332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23 (6):657–663. [DOI] [PubMed] [Google Scholar]
- 24. Grams MP, Wilson ZC, Sio TT, et al. Design and characterization of an economical (192)Ir hemi-brain small animal irradiator. Int J Radiat Biol. 2014;90 (10):936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wanshura LEC, Galvin KE, Ye H, et al. Sequential Activation of Snail1 and N-Myc Modulates Sonic Hedgehog-Induced Transformation of Neural Cells. Cancer Res. 2011;71 (15):5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mead R. The design of experiments: statistical principles for practical applications. Cambridge England; New York: Cambridge University Press; 1988. [Google Scholar]
- 27. Middlemas DS, Stewart CF, Kirstein MN, et al. Biochemical correlates of temozolomide sensitivity in pediatric solid tumor xenograft models. Clin Cancer Res. 2000;6 (3):998–1007. [PubMed] [Google Scholar]
- 28. Kummar S, Kinders R, Gutierrez ME, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27 (16):2705–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hussain M, Carducci MA, Slovin S, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Invest New Drugs. 2014;32 (5):904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kitange GJ, Mladek AC, Carlson BL, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res. 2012;18 (15):4070–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tentori L, Leonetti C, Scarsella M, et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res. 2003;9 (14):5370–5379. [PubMed] [Google Scholar]
- 32. Tentori L, Leonetti C, Scarsella M, et al. Inhibition of poly(ADP-ribose) polymerase prevents irinotecan-induced intestinal damage and enhances irinotecan/temozolomide efficacy against colon carcinoma. Faseb J. 2006;20 (10):1709–1711. [DOI] [PubMed] [Google Scholar]
- 33. Daniel RA, Rozanska AL, Mulligan EA, et al. Central nervous system penetration and enhancement of temozolomide activity in childhood medulloblastoma models by poly(ADP-ribose) polymerase inhibitor AG-014699. Br J Cancer. 2010;103 (10):1588–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Curtin NJ, Wang LZ, Yiakouvaki A, et al. Novel poly(ADP-ribose) polymerase-1 inhibitor, AG14361, restores sensitivity to temozolomide in mismatch repair-deficient cells. Clin Cancer Res. 2004;10 (3):881–889. [DOI] [PubMed] [Google Scholar]
- 35. Horton TM, Jenkins G, Pati D, et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol Cancer Ther. 2009;8 (8):2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Erice O, Smith MP, White R, et al. MGMT expression predicts PARP-mediated resistance to temozolomide. Mol Cancer Ther. 2015;14 (5):1236–1246. [DOI] [PubMed] [Google Scholar]
- 37. Agarwal S, Sane R, Oberoi R, et al. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med. 2011;13:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agarwal S, Manchanda P, Vogelbaum MA, et al. Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: findings in an orthotopic rat xenograft model of glioma. Drug Metab Dispos. 2013;41 (1):33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palma JP, Rodriguez LE, Bontcheva-Diaz VD, et al. The PARP inhibitor, ABT-888 potentiates temozolomide: correlation with drug levels and reduction in PARP activity in vivo. Anticancer Res. 2008;28(5A):2625–2635. [PubMed] [Google Scholar]
- 40. Palma JP, Wang YC, Rodriguez LE, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15 (23):7277–7290. [DOI] [PubMed] [Google Scholar]
- 41. Barazzuol L, Jena R, Burnet NG, et al. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat Oncol. 2013;8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin F, de Gooijer MC, Roig EM, et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res. 2014;20 (10):2703–2713. [DOI] [PubMed] [Google Scholar]
- 43. Isakoff SJ, Overmoyer B, Tung NM, et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J Clin Oncol. 2010;28 (15). [Google Scholar]
- 44. Pishvaian MJ, Slack R, Witkiewicz A, et al. A phase II study of the PARP inhibitor ABT-888 plus temozolomide in patients with heavily pretreated, metastatic colorectal cancer. J Clin Oncol. 2011;29 (15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He AR, Tesfaye A, Smith D, et al. Phase II trial of temozolomide and veliparib combination therapy for sorafenib-refractory advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2014;32 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith MA, Reynolds CP, Kang MH, et al. Synergistic Activity of PARP Inhibition by Talazoparib (BMN 673) with Temozolomide in Pediatric Cancer Models in the Pediatric Preclinical Testing Program. Clin Cancer Res. 2015;21 (4):819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Preusser M, Charles Janzer R, Felsberg J, et al. Anti-O6-methylguanine-methyltransferase (MGMT) immunohistochemistry in glioblastoma multiforme: observer variability and lack of association with patient survival impede its use as clinical biomarker. Brain Pathol. 2008;18 (4):520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodriguez FJ, Thibodeau SN, Jenkins RB, et al. MGMT immunohistochemical expression and promoter methylation in human glioblastoma. Appl Immunohistochem Mol Morphol. 2008;16 (1):59–65. [DOI] [PubMed] [Google Scholar]
- 49. Weller M, Stupp R, Reifenberger G, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6 (1):39–51. [DOI] [PubMed] [Google Scholar]
- 50. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352 (10):997–1003. [DOI] [PubMed] [Google Scholar]
- 51. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13 (9):916–926. [DOI] [PubMed] [Google Scholar]
- 52. Wick W, Platten M, Meisner C, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13 (7):707–715. [DOI] [PubMed] [Google Scholar]
- 53. Gilbert MR, Wang M, Aldape KD, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31 (32):4085–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wick W, Steinbach JP, Platten M, et al. Enzastaurin before and concomitant with radiation therapy, followed by enzastaurin maintenance therapy, in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation. Neuro Oncol. 2013;15 (10):1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gilbert MR, Sulman EP, Mehta MP. Bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370 (21):2048–2049. [DOI] [PubMed] [Google Scholar]
- 56. Stupp R, Hegi ME, Gorlia T, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15 (10):1100–1108. [DOI] [PubMed] [Google Scholar]
- 57. Hennessey PT, Ochs MF, Mydlarz WW, et al. Promoter methylation in head and neck squamous cell carcinoma cell lines is significantly different than methylation in primary tumors and xenografts. PloS One. 2011;6 (5):e20584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. De Witt Hamer PC Van Tilborg AA Eijk PP et al. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene. 2008;27 (14):2091–2096. [DOI] [PubMed] [Google Scholar]
- 59. Li A, Walling J, Kotliarov Y, et al. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res. 2008;6 (1):21–30. [DOI] [PubMed] [Google Scholar]
- 60. Diede SJ, Yao Z, Keyes CC, et al. Fundamental differences in promoter CpG island DNA hypermethylation between human cancer and genetically engineered mouse models of cancer. Epigenetics. 2013;8 (12):1254–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sarkaria JN, Yang L, Grogan PT, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6 (3):1167–1174. [DOI] [PubMed] [Google Scholar]
- 62. Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1 (6):508–523. [DOI] [PubMed] [Google Scholar]
- 63. Krumbach R, Schuler J, Hofmann M, et al. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer. 2011;47 (8):1231–1243. [DOI] [PubMed] [Google Scholar]
- 64. Julien S, Merino-Trigo A, Lacroix L, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18 (19):5314–5328. [DOI] [PubMed] [Google Scholar]
- 65. Robins HI, Wang MH, Gilbert MR, et al. Phase I Results from Rtog 0929, a Randomized Phase I/Ii Study of Abt-888 (Veliparib) in Combination with Temozolomide (Tmz) in Recurrent, Tmz-Resistant Glioblastoma. Neuro-oncology. 2012;14:67–67. [Google Scholar]
- 66. Kleinberg L, Supko JG, Mikkelsen T, et al. Phase I adult brain tumor consortium (ARTC) trial of ABT-888 (veliparib), temozolomide (TMZ), and radiotherapy (RT) for newly diagnosed glioblastoma multiforme (GBM) including pharmacokinetic (PK) data. J Clin Oncol. 2013;31 (15). [Google Scholar]
- 67. Su JM, Thompson P, Adesina A, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a pediatric brain tumor consortium report. Neuro Oncol. 2014;16 (12):1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Houghton PJ, Morton CL, Tucker C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49 (7):928–940. [DOI] [PubMed] [Google Scholar]
- 69. Neale G, Su X, Morton CL, et al. Molecular characterization of the pediatric preclinical testing panel. Clin Cancer Res. 2008;14 (14):4572–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.