

ABSTRACT
Recent evidence indicates that protein aggregates can spread between neurons in several neurodegenerative diseases but much remains unknown regarding the underlying mechanisms responsible for this spreading and its role in disease progression. We recently demonstrated that mutant Huntingtin aggregates spread between cells within the Drosophila brain resulting in non-cell autonomous loss of a pair of large neurons in the posterior protocerebrum. However, the full extent of neuronal loss throughout the brain was not determined. Here we examine the effects of driving expression of mutant Huntingtin in Olfactory Receptor Neurons (ORNs) by using a marker for cleaved caspase activity to monitor neuronal apoptosis as a function of age. We find widespread caspase activity in various brain regions over time, demonstrating that non-cell autonomous damage is widespread. Improved understanding of which neurons are most vulnerable and why should be useful in developing treatment strategies for neurodegenerative diseases that involve transcellular spreading of aggregates.
Keywords: aggregate spreading, apoptosis, cell death, drosophila, huntington's disease, neurodegeneration
Non-Cell-Autonomous Damage from Htt.RFP Aggregate Spreading
We previously reported that the spreading of mutant Huntingtin aggregates resulted in non-cell autonomous loss of neurons in the central brain. When mutant Huntingtin is expressed in Olfactory Receptor Neurons (ORNs), we found that a pair of large neurons in the posterior protocerebrum that are labeled using R44H11-LexA are lost by day 10 after eclosion. Loss of these neurons was dependent on uptake of aggregates, because their loss was prevented when endocytosis was blocked in these neurons.1
Although these results demonstrated non-cell autonomous loss of this pair of neurons, the full extent of damage to other neurons was not examined. To investigate neuronal loss more broadly, we expressed a polyglutamine (polyQ)-expanded version of the human Huntingtin protein with a Red Fluorescent Protein tag (UAS-htt.138Q.mRFP)2 in ORNs using the Or83b-Gal4 driver,3 and searched for apoptotic cells in the brain by immunostaining for activated (cleaved) Drosophila effector caspase (Dcp-1) over time. We also used a temperature sensitive Gal80 (tubulin-Gal80ts1)4 to repress Gal4 activation during development. Flies were raised at the permissive temperature of 18°C, then shifted to 29°C upon eclosion, inactivating Gal80 and allowing Gal4 activation. This protocol prevents onset of aggregate expression and spreading prior to eclosion. In one-day-old flies, Htt.RFP aggregates are seen at ORN terminals in the olfactory lobe of the central brain. However, no Dcp-1 activation is seen at this point (Fig. 1A). After 15 days, activated Dcp-1 staining is evident in both the central brain (Fig. 1B, arrowheads) and the optic lobes (Fig. 1B, arrows). By day 30, Dcp-1 staining is more widespread (Fig. 1C). While our previous findings demonstrated the loss of a specific pair of neurons, these results demonstrate that the damage resulting from the spread of Huntingtin aggregates is widespread throughout the brain.
Figure 1.
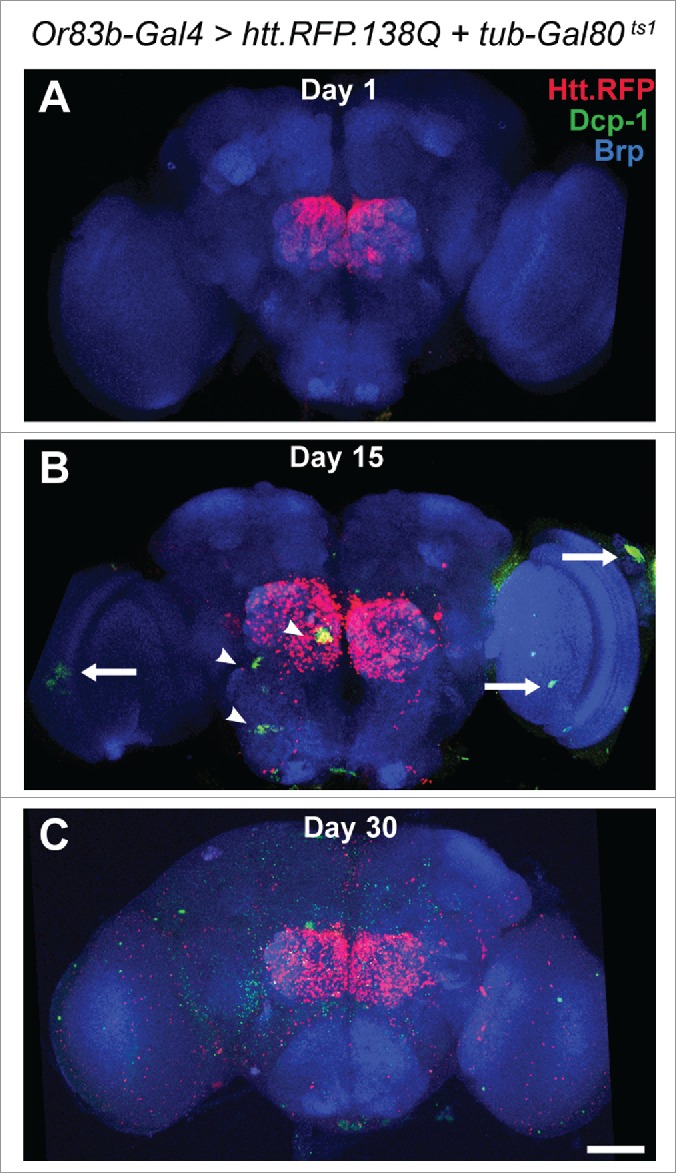
Distribution of activated death caspase, Dcp-1, after expression of Htt.RFP.138Q in ORNs. Immunostaining for cleaved Dcp-1 (green), an indicator of apoptosis, within the brain after expressing Htt.RFP (red) in ORNs for 1 (A), 15 (B), or 30 (C) days after eclosion. There is clear evidence for agedependent cell death in brain regions beyond the boundaries of Htt.RFP expression. Neuropil is marked by anti-Bruchpilot (Brp) (blue). Scale bar D 50 mm.
Why are Certain Cells Vulnerable to Aggregate Transmission?
The current data demonstrate that a number of additional cells in the brain are impacted by the spread of Htt.RFP aggregates from ORN-terminals. Future studies should focus on identifying those neurons that are particularly vulnerable to aggregate-induced cell death. In addition to neurons, it is also possible that certain populations of glia are also involved. Phagocytic glia play an important role in the clearance of Htt aggregates in the Drosophila brain.5 Furthermore, it has been shown that transfer of α-Synuclein aggregates to glia can initiate an inflammatory response, which in turn could exacerbate neurodegeneration.6,7 Indeed, hyperactivation of the immune response is sufficient to cause neurodegeneration in Drosophila.8
One of the most interesting findings regarding the spreading of aggregates is the differential sensitivity between neurons that accumulate aggregates but remain viable and other neurons that die rapidly after spreading begins. In our experiments these differences in neuronal response to aggregates were most apparent by comparing the R44H11-LxA-positive neurons that are lost relatively early, and the nb169-positive neurons that accumulate a large number of aggregates but do not degenerate. It is possible that the aggregates tend to accumulate over time in certain neurons merely because these neurons are somehow resistant to the aggregates and remain viable. However, because it remains unknown whether it is the aggregates or the soluble forms of the Htt.138Q protein that are more toxic, it is also possible that the “resistant” neurons are simply better at sequestering the proteins into aggregates. Identifying the differences between these various populations of neurons will be crucial for determining why certain neurons are more vulnerable and for developing strategies aimed at preventing loss of susceptible neurons. The ability to genetically manipulate these neuronal populations independently in vivo will enable these issues to be rigorously investigated in Drosophila.
What Determines the Spreading Capacity of Protein Aggregates?
Although the 588-amino acid form of polyQ-expanded huntingtin spread throughout the brain, a smaller Exon 1 fragment that is also polyQ-expanded did not spread beyond the terminals of ORNs in which they were expressed.1 PolyQ-expanded Exon 1 fragments are commonly used to model Huntington's Disease, and although they have been shown to form aggregates as well as induce cellular toxicity, these fragments could potentially be missing key domains that contribute to disease progression. One explanation for the differential effects of the Exon 1 fragment compared with the 588-amino acid form is that regions of the protein required for spreading are missing in the shorter fragments. These regions could include sites of interaction with other proteins or cellular components that have an impact on whether spreading will occur. The human Huntingtin protein, for example, interacts with a large variety of other proteins. Some of these interaction sites, including the HEAT repeat-containing regions, are not present in many of the shorter Exon 1 fragments. As several proteins that interact with these regions are involved in endocytosis and vesicle trafficking,9 perhaps it is these interactions that provide the capacity for spreading. We observed similar results when expressing a truncated version of poly-Q expanded Ataxin 3, which formed aggregates but did not spread to other cells. It will be informative to test full-length versions of these proteins or versions with different domains to determine whether particular domains of these proteins are necessary for spreading.
Aggregate Transmission and the Seeding of New Aggregates
Our results demonstrate the cell-to-cell transmission of mutant Huntingtin aggregates throughout the brain. However, a distinct mode of aggregate propagation is the “prion-like” seeding that involves conversion of wild-type soluble protein into new aggregates. Indeed, a separate study found that expression of PolyQ-expanded Huntingtin in neurons could convert wild-type protein into aggregates within the cytoplasm of phagocytic glia.5 This finding demonstrates that prion-like “seeding” can also occur in the Drosophila brain. While aggregate spreading and conversion of wild-type protein into new aggregates both appear to be important components of disease progression, Drosophila models offer a unique opportunity to distinguish between these mechanisms. For example, it is possible to express expanded human Huntingtin either alone or in combination with wild-type Huntingtin in the same cells or in different cells. Whether endogenous Drosophila proteins interact with these aggregates can also be investigated. A better understanding of the nature of the aggregates and their associated proteins, the different ways in which the aggregates are transmitted from cell-to-cell, and how these aggregates affect cell viability may offer new opportunities for therapeutic intervention by limiting aggregate spreading to prevent disease progression.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health grants F32 NS078958 (DTB), R01 AG033620, and R01 NS15390 (BG).
References
- 1.Babcock DT, Ganetzky B. Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc Natl Acad Sci U S A 2015; 112:E5427-33; PMID:26351672; http://dx.doi.org/ 10.1073/pnas.1516217112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss KR, Kimura Y, Lee WC, Littleton JT. Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington's disease model. Genetics 2012; 190:581-600; PMID:22095086; http://dx.doi.org/ 10.1534/genetics.111.133710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kreher SA, Kwon JY, Carlson JR. The molecular basis of odor coding in the Drosophila larva. Neuron 2005; 46:445-56; PMID:15882644; http://dx.doi.org/ 10.1016/j.neuron.2005.04.007 [DOI] [PubMed] [Google Scholar]
- 4.McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL. Spatiotemporal rescue of memory dysfunction in Drosophila. Science 2003; 302:1765-8; PMID:14657498; http://dx.doi.org/ 10.1126/science.1089035 [DOI] [PubMed] [Google Scholar]
- 5.Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun 2015; 6:6768; PMID:25866135; http://dx.doi.org/ 10.1038/ncomms7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci 2008; 28:7687-98; PMID:18650345; http://dx.doi.org/ 10.1523/JNEUROSCI.0143-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al.. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 2010; 285:9262-72; PMID:20071342; http://dx.doi.org/ 10.1074/jbc.M109.081125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao Y, Chtarbanova S, Petersen AJ, Ganetzky B. Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc Natl Acad Sci U S A 2013; 110:E1752-60; PMID:23613578; http://dx.doi.org/ 10.1073/pnas.1306220110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 2003; 28:425-33; PMID:12932731; http://dx.doi.org/ 10.1016/S0968-0004(03)00168-3 [DOI] [PubMed] [Google Scholar]