

Abstract
A library of compounds covering a broad chemical space was selected from a tuberculosis drug development program and was screened in a whole-cell assay against Mycobacterium ulcerans, the causative agent of the necrotizing skin disease Buruli ulcer. While a number of potent antitubercular agents were only weakly active or inactive against M. ulcerans, five compounds showed high activity (90% inhibitory concentration [IC90], ≤1 μM), making screening of focused antitubercular libraries a good starting point for lead generation against M. ulcerans.
TEXT
Buruli ulcer (BU) is a neglected tropical disease that is characterized by chronic necrotizing skin lesions. It is caused by Mycobacterium ulcerans, a slow-growing mycobacterium that produces the potent exotoxin mycolactone, which is the main virulence factor responsible for the necrotizing pathology of BU (1, 2). Standard specific treatment of BU involves 8 weeks of combination therapy with streptomycin and rifampin, which may have ototoxic, nephrotoxic, and hepatotoxic side effects (3). While streptomycin can in principal be replaced by clarithromycin or moxifloxacin (4), no replacement for rifampin is currently available (5, 6). As a consequence, antibiotic therapy of BU may become impossible if rifampin resistance in M. ulcerans emerges.
Since M. ulcerans is closely related to Mycobacterium tuberculosis (7), drug molecular targets may be conserved between the two species (8). Therefore, repurposing tuberculosis (TB) drug candidates for BU represents an attractive approach to overcome the problem of very limited financial resources for BU drug discovery and development. Here, we have tested a set of 83 compounds from the tuberculosis lead generation and lead optimization drug discovery programs of AstraZeneca. The set was selected based on activity against M. tuberculosis and on chemical diversity comprising advanced candidates and novel leads. For activity testing against M. ulcerans, resazurin-based assays were performed essentially as described previously (9). A good correlation between this type of metabolic activity testing and the enumeration of CFU was shown in a previous study (10). In brief, bacteria were incubated with the compounds at a concentration of 10 μM for 1 week at 30°C. Then, resazurin was added (10%, vol/vol), and the plates were further incubated overnight at 37°C followed by measurement of fluorescence intensities. Subsequently, MICs corresponding to 90% inhibition (IC90) of compounds active in the prescreen were assessed by testing 2-fold serial dilutions. Values were independently determined twice using two different low-passaged African M. ulcerans strains (S1013 and S1047) (11).
The compound set, encompassing established tuberculosis drugs and antimicrobials as well as advanced development compounds, represented a broad chemical space of 54 clusters. Based on the IC90 values, the compounds were classified into six different activity categories that ranged from <0.1 μM to >10 μM. These values were compared with the corresponding values obtained earlier for M. tuberculosis, revealing that the activity patterns for M. ulcerans and M. tuberculosis vary substantially (Fig. 1). Only three compounds (represented by black dots in the gray-shaded area in Fig. 1), the two aminopyrazoles 9 and 10 and the pyrazolopyrimidine 11, were more active against M. ulcerans than they were against M. tuberculosis. Twenty-one compounds (represented by red dots in Fig. 1) belonged to the same activity category for the two mycobacterial pathogens, whereas the majority (59/83, 71%) of compounds (represented by black dots in the white area) were more active against M. tuberculosis than they were against M. ulcerans. While none of the compounds tested had an IC90 value of ≤0.3 μM for M. ulcerans, 20/83 displayed activities of ≤0.3 μM for M. tuberculosis (Fig. 1). This lower sensitivity of M. ulcerans may be related to factors such as low growth rate, the extremely hydrophobic cell surface, and the production of an extracellular matrix. However, no general correlation between physicochemical properties of the compounds and differential IC90 values of M. ulcerans/M. tuberculosis was found (Fig. 2). M. ulcerans has undergone drastic genome reduction after emergence from the environmental mycobacterium Mycobacterium marinum (12, 13), and loss of genes and massive pseudogene accumulation may have caused loss of targets for some of the highly active anti-TB compounds.
FIG 1.

Comparison of activities of all 83 compounds tested on M. ulcerans and M. tuberculosis with MIC values grouped into six categories. The size of the individual circles indicates the number of compounds identified within the specific MIC range combination for M. tuberculosis and M. ulcerans. Compounds belonging to the same activity category for the two pathogens are marked in red. The three compounds showing higher activity against M. ulcerans than against M. tuberculosis are located in the gray-shaded area.
FIG 2.

Physicochemical properties of the compounds analyzed in relation to activity on M. tuberculosis and M. ulcerans. Displayed are scatter plots of ClogP, molecular weight (MW), polar surface area (PSA), and ion class of all compounds tested. The M. tuberculosis activities are shown in log scale along the x axes, while the M. ulcerans activity ranges are depicted in different colors.
The 11 compounds with IC90 values of ≤3 μM against M. ulcerans originated from seven chemically diverse compound families of early and advanced stages of drug development (Table 1). As for the early stage compounds, 3/8 tested diarylthiazoles (Table 1, compounds 1 to 8) showed IC90 values of ≤3 μM against M. ulcerans. In M. tuberculosis, diarylthiazoles seem to target the two-component system PrrB/PrrA (14); they also appeared as a hit in a high-throughput screen against Mycobacterium bovis BCG (15). The diarylthiazole fatostatin (compound 7) acts as an inhibitor of sterol regulatory element-binding proteins (SREBPs) (16, 17). In addition, 1/2 aminopyrazoles (compound 9), the only pyrazolopyrimidine (compound 11), and 1/2 hydroxyquinolones (compound 22) tested were highly active against M. ulcerans. For quinolinyl pyrimidines and phenothiazines, inhibitors of the alternative type II NADH dehydrogenases in M. tuberculosis (18, 19), no activity against M. ulcerans was observed. Since the target enzymes are also present in M. ulcerans, the weaker activity of M. tuberculosis NADH dehydrogenase inhibitors against M. ulcerans may be attributed to reduced permeability.
TABLE 1.
Structure, activity, and properties of the compound families active against M. ulcerans
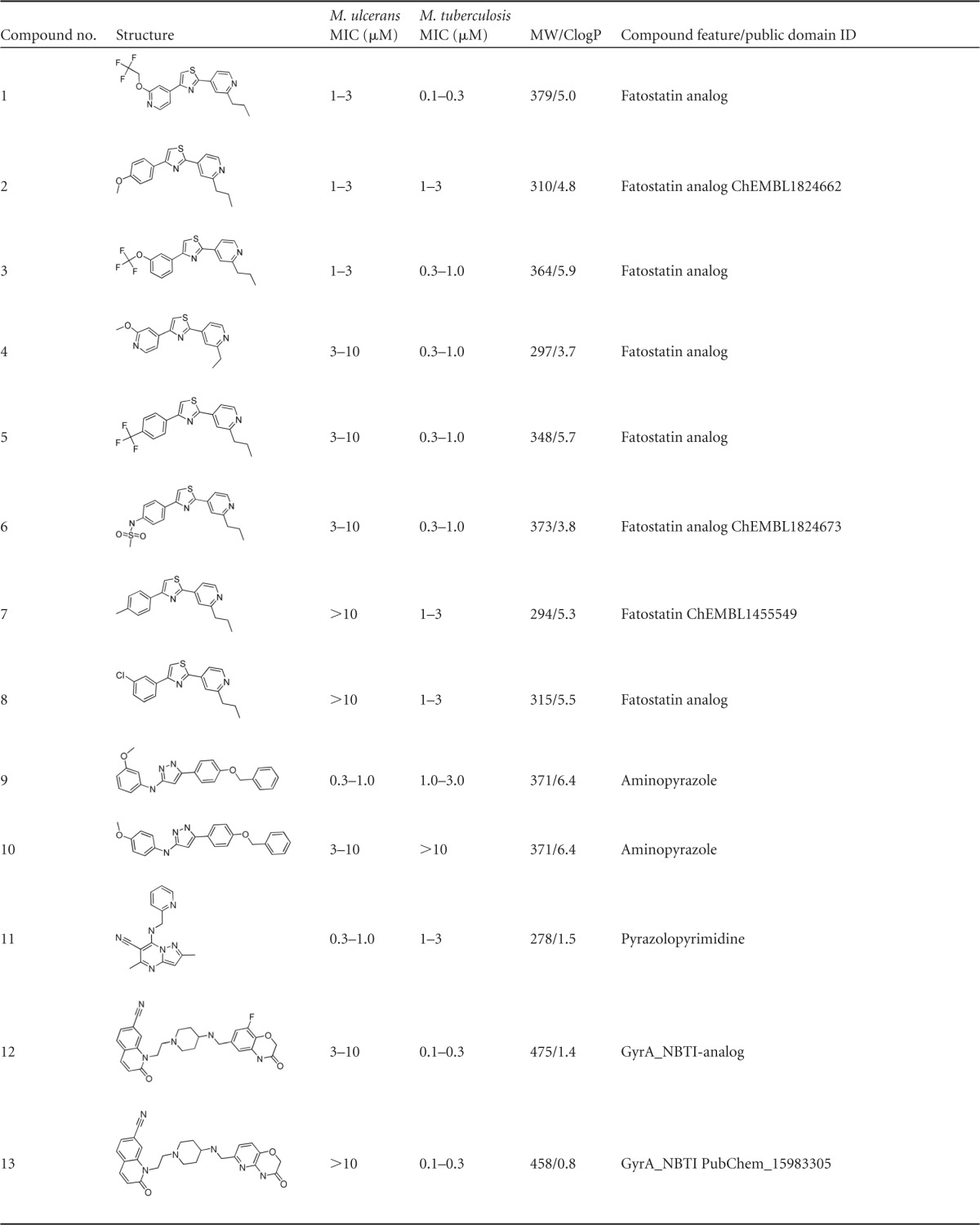
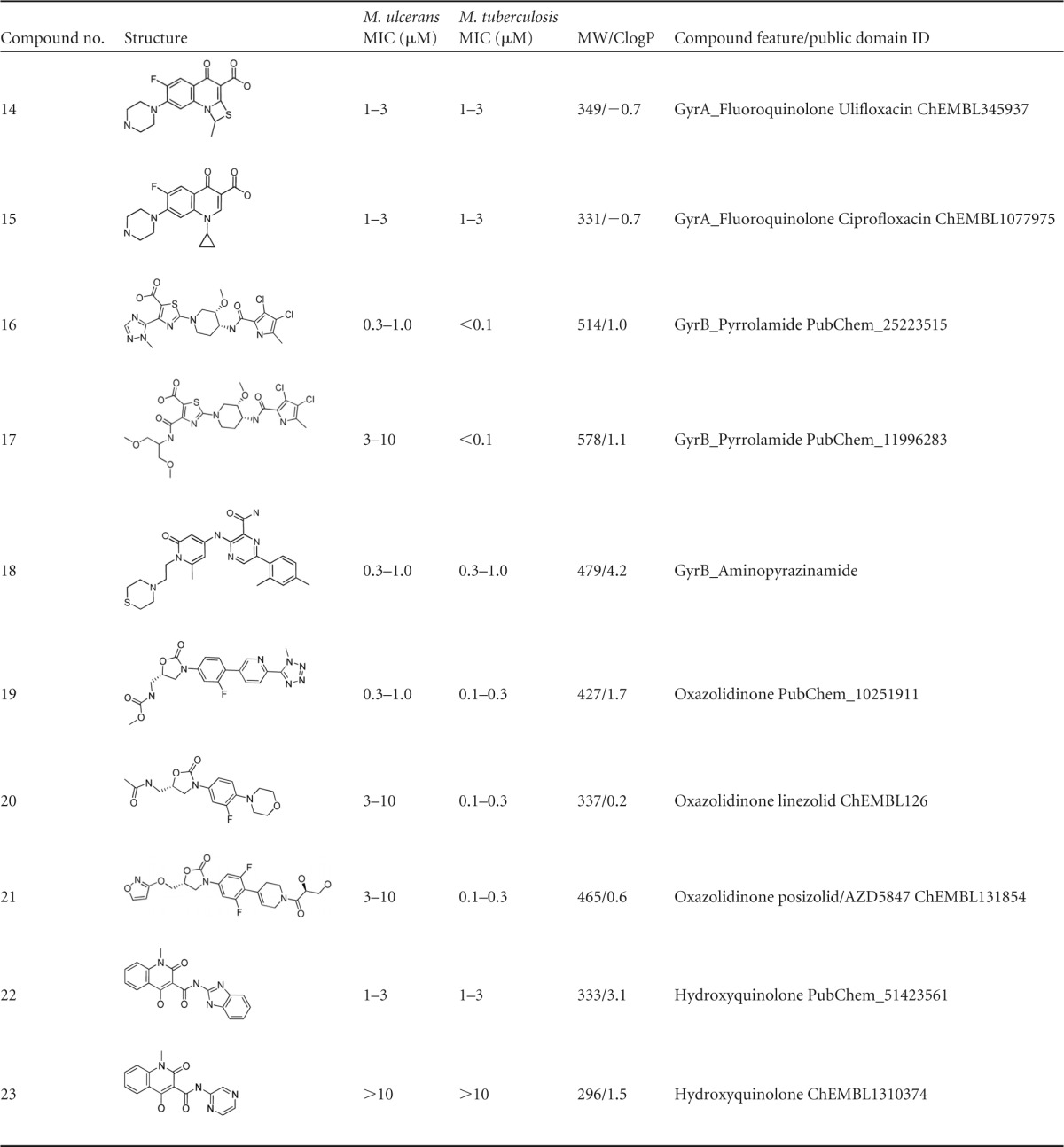
Of the advanced antibacterial agents tested, the quinolones had differential activity on M. ulcerans. While a prulifloxacin analogue (compound 14) and ciprofloxacin (compound 15) exhibited good activity, this was not the case for two novel bacterial topoisomerase II inhibitors (NBTI; compounds 12 and 13) (20–22). Furthermore, two GyrB inhibitors, a pyrrolamide (compound 16) (23, 24), an aminopyrazinamide (compound 18) (25), and 1/3 of the tested oxazolidinones (compound 19) showed activity of ≤1 μM. The other two oxazolidinones, compounds 20 and 21, with compound 21 being a recent anti-TB clinical candidate interfering with protein translation by binding to the mycobacterial 50S ribosomal subunit (26), displayed only moderate activity (3 to 10 μM) against M. ulcerans. The natural product doxycycline, belonging to the family of tetracyclines, was not active against M. ulcerans. Members of the quinolone family targeting GyrA have already been evaluated in vitro and in vivo, and for ofloxacin, ciprofloxacin, sparfloxacin, moxifloxacin, and sitafloxacin, activity against M. ulcerans was shown (5, 27–31). Our results provide further evidence that the M. ulcerans DNA gyrase is a vulnerable target. In contrast, selected compounds of the nitroimidazole family (32, 33), including PA824 (34), did not show any activity against M. ulcerans, reconfirming published results (5).
Most of the compounds active against M. ulcerans were reported to be specific inhibitors and noncytotoxic. As a class, the diarylthiazoles (compounds 1 to 8) were shown to be inactive in cytotoxicity assays (14). Similarly, the gyrase inhibitors, such as quinolones (compounds 12 to 15) (20–22) and pyrrolocarboxamides (compound 16 and 17) (23, 24), are advanced leads from tuberculosis drug discovery programs and were demonstrated to be noncytotoxic. The other well-characterized advanced antibacterial compounds, oxazolidinones (compounds 19 to 21) (26), are specific inhibitors of bacterial protein biosynthesis and do not possess any cytotoxicity. Further profiling and structure activity relationship exploration is required to assess the full potential of these chemical classes as anti-BU candidates.
With the TB drug pipeline currently being filled with more potent and novel drug candidates, our results demonstrate that repurposing may in fact lead to the development of new treatment options for BU.
ACKNOWLEDGMENTS
The M. tuberculosis MIC data of the compounds reported were generated by a microbiology team led by Vasanthi Ramachandran at AstraZeneca, Bangalore, as part of M. tuberculosis lead generation and lead optimization programs. We acknowledge the team for the same. We acknowledge the untiring efforts of Sunita de Sousa from AstraZeneca, Bangalore, and Kevin Pritchard from AstraZeneca, United Kingdom, to encourage and manage this collaboration between AstraZeneca and the Swiss Tropical and Public Health Institute. In addition, we thank BIO Ventures for Global Health (BVGH) for catalyzing this collaboration through WIPO Re:Search.
REFERENCES
- 1.George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, Small PL. 1999. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283:854–857. doi: 10.1126/science.283.5403.854. [DOI] [PubMed] [Google Scholar]
- 2.George KM, Pascopella L, Welty DM, Small PL. 2000. A Mycobacterium ulcerans toxin, mycolactone, causes apoptosis in guinea pig ulcers and tissue culture cells. Infect Immun 68:877–883. doi: 10.1128/IAI.68.2.877-883.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan-Tompkins NH. 1995. Toxic effects and drug interactions of antimycobacterial therapy. Clin Dermatol 13:223–233. doi: 10.1016/0738-081X(95)00022-8. [DOI] [PubMed] [Google Scholar]
- 4.Gordon CL, Buntine JA, Hayman JA, Lavender CJ, Fyfe JA, Hosking P, Starr M, Johnson PD. 2010. All-oral antibiotic treatment for Buruli ulcer: a report of four patients. PLoS Negl Trop Dis 4:e770. doi: 10.1371/journal.pntd.0000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji B, Lefrançois S, Robert J, Chauffour A, Truffot C, Jarlier V. 2006. In vitro and in vivo activities of rifampin, streptomycin, amikacin, moxifloxacin, R207910, linezolid, and PA-824 against Mycobacterium ulcerans. Antimicrob Agents Chemother 50:1921–1926. doi: 10.1128/AAC.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji B, Chauffour A, Robert J, Lefrançois S, Jarlier V. 2007. Orally administered combined regimens for treatment of Mycobacterium ulcerans infection in mice. Antimicrob Agents Chemother 51:3737–3739. doi: 10.1128/AAC.00730-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. 2006. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol 6:95. doi: 10.1186/1471-2148-6-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potet J, Casenghi M, Charitha G, Mueller Y, Etard JF, Comte E. 2013. Buruli ulcers: an advocacy agenda to improve its control. Med Sante Trop 23:227–228. (In French.) [DOI] [PubMed] [Google Scholar]
- 9.Scherr N, Röltgen K, Witschel M, Pluschke G. 2012. Screening of antifungal azole drugs and agrochemicals with an adapted alamarBlue-based assay demonstrates antibacterial activity of croconazole against Mycobacterium ulcerans. Antimicrob Agents Chemother 56:6410–6413. doi: 10.1128/AAC.01383-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scherr N, Pluschke G, Thompson CJ, Ramón-García S. 2015. Selamectin is the avermectin with the best potential for Buruli ulcer treatment. PLoS Negl Trop Dis 9:e0003996. doi: 10.1371/journal.pntd.0003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bratschi MW, Bolz M, Grize L, Kerber S, Minyem JC, Um Boock A, Yeboah-Manu D, Ruf MT, Pluschke G. 2014. Primary cultivation: factors affecting contamination and Mycobacterium ulcerans growth after long turnover time of clinical specimens. BMC Infect Dis 14:636. doi: 10.1186/s12879-014-0636-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stinear T, Seemann T, Pidot S, Frigui W, Reysset G, Garnier T, Meurice G, Simon D, Bouchier C, Ma L, Tichit M, Porter J, Ryan J, Johnson P, Davies J, Jenkin G, Small P, Jones L, Tekaia F, Laval F, Daffé M, Parkhill J, Cole S. 2007. Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res 17:192–200. doi: 10.1101/gr.5942807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doig KD, Holt KE, Fyfe JA, Lavender CJ, Eddyani M, Portaels F, Yeboah-Manu D, Pluschke G, Seemann T, Stinear TP. 2012. On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genomics 13:258. doi: 10.1186/1471-2164-13-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellale E, Naik M, V BV, Ambady A, Narayan A, Ravishankar S, Ramachandran V, Kaur P, McLaughlin R, Whiteaker J, Morayya S, Guptha S, Sharma S, Raichurkar A, Awasthy D, Achar V, Vachaspati P, Bandodkar B, Panda M, Chatterji M. 2014. Diarylthiazole: an antimycobacterial scaffold potentially targeting PrrB-PrrA two-component system. J Med Chem 57:6572–6582. doi: 10.1021/jm500833f. [DOI] [PubMed] [Google Scholar]
- 15.Ballell L, Bates RH, Young RJ, Alvarez-Gomez D, Alvarez-Ruiz E, Barroso V, Blanco D, Crespo B, Escribano J, González R, Lozano S, Huss S, Santos-Villarejo A, Martín-Plaza JJ, Mendoza A, Rebollo-Lopez MJ, Remuiñan-Blanco M, Lavandera JL, Pérez-Herran E, Gamo-Benito FJ, García-Bustos JF, Barros D, Castro JP, Cammack N. 2013. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. Chem Med Chem 8:313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamisuki S, Shirakawa T, Kugimiya A, Abu-Elheiga L, Choo HY, Yamada K, Shimogawa H, Wakil SJ, Uesugi M. 2011. Synthesis and evaluation of diarylthiazole derivatives that inhibit activation of sterol regulatory element-binding proteins. J Med Chem 54:4923–4927. doi: 10.1021/jm200304y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, Shinohara T, Kawazoe Y, Sato S, Asakura K, Choo HY, Sakai J, Wakil SJ, Uesugi M. 2009. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol 16:882–892. doi: 10.1016/j.chembiol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Shirude PS, Paul B, Roy Choudhury N, Kedari C, Bandodkar B, Ugarkar BG. 2012. Quinolinyl pyrimidines: potent inhibitors of NDH-2 as a novel class of anti-TB agents. ACS Med Chem Lett 3:736–740. doi: 10.1021/ml300134b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teh JS, Yano T, Rubin H. 2007. Type II NADH: menaquinone oxidoreductase of Mycobacterium tuberculosis. Infect Disord Drug Targets 7:169–181. doi: 10.2174/187152607781001781. [DOI] [PubMed] [Google Scholar]
- 20.Dougherty TJ, Nayar A, Newman JV, Hopkins S, Stone GG, Johnstone M, Shapiro AB, Cronin M, Reck F, Ehmann DE. 2014. NBTI 5463 is a novel bacterial type II topoisomerase inhibitor with activity against Gram-negative bacteria and in vivo efficacy. Antimicrob Agents Chemother 58:2657–2664. doi: 10.1128/AAC.02778-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shirude PS, Hameed S. 2012. Nonfluroquinolone-based inhibitors of mycobacterial type II topoisomerase as potential therapeutic agents for TB. Ann Rep Med Chem 47:319–330. doi: 10.1016/B978-0-12-396492-2.00021-7. [DOI] [Google Scholar]
- 22.Bisacchi GS, Dumas J. 2009. Recent advances in the inhibition of bacterial type II topoisomerases. Ann Rep Med Chem 44:379–396. doi: 10.1016/S0065-7743(09)04419-4. [DOI] [Google Scholar]
- 23.Reck F, Alm R, Brassil P, Newman J, Dejonge B, Eyermann CJ, Breault G, Breen J, Comita-Prevoir J, Cronin M, Davis H, Ehmann D, Galullo V, Geng B, Grebe T, Morningstar M, Walker P, Hayter B, Fisher S. 2011. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J Med Chem 54:7834–7847. doi: 10.1021/jm2008826. [DOI] [PubMed] [Google Scholar]
- 24.P SH, Solapure S, Mukherjee K, Nandi V, Waterson D, Shandil R, Balganesh M, Sambandamurthy VK, Raichurkar AK, Deshpande A, Ghosh A, Awasthy D, Shanbhag G, Sheikh G, McMiken H, Puttur J, Reddy J, Werngren J, Read J, Kumar M, R M, Chinnapattu M, Madhavapeddi P, Manjrekar P, Basu R, Gaonkar S, Sharma S, Hoffner S, Humnabadkar V, Subbulakshmi V, Panduga V. 2014. Optimization of pyrrolamides as mycobacterial GyrB ATPase inhibitors: structure-activity relationship and in vivo efficacy in a mouse model of tuberculosis. Antimicrob Agents Chemother 58:61–70. doi: 10.1128/AAC.01751-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirude PS, Madhavapeddi P, Tucker JA, Murugan K, Patil V, Basavarajappa H, Raichurkar AV, Humnabadkar V, Hussein S, Sharma S, Ramya VK, Narayan CB, Balganesh TS, Sambandamurthy VK. 2013. Aminopyrazinamides: novel and specific GyrB inhibitors that kill replicating and nonreplicating Mycobacterium tuberculosis. ACS Chem Biol 8:519–523. doi: 10.1021/cb300510w. [DOI] [PubMed] [Google Scholar]
- 26.Balasubramanian V, Solapure S, Iyer H, Ghosh A, Sharma S, Kaur P, Deepthi R, Subbulakshmi V, Ramya V, Ramachandran V, Balganesh M, Wright L, Melnick D, Butler SL, Sambandamurthy VK. 2014. Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob Agents Chemother 58:495–502. doi: 10.1128/AAC.01903-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dhople AM. 2001. In vitro activity of KRM-1648, either singly or in combination with ofloxacin, against Mycobacterium ulcerans. Int J Antimicrob Agents 17:57–61. doi: 10.1016/S0924-8579(00)00306-X. [DOI] [PubMed] [Google Scholar]
- 28.Dhople AM, Namba K. 2002. In vitro activity of sitafloxacin (DU-6859a) alone, or in combination with rifampicin, against Mycobacterium ulcerans. J Antimicrob Chemother 50:727–729. doi: 10.1093/jac/dkf218. [DOI] [PubMed] [Google Scholar]
- 29.Thangaraj HS, Adjei O, Allen BW, Portaels F, Evans MR, Banerjee DK, Wansbrough-Jones MH. 2000. In vitro activity of ciprofloxacin, sparfloxacin, ofloxacin, amikacin and rifampicin against Ghanaian isolates of Mycobacterium ulcerans. J Antimicrob Chemother 45:231–233. doi: 10.1093/jac/45.2.231. [DOI] [PubMed] [Google Scholar]
- 30.Bentoucha A, Robert J, Dega H, Lounis N, Jarlier V, Grosset J. 2001. Activities of new macrolides and fluoroquinolones against Mycobacterium ulcerans infection in mice. Antimicrob Agents Chemother 45:3109–3112. doi: 10.1128/AAC.45.11.3109-3112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefrançois S, Robert J, Chauffour A, Ji B, Jarlier V. 2007. Curing Mycobacterium ulcerans infection in mice with a combination of rifampin-streptomycin or rifampin-amikacin. Antimicrob Agents Chemother 51:645–650. doi: 10.1128/AAC.00821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barry CE III, Boshoff HI, Dowd CS. 2004. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr Pharm Des 10:3239–3262. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 33.Barry CE. 2011. Lessons from seven decades of antituberculosis drug discovery. Curr Top Med Chem 11:1216–1225. doi: 10.2174/156802611795429158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. 2000. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]