

Abstract
Mycobacterium abscessus causes chronic pulmonary infections that are extremely difficult to cure. The currently recommended combination therapy is associated with high failure rates and relapse. Tigecycline has been explored in salvage regimens, with a response rate of 43% in those who received at least a month of therapy. We performed a dose-response study in a hollow-fiber system model of pulmonary M. abscessus infection in which we recapitulated tigecycline human pulmonary concentration-time profiles of 8 different doses for 21 days. We identified the maximal kill or efficacy in CFU per milliliter and the ratio of the 0- to 24-h area under the concentration-time curve to MIC (AUC/MIC) associated with 80% efficacy (EC80). The tigecycline efficacy was 5.38 ± 2.35 log10 CFU/ml, and the drug achieved the unprecedented feat of a bacterial level of 1.0 log10 CFU/ml below the pretreatment inoculum (1-log kill) of M. abscessus in the hollow-fiber system. The EC80 AUC/MIC ratio was 36.65, while that for a 1-log kill was 44.6. Monte Carlo experiments with 10,000 patients were used to identify the clinical dose best able to achieve the EC80 or 1-log kill. The standard dose of 100 mg/day had a cumulative fraction of response of 51% for the EC80 and 46% for 1-log kill. For both the EC80 target and 1-log kill, the optimal tigecycline clinical dose was identified as 200 mg/day. The susceptibility breakpoint was ≤0.5 mg/liter. Tigecycline is the most active single agent evaluated to date, and we propose that 200 mg/day be examined as the backbone of new combination therapy regimens to replace current treatment.
INTRODUCTION
Mycobacterium abscessus is, for all the correct reasons, considered an “antibiotic nightmare” (1). This pathogen primarily causes chronic pulmonary infections that are extremely difficult to cure owing to the extensive drug resistance intrinsic to this organism (2). Personalized treatment regimens based on in vitro drug susceptibility testing and aggressive surgical resection yielded “cure” rates of only 57% in one case series (3). Even then, over the years there is relapse and death among those who have been “cured.” Thus, there has been a continued search for new drugs, with the hope to craft a new effective regimen. Tigecycline, the first developed glycylcycline, was designed to overcome mechanisms of resistance that are known for older tetracyclines, such as the active efflux and the ribosome protection mechanisms. It is considered a bacteriostatic antibiotic, sometimes bactericidal, with demonstrated broad in vitro effects on several Gram-positive and Gram-negative bacteria, including some rapidly growing mycobacteria (4–7). This broad antimicrobial spectrum, its large volume of distribution (≥7 to 10 liters/kg), and the known intracellular accumulation represent a sound rationale for the clinical use of tigecycline (4, 5). Although tigecycline is FDA approved to be used for the treatment of complicated skin/soft tissue infections, complicated intra-abdominal infections, and community-acquired bacterial pneumonia, in a recent case series, tigecycline salvage regimens in 30 patients were associated with improvement in 43% of patients who received >1 month of therapy (8). These patients received doses of tigecycline designed for Gram-negative and Gram-positive bacterial infections. In patients treated with tigecycline for pneumonia caused by these other bacteria, the main determinant of a clinical response has been identified as a ratio of the free-drug area under the concentration-time curve from 0 to 24 h (fAUC0–24) to the MIC of >12.8 in plasma (9, 10). This suggests a concentration-dependent effect of tigecycline and that doses could be optimized based on the free-drug AUC0–24/MIC ratios. Here, we wanted to identify the efficacy of tigecycline and utilize it to identify optimal doses for treatment of pulmonary disease caused by M. abscessus.
MATERIALS AND METHODS
Materials.
The test strain was M. abscessus subsp. abscessus ATCC 19977 (American Type Culture Collection, Manassas, VA). Stocks of the mycobacteria were kept at −80°C in Middlebrook 7H9 broth (Remel, Thermo Fisher Scientific, Lenexa, KS) supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC) and 15% glycerol. Twenty-four hours before each experiment, one vial was thawed and incubated at 30°C to achieve logarithmic growth phase (log phase). Tigecycline powder was purchased from Baylor University Medical Center pharmacy. For each experiment, the antibiotic was reconstituted, sterile filtered, and then diluted in sterile water to the desired concentrations. Hollow-fiber cartridges were purchased from FiberCell Systems (Frederick, MD).
Drug susceptibility testing.
The MIC was determined two times by broth macrodilution in Middlebrook 7H9 broth (here referred to as “broth”) read at 72 h (11). In addition, CFU per ml were enumerated from each concentration evaluated in the broth macrodilution test to determine the lowest concentration associated with >99% inhibition of growth, corresponding to the MIC.
Hollow-fiber study.
The hollow-fiber system (HFS) model of pulmonary M. abscessus infection was previously developed and used to perform pharmacokinetic/pharmacodynamic (PK/PD) evaluation of amikacin and moxifloxacin (12, 13). The peripheral compartments of 8 HFSs were each inoculated with 20 ml of 6 log10 CFU/ml M. abscessus. Treatment was administered daily for 21 days at doses that mimicked the non-protein-bound or free AUC0–24, peak concentrations, and times to maximum concentration achieved in the lungs of humans treated with tigecycline doses of 0, 12.5, 25, 50, 100, 200, 400, and 800 mg, assuming linear increases in AUC0–24 with dose since clearance is constant over a range of doses (14–16). The doses were administered to the central compartment of each HFS once daily via computerized syringe pumps. Tigecycline concentrations achieved in all the systems were validated by repetitive sampling from the central compartment of each HFS at 0, 1, 6, 9, 12, 18, 23.5, 25, 30, 33, 36, 42, and 47.5 h after the first dose. The M. abscessus burden was quantified by taking 1-ml samples from the peripheral compartment culture contents of each system on days 0, 1, 2, 3, 5, 7, 10, 14, and 21 of treatment. After washing by centrifugation with saline to avoid antibiotic carryover, samples were serially 10-fold diluted and cultured on Middlebrook 7H10 agar. The tigecycline MIC was identified at the end of the experiment in each HFS by broth macrodilution.
Drug assay.
Tigecycline concentrations in the samples collected from the central compartment of each HFS were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Tigecycline and tigecycline-D9 (internal standard) were purchased from Toronto Research Chemicals (Toronto, Canada). A calibrator, controls, and the internal standard were included in each analytical run for quantitation. Stock solutions of tigecycline and internal standard were prepared in 80:20 methanol-water at a concentration of 1 mg/ml and stored at −20°C. A seven-point calibration curve was prepared by diluting tigecycline stock solution in drug-free medium (0.1, 0.2, 1, 2, 5, 10, and 20 mg/liter). Quality control samples were prepared by spiking the medium with stock standards for two levels of controls. Samples were prepared in 96-well microtiter plates by the addition of 10 μl of calibrator, quality controls, or sample to 190 μl 0.1% formic acid in water containing 10 mg/liter internal standard followed by vortex mixing. Chromatographic separation was achieved on an Acquity ultraperformance liquid chromatography (UPLC) HSS T3 analytical column (1.8 µm, 50 by 2.1 mm; Waters) maintained at 30°C at a flow rate of 0.2 ml/min with a binary gradient and a total run time of 6 min. The observed (m/z) values of the fragment ions were 586.33 to 569.3 for tigecycline and 595.4 to 578.4 for tigecycline-13CD. Sample injection and separation was performed with an Acquity UPLC interfaced with a Xevo TQ mass spectrometer (Waters). All data were collected using Mass Lynx version 4.1 SCN810. The limit of quantitation for this assay was 0.1 mg/liter. The inter- and intraday variations were 12.1% and 11.8%, respectively.
Pharmacokinetic analysis and PK/PD modeling.
Tigecycline concentrations from each of the HFS units at all time points were comodeled using the ADAPT 5 software (Biomedical Simulations Resource, University of Southern California) following steps for the pharmacokinetic parameter analysis described in detail in prior studies (17–19). The observed AUC0–24 and AUC0–24/MIC ratios were calculated from the pharmacokinetic parameter estimates identified. The dose response was modeled using the inhibitory sigmoid Emax model with the total bacterial burden used as the response parameter and the steady-state AUC0–24/MIC ratio as drug exposure, with maximal kill (Emax) or efficacy and effect in log10 CFU/ml. This relationship was used to calculate the EC80, which is the exposure mediating 80% of Emax, since 100% Emax lies on an asymptote of the inhibitory sigmoid Emax model. In addition, we also calculated the AUC0–24/MIC ratio at which the bacterial burden is similar to that at time zero (“stasis”), as well as that associated with a 1-log10 CFU/ml decrease compared to time zero (“1-log kill”).
Monte Carlo simulations.
Monte Carlo simulations were performed in order to translate the HFS-derived AUC/MIC exposures to an optimal clinical dose. Steps for the Monte Carlo experiments are detailed in recommendations and in our prior work (19, 20) and were followed to identify the clinical dose best able to achieve or exceed specific target exposures in lungs of 10,000 patients with pulmonary M. abscessus. Three target exposures were examined: (i) EC80, (ii) AUC0–24/MIC ratio associated with stasis, and (iii) AUC0–24/MIC ratio associated with 1-log kill. The same exercise was also used to identify the PK/PD-derived susceptibility breakpoint, which we consider to be the MIC below which >10% of patients achieve the EC80 with standard dosing or, alternatively, with the optimal dose that can be tolerated by patients, based on prior work (19–23). We examined doses of 50 mg, 100 mg, 150 mg, and 200 mg per day. The population pharmacokinetic parameters and covariance used as prior data were those from Rubino et al. (14). In addition, we took into account the fact that the tigecycline AUC in epithelial lining fluid (ELF) in 1.32-fold higher than that in plasma (15). Drug concentrations in ELF are considered concentration surrogates at the site of effect for pneumonias, including those caused by mycobacteria (14, 15, 17–26). Since the concentration of proteins in ELF in patients is low (25–27), we considered ELF protein binding negligible. An alternate view could be that a large portion of M. abscessus in lungs is intracellular (this is by no means resolved), in which case it would be the intracellular tigecycline concentration that should be simulated. Since tigecycline's concentration in alveolar macrophages is >58-fold than that in ELF, our use of ELF exposures would constitute a worst-case scenario (15). We also examined the probability that these doses would be likely to achieve a plasma AUC of >6.87 mg · h/liter, which was identified by Rubino et al. as being associated with greater incidence of nausea and vomiting (10). The tigecycline MIC distribution was from isolates of patients treated in northern Texas, as reported by Wallace and colleagues (7). The cumulative fraction of response (CFR) is a summation calculated as , where PTA is the probability of target attainment at each MIC and F is the proportion of isolates at each MIC (19, 28).
RESULTS
MIC and pharmacokinetic analysis.
The tigecycline MIC for the M. abscessus strain was 3.12 mg/liter. Tigecycline pharmacokinetics in the HFS model of pulmonary M. abscessus infection were best described by a two-compartment model, based on Akaike information criterion and Bayesian information criterion scores, as intended in the study design. The pharmacokinetic parameters achieved in the HFS model were a total clearance of 20.6 ± 29.6 liters/h, a volume of the central compartment of 459 ± 13.1 liters, an intercompartmental clearance of 3.41 liters/h, and a peripheral volume of 0.80 ± 0.45 liters.
Time-kill curve in the HFS model.
Figure 1 shows time-kill curves for the HFS model of pulmonary M. abscessus infection, which show the bacterial burden over time for each tigecycline AUC0–24/MIC exposure. The AUC0–24/MIC ratios of 44.60 and 79.85 achieved considerable killing of the M. abscessus population. Figure 1 also shows that the maximum microbial kill was on day 7, when tigecycline achieved a microbial kill of 1.37 log10 CFU/ml below the pretreatment bacterial burden (stasis). Regrowth was observed after 14 days and occurred with all doses. However, there was no change in the tigecycline MIC in any of the HFSs treated with tigecycline or in the nontreated control systems at the end of therapy.
FIG 1.
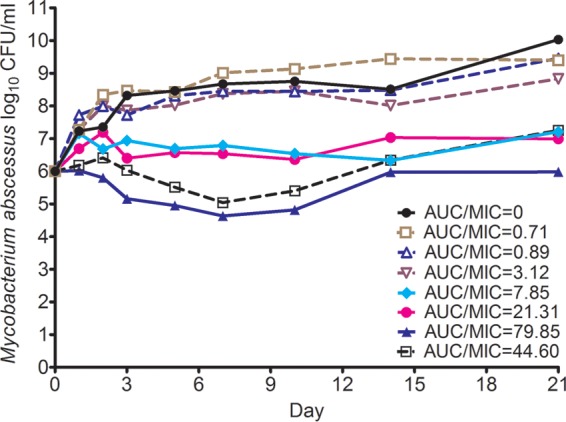
Tigecycline time-kill curves in the hollow-fiber system model of M. abscessus infection. The two higher exposures lead to considerable killing, which was maximum at 7 days. Regrowth started at day 10 and after day 14 was observed in all systems.
PK/PD modeling.
The inhibitory sigmoid Emax model curves for each sampling day before regrowth are shown in Fig. 2. The day 7 parameters were an Emax of 5.38 ± 2.35 log10 CFU/ml, a Hill slope of 0.96 ± 0.52, and an EC50 that was an AUC0–24/MIC ratio of 20.42 ± 19.75 (r2 = 0.96). On day 10, which we took as the end of the bactericidal effect since all treated systems started regrowth, the Hill slope was 1.12 ± 0.64, the EC50 was an AUC0–24/MIC ratio of 10.63 ± 6.68 (r2 = 0.95), and the EC80 was 36.65. The AUC0–24/MIC ratio associated with stasis (i.e., no growth in bacterial burden compared to day 0) was 21.3, while that associated with a 1-log kill was 44.60.
FIG 2.
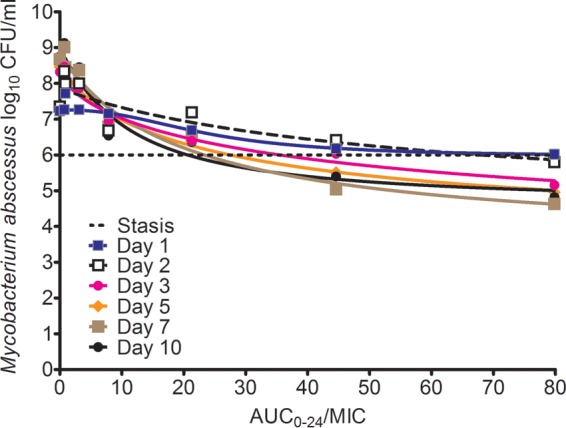
Inhibitory sigmoid Emax relationships between tigecycline AUC0–24/MIC ratios and Mycobacterium abscessus burden. From day 2 on, the kill went below the stasis level, but the highest efficacy was at day 7.
Translation of laboratory findings to the clinical world.
Next we performed Monte Carlo simulations to identify the probability of doses of tigecycline between 50 mg and 200 mg per day achieving or exceeding either the AUC0–24/MIC ratio associated with stasis, the EC80, or the AUC0–24/MIC ratio associated with a 1-log kill, in 10,000 patients. The 50-mg/day dose had CFRs of 48.41% of the 10,000 patients for stasis, 1.40% for the EC80, and <1% for a 1-log kill. Therefore, that dose would be considered unsatisfactory. Figure 3 shows the performance of the standard 100-mg/day dose for each of the three target exposures. Notably, the EC80 target shows that the 100 mg was associated with >90% target attainment up to an MIC of 0.125 mg/liter, after which it plummeted (Fig. 3B). The dose achieved a CFR of 46.7% for the 1-log kill target.
FIG 3.
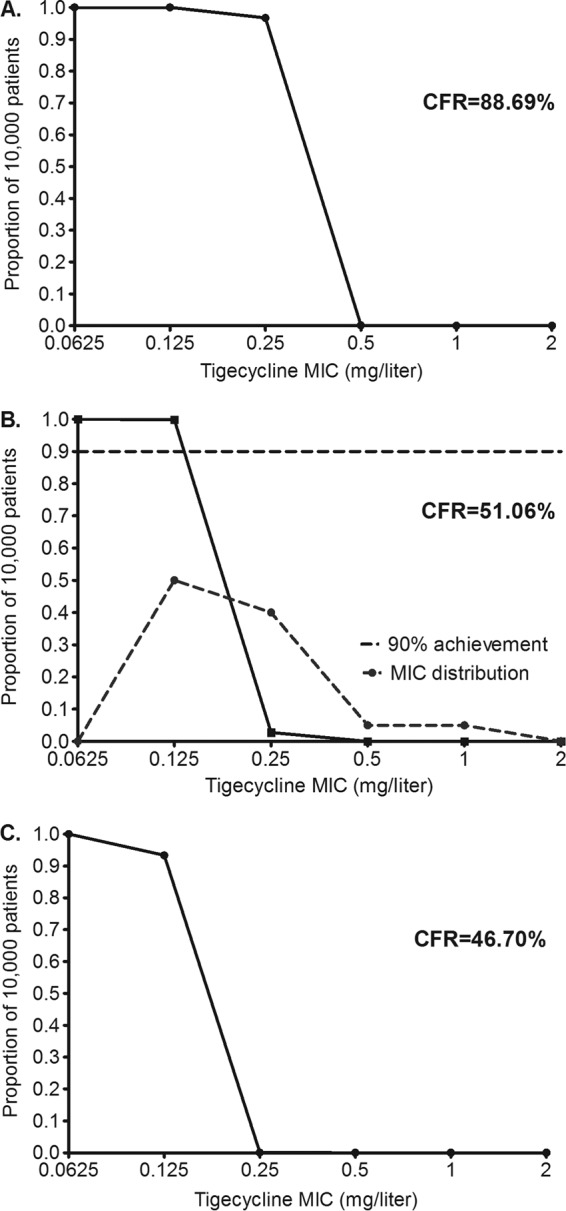
Probability of target attainment in patients treated with 100 mg/day. Proportions of 10,000 patients achieving the AUC/MIC ratio associated with stasis (A) EC80 target (B), and the target AUC/MIC ratio associated with a 1-log kill (C) are shown. The cumulative fraction of response (CFR) is low for the EC80 and 1-log kill targets. Moreover, in panel A, the standard dose will not achieve even stasis if the MIC is 0.5 mg/liter.
The performance of the 150-mg/day dose is shown in Fig. 4. More than 90% of patients would achieve stasis at this dose (Fig. 4A). In regard to the EC80, the MIC above which >10% would fail to achieve that exposure was 0.25 mg/liter; at 0.5 mg/liter, the proportion fell to 0%. Figure 4C shows that the proportion that achieved an AUC0–24/MIC ratio associated with a 1-log kill at 150 mg/day was approximately 63%.
FIG 4.
Probability of target attainment in patients treated with 150 mg/day for the targets AUC/MIC ratio associated with stasis (A), EC80(B), and AUC/MIC ratio associated with a 1-log kill (C). The CFRs for the EC80 and 1-log kill targets are below 90%, and thus the dose should be considered suboptimal.
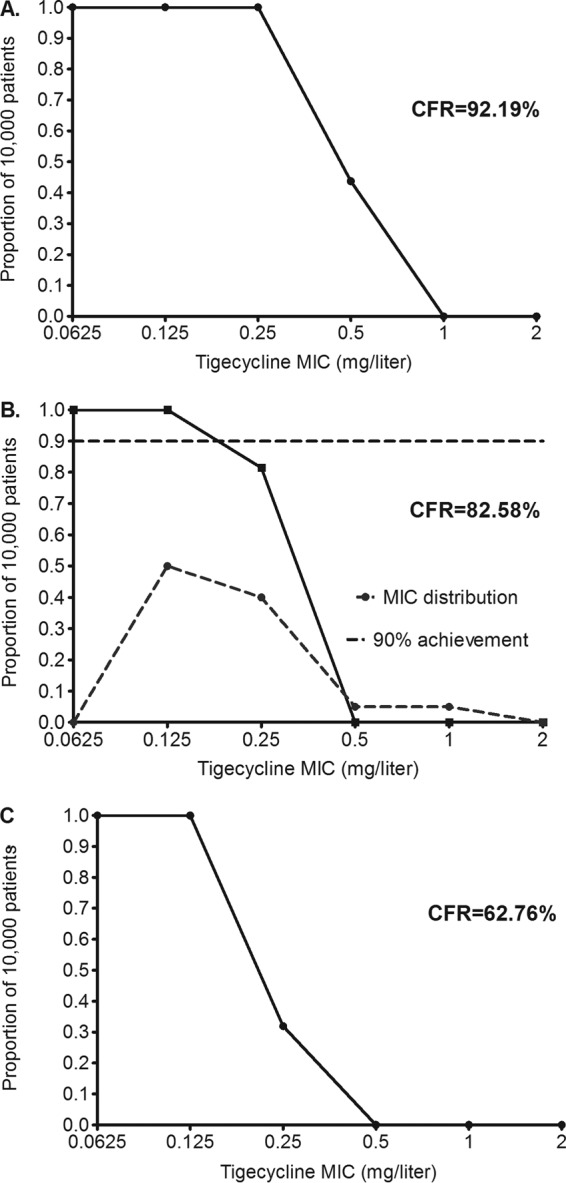
The 200-mg/day dose achieved target exposures as shown in Fig. 5. Both stasis (Fig. 5A) and EC80 exposures (Fig. 5B) were achieved in >90% of patients. Figure 5B shows target attainment of >98% at an MIC of 0.25 mg/liter, which then fell to 2.80% at an MIC of 0.5 mg/liter. If this dose was adopted for pulmonary M. abscessus infection, the pharmacokinetic/pharmacodynamic-based susceptibility breakpoint would be 0.5 mg/liter, as shown in Fig. 5B. The AUC0–24/MIC ratio associated with a 1-log kill was achieved or exceeded in 87.34% of patients, which though short of 90% is close, suggesting that this is the optimal dose for rapid microbial kill in pulmonary M. abscessus disease.
FIG 5.
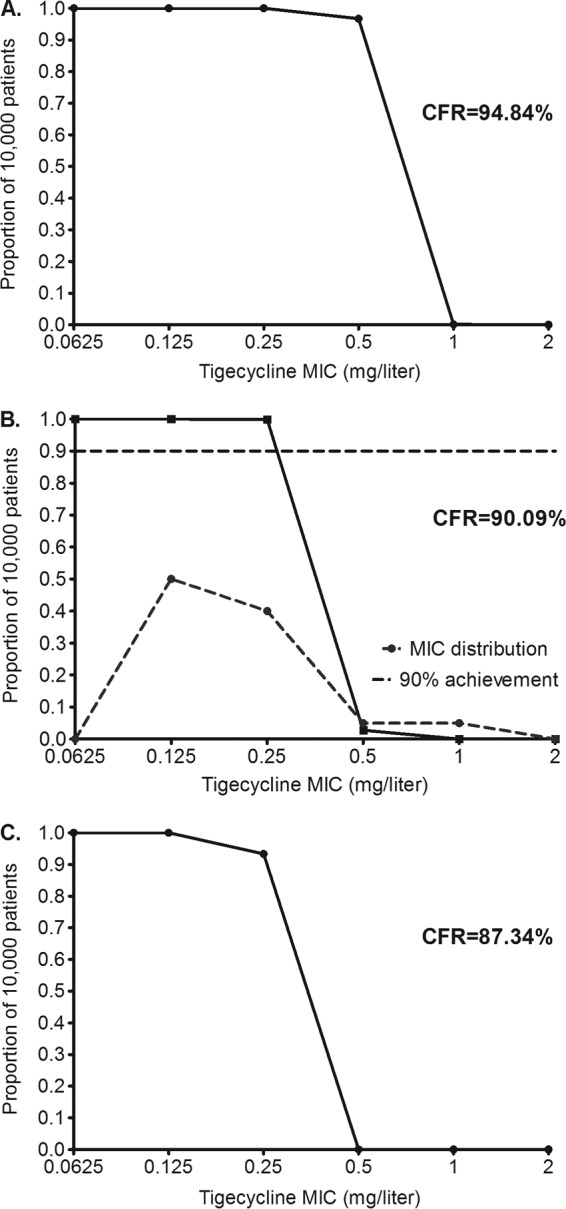
Probability of target attainment in patients treated with 200 mg/day. The dose of 200 mg/day achieved CFRs of >90% for stasis (A) and EC80 (B) and >87% for a 1-log kill (C). Panel A also shows that even at this dose, if the MIC is >0.5 mg/liter, 0% of patients would achieve the concentration associated with stasis. Thus, susceptibility breakpoints should be below an MIC of 1 mg/liter.
Since the tigecycline AUC0–24 threshold that predicts nausea and vomiting is a plasma AUC of >6.87 mg · h/liter, we also examined the probability of achieving that plasma concentration (as opposed to lung concentrations as described above) at the different doses. Figure 6 shows a steep increase in the proportion of the patients achieving this at above 150 mg/day, achieving a probability of close to 1 at 200 mg/day.
FIG 6.
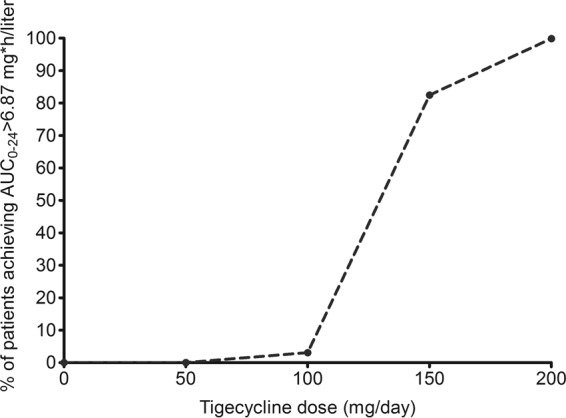
Proportion of 10,000 patients who achieve the tigecycline AUC threshold predictive of nausea and vomiting.
DISCUSSION
Tigecycline is efficacious against the notoriously resistant M. abscessus in the hollow-fiber model system for pulmonary disease. For the first time in the HFS model of pulmonary M. abscessus infection, a drug exhibited a considerable microbial kill below the stasis line, exceeding what has been previously observed for amikacin or moxifloxacin (12, 13). This magnitude of the efficacy means that tigecycline could enter a regimen as the first truly bactericidal drug for M. abscessus. This alone should qualify this drug to be considered as a first-line agent early during therapy and the backbone for new combination regimens. Perhaps aggressive regimens, including combination of tigecycline with an extended group of drugs, should be evaluated in order to overcome the limited efficacy of the current regimens used to treat M. abscessus pulmonary disease.
The tigecycline EC80 exposure for M. abscessus microbial kill was calculated as an AUC0–24/MIC ratio of 36.65. In patients with community-acquired pneumonia due to a variety of Gram-positive and Gram-negative pathogens, the ratio associated with faster time to fever resolution for tigecycline was an fAUC0–24/MIC ratio of ≥12.8 (10). Thus, the exposure associated with optimal efficacy that we identified is much higher than that for other pathogens. Nevertheless, the microbial kill indices (either EC80, stasis, or 1-log kill) that translate from the HFS model of pulmonary M. abscessus infection to patients are still unclear. However, in our simulations, it is interesting that the CFR of 46.70% based on the 1-log kill target was the one closet to clinical response rates of 43% in tigecycline-containing regimens in pulmonary M. abscessus disease studies by Wallace et al. (8). This is consistent with the notion that other antibiotics currently used exert very limited antimicrobial effects, as seen in our HFS model and with clinical experience (1–3, 8, 12, 13). The 200-mg/day dose achieved this 1-log kill target exposure in close to 90% of patients, suggesting that it is the optimal dose for a bactericidal effect in these patients. Indeed, this dose has been shown to be more efficacious in studies with treatment of other bacterial infections (29, 30). Administering 200 mg/day will unfortunately also mean that virtually all patients will achieve AUCs of >6.87 mg · h/liter, predictive of development of adverse events. It may be that this large dose will be administered together with an antiemetic agent, which is in fact common practice already, during the first few months of therapy in a multidrug regimen to rapidly drive down bacterial the burden and then will be dropped from the regimen (8). This strategy, however, sacrifices tolerability of the treatment for efficacy, a difficult trade-off. It could be that in combination therapy, lower tigecycline exposures, and hence doses, are needed for optimal microbial kill. The lessons from tuberculosis, however, have been that most of the time, the optimal exposures identified in monotherapy tend to be the same in combination therapy, likely because most combinations exhibit additivity as opposed to synergy (19, 20, 31).
Finally, no susceptibility breakpoint has been established yet for tigecycline against rapidly growing mycobacteria, including M. abscessus. Based on our findings here, the PK/PD-derived susceptibility breakpoint is ≤0.5 mg/liter with the proposed dose, as well as with the current standard dose. Indeed, at MICs higher than this, even the dose of 200 mg/day achieves stasis in ∼0% of patients. These breakpoints that we propose differ from the epidemiologic cutoff values used by EUCAST. The proposed breakpoints are, rather, designed to identify which patients would fail therapy even at the 200-mg dose and are therefore designed for clinical decision-making. This gives clinicians a tool to decide whether to use tigecycline, given higher probabilities of failure even to achieve stasis, with an agent that would be associated with high rates of adverse events.
This study has some limitations. First, the assay to monitor and detect the size of the resistant subpopulation was not optimized. Thus, we could determine neither the mutation frequency nor the proportion of the resistant subpopulation throughout the study. Second, only the M. abscessus type strain was examined. Ideally several clinical isolates should be evaluated. Nevertheless, taking into account a wider MIC distribution in our simulations should in part compensate for that. Third, these are results from an HFS model for extracellular pulmonary M. abscessus infection. It is unknown what proportion of the pathogen is extracellular or intracellular in lungs of patients. However, given tigecycline's high ratios of penetration into alveolar macrophages, reflected by an alveolar cell-to-serum AUC ratio of 77.5 times (15), even the standard dose would achieve either the EC80 or 1-log kill in patients. Thus, our dose recommendations may be too conservative. Nevertheless, in other mycobacterial pneumonias, the ELF has been accurate as a surrogate of target site concentrations in pneumonia patients (31–33).
In summary, tigecycline has the potential to improve treatment outcomes in M. abscessus disease. To date, it is one of the most active drugs evaluated in the HFS model of pulmonary M. abscessus infection. We recommend 200 mg; however, the caveat is that this is based on the assumption of a predominantly extracellular bacterial location. The role of tigecycline in regimens to treat pulmonary M. abscessus infection deserves further exploration, including development of combination therapy regimens.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant R56 AI111985 to T.G.) and by a doctoral fellowship from the Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología Francisco José de Caldas, Colciencias, Colombian government (grant 529-2012 to B.E.F.).
T.G. is a consultant for Astellas Pharma USA and LuminaCare solutions; he founded Jacaranda Biomed Inc. All other authors have no conflicts of interest.
REFERENCES
- 1.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother 67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 2.Griffith DE. 2011. The talking Mycobacterium abscessus blues. Clin Infect Dis 52:572–574. doi: 10.1093/cid/ciq252. [DOI] [PubMed] [Google Scholar]
- 3.Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. 2011. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis 52:565–571. doi: 10.1093/cid/ciq237. [DOI] [PubMed] [Google Scholar]
- 4.Stein GE, Craig WA. 2006. Tigecycline: a critical analysis. Clin Infect Dis 15:518–524. [DOI] [PubMed] [Google Scholar]
- 5.Estes KS, Derendorf H. 2010. Comparison of the pharmacokinetic properties of vancomycin, linezolid, tigecyclin and daptomycin. Eur J Med Res 15:533–543. doi: 10.1186/2047-783X-15-12-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giamarellou H, Poulakou G. 2011. Pharmacokinetic and pharmacodynamic evaluation of tigecycline. Expert Opin Drug Metab Toxicol 7:1459–1470. doi: 10.1517/17425255.2011.623126. [DOI] [PubMed] [Google Scholar]
- 7.Wallace RJ Jr, Brown-Elliott BA, Crist CJ, Mann L, Wilson RW. 2002. Comparison of the in vitro activity of the glycylcycline tigecycline (formerly GAR-936) with those of tetracycline, minocycline, and doxycycline against isolates of nontuberculous mycobacteria. Antimicrob Agents Chemother 46:3164–3167. doi: 10.1128/AAC.46.10.3164-3167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace RJ Jr, Dukart G, Brown-Elliott BA, Griffith DE, Scerpella EG, Marshall B. 2014. Clinical experience in 52 patients with tigecycline-containing regimens for salvage treatment of Mycobacterium abscessus and Mycobacterium chelonae infections. J Antimicrob Chemother 69:1945–1953. doi: 10.1093/jac/dku062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhavnani SM, Rubino CM, Hammel JP, Forrest A, Dartois N, Cooper CA, Korth-Bradley J, Ambrose PG. 2012. Pharmacological and patient-specific response determinants in patients with hospital-acquired pneumonia treated with tigecycline. Antimicrob Agents Chemother 56:1065–1072. doi: 10.1128/AAC.01615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubino CM, Bhavnani SM, Forrest A, Dukart G, Dartois N, Cooper A, Korth-Bradley J, Ambrose PG. 2012. Pharmacokinetics-pharmacodynamics of tigecycline in patients with community-acquired pneumonia. Antimicrob Agents Chemother 56:130–136. doi: 10.1128/AAC.00277-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clinical and Laboratory Standards Institute. 2003. Susceptibility testing of mycobacteria, nocardiae, and other aerobic actinomycetes; approved standard. Clinical and Laboratory Standards Institute, Wayne, PA. [PubMed] [Google Scholar]
- 12.Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow-fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother 60:1242–1248. doi: 10.1128/AAC.02282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2015. Moxifloxacin efficay in a hollow-fiber model of Mycobacterium abscessus disease, abstr A-014c Program Abstr 55th Intersci Conf Antimicrob Agents Chemother-28th Int Congr Chemother Meet, San Diego, CA. [Google Scholar]
- 14.Rubino CM, Forrest A, Bhavnani SM, Dukart G, Cooper A, Korth-Bradley J, Ambrose PG. 2010. Tigecycline population pharmacokinetics in patients with community- or hospital-acquired pneumonia. Antimicrob Agents Chemother 54:5180–5186. doi: 10.1128/AAC.01414-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conte JE Jr, Golden JA, Kelly MG, Zurlinden E. 2005. Steady-state serum and intrapulmonary pharmacokinetics and pharmacodynamics of tigecycline. Int J Antimicrob Agents 25:523–529. doi: 10.1016/j.ijantimicag.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Barbour A, Schmidt S, Ma B, Schiefelbein L, Rand KH, Burkhardt O, Derendorf H. 2009. Clinical pharmacokinetics and pharmacodynamics of tigecycline. Clin Pharmacokinet 48:575–584. doi: 10.2165/11317100-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 17.Deshpande D, Srivastava S, Meek C, Leff R, Hall GS, Gumbo T. 2010. Moxifloxacin pharmacokinetics/pharmacodynamics and optimal dose and susceptibility breakpoint identification for treatment of disseminated Mycobacterium avium infection. Antimicrob Agents Chemother 54:2534–2539. doi: 10.1128/AAC.01761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musuka S, Srivastava S, Siyambalapitiyage Dona CW, Meek C, Leff R, Pasipanodya J, Gumbo T. 2013. Thioridazine pharmacokinetic-pharmacodynamic parameters “wobble” during treatment of tuberculosis: a theoretical basis for shorter-duration curative monotherapy with congeners. Antimicrob Agents Chemother 57:5870–5877. doi: 10.1128/AAC.00829-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gumbo T, Angulo-Barturen I, Ferrer-Bazaga S. 2015. Pharmacokinetic-pharmacodynamic and dose-response relationships of antituberculosis drugs: recommendations and standards for industry and academia. J Infect Dis 211(Suppl 3):S96–S106. doi: 10.1093/infdis/jiu610. [DOI] [PubMed] [Google Scholar]
- 20.Pasipanodya J, Gumbo T. 2011. An oracle: antituberculosis pharmacokinetics-pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother 55:24–34. doi: 10.1128/AAC.00749-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gumbo T, Pasipanodya JG, Wash P, Burger A, McIlleron H. 2014. Redefining multidrug-resistant tuberculosis based on clinical response to combination therapy. Antimicrob Agents Chemother 58:6111–6115. doi: 10.1128/AAC.03549-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gumbo T, Chigutsa E, Pasipanodya J, Visser M, van Helden PD, Sirgel FA, McIlleron H. 2014. The pyrazinamide susceptibility breakpoint above which combination therapy fails. J Antimicrob Chemother 69:2420–2425. doi: 10.1093/jac/dku136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gumbo T. 2010. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother 54:1484–1491. doi: 10.1128/AAC.01474-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Craig WA. 2007. Pharmacodynamics of antimicrobials: general concepts and applications, p 1–19. In Nightangle CH, Ambrose PG, Drusano GL, Murakawa T (ed), Antimicrobial pharmacodynamics in theory and practice 2nd ed, vol 44 Informa Healthcare USA, Inc., New York, NY. [Google Scholar]
- 25.Craig WA, Redington J, Ebert SC. 1991. Pharmacodynamics of amikacin in vitro and in mouse thigh and lung infections. J Antimicrob Chemother 27(Suppl C):29–40. [DOI] [PubMed] [Google Scholar]
- 26.Kiem S, Schentag JJ. 2008. Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob Agents Chemother 52:24–36. doi: 10.1128/AAC.00133-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gumbo T, Siyambalapitiyage Dona CS, Meek C, Leff R. 2009. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother 53:3197–3204. doi: 10.1128/AAC.01681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gumbo T. 2008. Integrating pharmacokinetics, pharmacodynamics and pharmacogenomics to predict outcomes in antibacterial therapy. Curr Opin Drug Discov Dev 11:32–42. [PubMed] [Google Scholar]
- 29.Falagas ME, Vardakas KZ, Tsiveriotis KP, Triarides NA, Tansarli GS. 2014. Effectiveness and safety of high-dose tigecycline-containing regimens for the treatment of severe bacterial infections. Int J Antimicrob Agents 44:1–7. doi: 10.1016/j.ijantimicag.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 30.De Pascale G, Montini L, Pennisi M, Bernini V, Maviglia R, Bello G, Spanu T, Tumbarello M, Antonelli M. 2014. High dose tigecycline in critically ill patients with severe infections due to multidrug-resistant bacteria. Crit Care 18:R90. doi: 10.1186/cc13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gumbo T, Pasipanodya JG, Romero K, Hanna D, Nuermberger E. 2015. Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin Infect Dis 61(Suppl 1):S25–S31. doi: 10.1093/cid/civ427. [DOI] [PubMed] [Google Scholar]
- 32.Chigutsa E, Pasipanodya JG, Visser ME, van Helden PD, Smith PJ, Sirgel FA, Gumbo T, McIlleron H. 2015. Impact of nonlinear interactions of pharmacokinetics and MICs on sputum bacillary kill rates as a marker of sterilizing effect in tuberculosis. Antimicrob Agents Chemother 59:38–45. doi: 10.1128/AAC.03931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasipanodya JG, McIlleron H, Burger A, Wash PA, Smith P, Gumbo T. 2013. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis 208:1464–1473. doi: 10.1093/infdis/jit352. [DOI] [PMC free article] [PubMed] [Google Scholar]