

Abstract
Acinetobacter baumannii is a Gram-negative bacterial pathogen responsible for a range of nosocomial infections. The recent rise and spread of multidrug-resistant A. baumannii clones has fueled a search for alternative therapies, including bacteriophage endolysins with potent antibacterial activities. A common feature of these lysins is the presence of a highly positively charged C-terminal domain with a likely role in promoting outer membrane penetration. In the present study, we show that the C-terminal amino acids 108 to 138 of phage lysin PlyF307, named P307, alone were sufficient to kill A. baumannii (>3 logs). Furthermore, P307 could be engineered for improved activity, the most active derivative being P307SQ-8C (>5-log kill). Both P307 and P307SQ-8C showed high in vitro activity against A. baumannii in biofilms. Moreover, P307SQ-8C exhibited MICs comparable to those of levofloxacin and ceftazidime and acted synergistically with polymyxin B. Although the peptides were shown to kill by disrupting the bacterial cytoplasmic membrane, they did not lyse human red blood cells or B cells; however, serum was found to be inhibitory to lytic activity. In a murine model of A. baumannii skin infection, P307SQ-8C reduced the bacterial burden by ∼2 logs in 2 h. This study demonstrates the prospect of using peptide derivatives from bacteriophage lysins to treat topical infections and remove biofilms caused by Gram-negative pathogens.
INTRODUCTION
Acinetobacter baumannii is an increasingly significant nosocomial pathogen worldwide (1). Arising from both intrinsic and acquired antibiotic resistance, multi- and pan-drug-resistant clones of A. baumannii can readily be isolated from hospital environments (2). A. baumannii has been shown to develop resistance to several classes of antibiotics, including aminoglycosides, cephalosporins, carbapenems, tigecycline, and colistin (3). The reasons for this high resistance include a high degree of genetic plasticity combined with an intrinsic resistance to certain antibiotics due to the presence of β-lactamases, the low permeability of the outer membrane, and highly efficient efflux pump systems (4). Furthermore, A. baumannii is prone to develop biofilms on solid surfaces, including medical devices (5). Thus, A. baumannii is not only problematic as an infectious agent but also increasingly difficult to be removed from hospital environments, a phenomenon similar to that observed with the Gram-positive nosocomial pathogen Staphylococcus aureus.
One of the last-resort antibiotics for A. baumannii is the antimicrobial peptide polymyxin B (6). The bactericidal effect of polymyxin B is mediated through its positively charged DAB (α,γ-diaminobutyric acid) residues interacting with lipopolysaccharide and destabilizing the outer membrane (7). Many antimicrobial peptides kill in a similar way: clustered cationic residues permeabilize the bacterial membrane to cause lysis and death (8). Due to this mechanism of action, most of the membrane-acting antimicrobial peptides commonly have cytotoxic effects on eukaryotic cells (9). Indeed, polymyxin B has severe side effects: cytotoxicity, nephrotoxicity, and neurotoxicity (10). Since careful administration is required to avoid its toxicity, the dose range of polymyxin B is limited, and resistant strains of A. baumannii have been documented (11).
Recently, there has been a growing interest in the use of bacterial viruses (i.e., bacteriophage therapy) to treat infections by Gram-negative bacteria, including A. baumannii (12–14). Several phages that infect A. baumannii have been identified and characterized. However, their restricted spectrum (killing only ∼60% of A. baumannii isolates) limits the effectiveness of such phages as therapeutic agents (12, 13). Using an alternative bacteriophage-based approach, our group and others have taken advantage of the lytic enzymes (lysins) encoded and produced by bacteriophages during lytic proliferation (15–18). Bacteriophage lysins are classified as peptidoglycan hydrolases, being able to cleave a variety of bonds in the bacterial peptidoglycan. Cleavage of the cell wall by lysins destabilizes the peptidoglycan and weakens the structural framework, resulting in hypotonic lysis. Although purified lysins are effective at killing Gram-positive bacteria (19), the outer membrane of Gram-negative bacteria largely limits lysins from accessing and cleaving the subjacent peptidoglycan. Different strategies have been used to increase the efficiency of lysins against Gram-negative bacteria, including the use of the chelating agent EDTA (16, 17), and the genetic engineering of lysins to add either highly charged/hydrophobic N-/C-terminal extensions (20) or other membrane-translocating domains (21, 22). However, there has been little focus on the intrinsic features of certain active lysins against Gram-negative bacteria and how they function to allow the lysins to cross the outer membrane and reach the subjacent peptidoglycan substrate.
Here, we have identified a highly cationic C-terminal domain within an A. baumannii phage lysin as a peptide with potent antibacterial activity. We have modified the peptide to further improve its activity and have proven the high efficiency of such peptides to kill A. baumannii both in vitro and in vivo in a skin infection model.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Acinetobacter baumannii strains in this study include clinical isolates from Hospital for Special Surgery in New York (isolates 1775 to 1799) (18), Ohio State University (S1, S3, and S5 provided by Vijay Pancholi), and ATCC 17978 from the American Type Culture Collection. Bacteria were cultured in Trypticase soy broth or brain heart infusion (Thermo Fisher Scientific, Waltham, MA) at 37°C with aeration (200 rpm). Stationary-phase bacteria were cultured overnight, while log-phase bacteria were grown for 3 h in fresh medium from 100× dilutions of overnight cultures. Strains for determining the specificity of the antimicrobial peptides were cultured under the same conditions, at the temperatures indicated: Bacillus anthracis ΔSterne (30°C), Escherichia coli DH5α (37°C), Klebsiella pneumoniae ATCC 700603 and 10031 (37°C), Pseudomonas aeruginosa PAO1 (30°C), and Staphylococcus aureus RN4220 (30°C).
Biofilms were set up as previously described (18). Briefly, an overnight culture of A. baumannii strain 1791 was diluted 1,000× (∼105 CFU/ml) in Trypticase soy broth containing 0.2% glucose, followed by incubation at 37°C for 72 h in ∼2.5-cm segments of polyvinyl chloride (PVC) catheter tubing (CareFusion) with the ends clamped shut to prevent evaporation. The catheter biofilms were washed with autoclaved Milli-Q water to remove planktonic cells before peptide treatment. Crystal violet (0.1%) was used to detect the presence of biofilm.
Peptide synthesis and preparation.
Peptides were synthesized at The Rockefeller University Proteomics Resource Center. All peptides were created using a Protein Technologies Symphony peptide synthesizer (PTI, Tucson, AZ) on precoupled Wang (p-alkoxy-benzyl alcohol) resin (Bachem, Torrance, CA). Reaction vessels were loaded at 25 μM, and peptides were elongated using Fmoc (9-fluorenylmethoxy carbonyl)-protected amino acids (Anaspec, San Jose, CA) (23). Deprotection of the amine was accomplished with 20% piperidine (Sigma-Aldrich) in NMP (N-methylpyrrolidinone). Repetitive coupling reactions were conducted using 0.6 M HATU/Cl-HOBT (azabenzotriazol tetramethyluronium hexafluorophosphate/6-chloro-1-hydroxybenzotriazole)(P3 Biosystems, Shelbyville, KY, USA) and 0.4 M NMM (N-methylmorpholine) using NMP (EMD) as the primary solvent (24). Resin cleavage and side chain deprotection were achieved by transferring to a 100-ml round-bottom flask and reacting with 4.0 ml of concentrated, sequencing-grade, trifluoroacetic acid (TFA; Fisher) with triisopropylsilane (Fluka), degassed water, and 3,6-dioxa-1,8-octanedithiol (DODT; Sigma-Aldrich) in a ratio of 95:2:2:1 over a 6-h time frame. This was followed by column filtration to a 50-ml round-bottom flask, and the TFA volume was reduced to 2 ml using a rotary evaporator. A standard ether precipitation was performed on the individual peptides by transferring to a 50-ml Falcon tube containing 40 ml of cold tert-butyl methyl ether (TBME; Sigma-Aldrich). Samples were placed in an ice bath for 2 h to aid precipitation, followed by pellet formation using centrifugation (3,300 rpm, 5 min). Excess ether was removed by vacuum aspiration, and the peptide pellets were allowed to dry overnight in a fume hood. Dried peptide pellets were resolved in 20% acetonitrile and 10 ml of high-pressure liquid chromatography (HPLC)-grade water, subsampled for liquid chromatography-mass spectrometry (LC/MS), and lyophilized. All crude products were subsequently analyzed by reversed-phase Aquity UPLC (Waters Chromatography, Milford, MA) using a Waters BEH C18 column. Individual peptide integrity was verified by tandem electrospray mass spectrometry using a ThermoFinnigan LTQ (Thermo Fisher, Waltham, MA) spectrometer system. Preparative chromatography was accomplished on a Vydac C18 RP preparative column on a Waters 600 Prep HPLC apparatus. Individual fractions were collected in 30 s intervals, characterized using LC/MS, and fractions containing the desired product were lyophilized. These were stored at −20°C until they were resuspended in autoclaved Milli-Q water for various assays. The stock solutions were then stored at 4°C.
Bactericidal assays.
Unless otherwise indicated, stationary-phase bacteria (overnight cultures) were used for the assays. Bacteria were washed in assay buffer, resuspended to ∼106 CFU/ml, mixed with each antimicrobial peptide, and incubated for 2 h at room temperature (22-25°C). After treatment, the reactions were serially diluted and plated for enumeration. The following factors influencing the activities of the peptides (P307 and P307SQ-8C) were examined: buffer (50 mM sodium phosphate or Tris-HCl, pH 6.8 to 8.8; NaCl, 0 to 200 mM), concentration (0 to 125 μg/ml P307), time (1 to 120 min), specificity (the bacterial strains mentioned above), and growth phase (log, stationary, and biofilm). The buffer system for the experiments with the latter five factors was 50 mM Tris-HCl (pH 7.5). The biofilm-associated bacteria were resuspended by thoroughly scraping the catheter tubing, followed by vortexing for 1 min.
To examine the mechanism(s) of action of the peptides, the following bactericidal assays were also conducted. The contribution of disulfide bond formation to the increased activity of P307SQ-8C was investigated by comparison to P307SQ-8A in the presence of the reducing agent TCEP [0.1 and 1 mM; tris(2-carboxyethyl) phosphine hydrochloride solution; Sigma]. To examine the importance of ionic interaction, the least sensitive Gram-negative bacterial species at pH 7.5 were treated with P307 and P307SQ-8C at pH 8.8. Experiments were conducted at least in triplicate, and representative data are shown as means ± standard deviations. The black horizontal lines mark the limit of detection.
MICs and synergy.
The broth microdilution method was used (25) to determine the MICs of levofloxacin, ceftazidime, polymyxin B, P307, and P307SQ-8C for A. baumannii strains 1791, 1798, S5, and ATCC 17978. For the antibiotics, 1.5- to 2-fold serial dilutions (three lower and three higher) of the MICs determined by Etest (18) were included. For the peptides, 2-fold serial dilutions (2,000 to 31.25 μg/ml) were tested. Overnight cultures were resuspended to ∼105 CFU/ml in Mueller-Hinton broth (pH 7.9). Antibiotics or peptides were added to a final volume of 100 μl for each dilution. Bacteria were allowed to grow at 37°C for 24 h at 220 rpm. The absorbance at 595 nm was then read in a SpectraMax Plus Reader (Molecular Devices). The MICs were determined as the lowest concentrations of antimicrobial agents that visibly inhibited bacterial growth. Each experiment was repeated at least twice in duplicate.
To investigate whether synergy exists between each peptide and antibiotic, checkerboard assays were conducted by mixing fractional inhibitory concentrations (FICs) of antibiotics (levofloxacin, ceftazidime, and polymyxin B) with those of peptides P307 and P307SQ-8C, as previously described (26). Isobolograms were constructed to determine the FIC indices (FICIs). Each experiment was repeated at least three times.
DNA-binding assay.
The affinity of peptide P307 for DNA was investigated by electrophoretic mobility shift assay (27). P307 was mixed with 50 ng of random DNA (1-kb amplicon of Bdellovibrio genomic DNA) at different peptide/DNA ratios (0:1 to 15:1) in a binding buffer (5 mM Tris-HCl [pH 8.0], 10 mM EDTA, 5% glycerol, 20 mM KCl, 50 μg/ml bovine serum albumin). As a positive control, a peptide from Bdellovibrio bacteriovorus that was previously found in our laboratory to have DNA-binding capacity (amino acid sequence MASKTKKTEYIRERKKATSGKKRKAANRTKGTTKSAKTLFKD) was used (unpublished results). After incubation for 1 h at 22 to 25°C, the peptide and DNA mixture was analyzed by agarose gel electrophoresis (27).
TEM.
An overnight culture of A. baumannii strain 1791 was collected, washed in 50 mM Tris-HCl (pH 7.5), and resuspended in the same buffer to ∼108 CFU/ml. The bacteria were treated with 300 μg/ml P307SQ-8C or buffer (control) for 5 min or 2 h. The samples were fixed with 2.5% glyceraldehyde in 0.1 M sodium cacodylate, and then transmission electron microscopy (TEM) images were obtained at a magnification of ×2,600 or ×5,000. Representative images are shown in the figures.
SYTOX green uptake assay.
The permeability of bacterial membrane upon peptide treatment was measured by SYTOX green uptake (28). Briefly, overnight cultures of bacteria were washed and resuspended to ∼107 CFU/ml in 50 mM Tris-HCl (pH 7.5). Benzonase nuclease (25 U/ml; Novagen) and SYTOX green (1 μM; Invitrogen) was added to the bacterial cells, followed by incubation for 15 min at 22 to 25°C in the dark. Peptides (50 μg/ml) were added, and polymyxin B (2 μg/ml; Sigma) was used as a control (29). Relative fluorescence units were measured in a SpectraMax Plus reader (Molecular Devices) at 22 to 25°C (excitation, 485 nm; emission, 520 nm) for 2 h. Experiments were carried out twice in duplicate, and representative data are shown as means ± standard deviations.
Cytotoxicity assays.
The hemolytic assays were conducted as previously described (28). Briefly, human blood was gathered in an EDTA tube, and red blood cells (RBCs) were collected through low-speed centrifugation. Cells were washed and resuspended to a 10% RBC solution in PBS and then mixed with 345 μg/ml P307SQ-8C. Phosphate-buffered saline (PBS) and 1% Triton X-100 were used as negative and positive controls, respectively. After 1 h of incubation at 37°C, the supernatant was collected, and the absorbance at 405 nm was recorded through a SpectraMax Plus Reader (Molecular Devices) to measure the release of hemoglobin. The reactions were carried out twice in triplicate, and representative data are shown as means ± standard deviations.
A human B lymphoblastoid cell line (LCL) obtained from The Rockefeller University Hospital was grown in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum. Cells were harvested by low-speed centrifugation, washed, and resuspended in PBS to ∼5 × 106 live cells/ml, as determined by trypan blue exclusion tests. LCL cells (∼5 × 105) were incubated with 172.5, 345, and 517.5 μg/ml P307SQ-8C at 37°C in a humidified 5% CO2 atmosphere for 1 h. The cells were then stained according to the manufacturer's instructions (CellTiter 96 nonradioactive cell proliferation assay; Promega), where live B cells reduced the dye to insoluble formazan for an additional 4 h. Solubilization/stop solution was added, followed by incubation at 37°C overnight. Next, the absorbance at 570 nm was measured in SpectraMax Plus Reader (Molecular Devices) to quantitate B cell survival. Triton X-100 was used as a positive control. The reactions were carried out in triplicate, and data are shown as means ± standard deviations.
Endotoxin release.
To detect the amount of endotoxin release, strain 1791 (∼107 CFU/ml) was incubated at 22 to 25°C for 2 h with different concentrations of P307 or P307SQ-8C. The samples were briefly centrifuged (2,000 × g) for 1 min to remove live cells. All reagents were prepared in endotoxin-free autoclaved Milli-Q water. An endpoint chromogenic LAL assay (Lonza) was conducted according to the manufacturer's instructions. Polymyxin B and 100% ethanol were used for comparison and for a positive control, respectively. The experiment was conducted twice in duplicate, and representative data are shown as means ± standard deviations.
Bactericidal activity in plasma and its components.
Human blood was collected in a heparin tube and centrifuged. The supernatant was filtered and stored at 4°C overnight. The filtrate was centrifuged and filtered again to remove any debris. The resulting solution was used as 100% plasma. The components of plasma examined for interference were monovalent cation (Na+), divalent cations (Ca2+ and Mg2+), and albumin. Chloride salts of the cations and albumin from human serum (lyophilized powder, ≥97%; Sigma) were prepared in Milli-Q water and sterile filtered. Bactericidal assays, performed as described above, were conducted in 100% plasma and in buffered solutions (50 mM Tris-HCl [pH 7.5]) containing 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, and 50 mg/ml human serum albumin using 100 μg/ml P307 or P307SQ-8C. The experiments were conducted at least in triplicate, and the data are shown as means ± standard deviations.
Mouse in vivo skin model.
The Rockefeller University Institutional Animal Care and Use Committee approved all in vivo protocols. Skin infection was induced with A. baumannii as previously described (30). Briefly, the backs of 30 female CD-1 mice (6 to 8 weeks of age; Charles River Laboratories) were shaved with an electric razor. Nair depilatory cream was applied to the shaved areas to remove any remaining hair. The areas were then disinfected with alcohol wipes and tape stripped with autoclave tape 20 times in succession, using a fresh piece of tape each time. The tape-stripped areas were disinfected again with alcohol wipes. An area of ∼1 cm2 was then marked and infected with 10 μl of ∼108 CFU/ml of A. baumannii strain 1791. The bacteria were allowed to colonize for 16 to 18 h, after which the infected area was either left untreated or treated with 200 μg of P307SQ-8C or 2 μg of polymyxin B in autoclaved Milli-Q water for 2 h. To harvest the remaining bacteria on the skin, the mice were sacrificed, and the infected skin was processed in 500 μl of PBS for 1 to 2 min in a Stomacher 80 Biomaster using a microbag (Seward, Ltd., United Kingdom). The solution was serially diluted and plated on LB agar containing 4 μg/ml levofloxacin and 12 μg/ml ampicillin for selection. The resulting CFU/ml values from each animal were plotted as individual points, and horizontal bars represent the mean values. Data were analyzed using Ordinary one-way analysis of variance (ANOVA) in GraphPad Prism 6.0. The dotted horizontal line marks the limit of detection.
RESULTS
Identification and modification of putative antibacterial peptides from phage lysin PlyF307.
Previously published works on phage lysins against Gram-negative bacteria (15, 18) suggested that the C-terminal portions of lysins might destabilize the bacterial outer membrane to facilitate access to the lysin's substrate, i.e., the peptidoglycan. Specifically, the highly cationic C-terminal region of A. baumannii phage lysin LysAB2 was speculated to be involved in permeabilizing the outer membrane (15). We were able to identify a similar highly positively charged sequence in one of our A. baumannii phage lysins, PlyF307 (18). This peptide, termed P307 (Table 1), extends from amino acids 108 to 138 in PlyF307 and partly resembles the LysAB2 peptide in terms of length and charge, but shares only 41.2% similarity and 17.6% identity (MacVector ClustalW alignment).
TABLE 1.
Peptide derivatives of PlyF307a
| Derivative | Amino acid sequenceb | pI | Molecular mass (kDa) |
|---|---|---|---|
| PlyF307 | 146 amino acids (GenBank accession number KJ740396) | 10.12 | 16.1 |
| P307 | NAKDYKGAAAEFPKWNKAGGRVLAGLVKRRK (amino acids 108 to 138) | 10.70 | 3.4 |
| P307AE-8 | NAKDYKGAAAEFPKWNKAGGRVLAGLVKRRKAEMELFLK (original) | 10.21 | 4.4 |
| P307SQ-8C | NAKDYKGAAAEFPKWNKAGGRVLAGLVKRRKSQSRESQC (from hepatitis B virus) | 10.38 | 4.3 |
| P307CS-8 | NAKDYKGAAAEFPKWNKAGGRVLAGLVKRRKCSQRQSES (scramble) | 10.38 | 4.3 |
| P307SQ-8A | NAKDYKGAAAEFPKWNKAGGRVLAGLVKRRKSQSRESQA (C39A) | 10.69 | 4.3 |
Amino acid sequences in single-letter code, theoretical isoelectric points (pI), and molecular masses are presented.
Different modifications to P307 are indicated by underscoring.
We modified P307 with the eight amino acids constituting the full C-terminal part of native PlyF307, forming peptide P307AE-8 (Table 1). In addition, we fused eight amino acids (SQSRESQC) to the C terminus of P307 and yielded P307SQ-8C (Table 1). This modification was based on the observation of an earlier study showing that the lack of these eight amino acids reduces the activity spectrum of the antimicrobial peptide from hepatitis B virus core protein (28). We also generated two additional modifications of P307SQ-8C: scrambling the sequence of the last eight amino acids to CSQRQSES (P307CS-8); and changing the last amino acid from C to A (P307SQ-8A) (Table 1). The sequence was scrambled to evaluate the importance of a specific sequence and the cysteine change was introduced to examine the importance of intermolecular disulfide bond formation for activity. A predicted structure of PlyF307, generated through I-TASSER (31–33), suggested that P307 and its derivatives formed a “hairpin-like” di-alpha-helical structure connected with a flexible linker region (Fig. 1).
FIG 1.
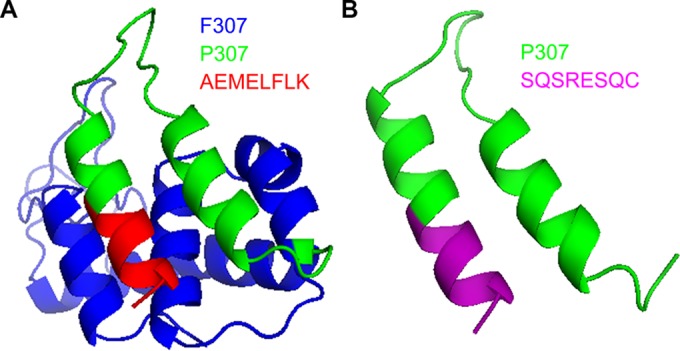
Structural predictions of PlyF307 and P307SQ-8C. (A) Full-length lysin PlyF307 is shown in blue, P307 fragment is shown in green, and the last eight amino acids (AEMELFLK) are shown in red. (B) P307SQ-8C is shown with P307 in green and the last eight amino acids (SQSRESQC) in purple. Structures were predicted using I-TASSER server (Zhang lab, University of Michigan) (31–33).
Comparison of the peptides.
We evaluated the bactericidal activities of the peptides P307, P307AE-8, and P307SQ-8C. P307SQ-8C was the most active, reducing ∼106 CFU/ml of bacteria to below the limit of detection (<10 CFU/ml). P307 was slightly more active than P307AE-8, but both peptides induced an ∼3.8-log decrease in viability (Fig. 2A). Since P307SQ-8C was the most active, we investigated the importance of the last eight amino acids SQSRESQC. To test whether the linkage between P307 and the 8-amino-acid peptide was important, we compared the activities of P307, P307SQ-8C, the combination of P307 and equimolar concentrations of the SQSRESQC peptide, and the SQSRESQC peptide alone. We found that the combination was only as active as P307, while the SQSRESQC peptide alone had no activity (Fig. 2B). Hence, the linkage was essential for the high bactericidal activity of P307SQ-8C. Next, we investigated the importance of sequence and composition by synthesizing P307CS-8 with the last eight amino acids in P307SQ-8C scrambled to CSQRQSES. The bactericidal activities of P307SQ-8C and P307CS-8 were comparable (Fig. 2C), indicating that the amino acid composition was more important than the sequence for the superior activity of P307SQ-8C. For further investigation, we used P307SQ-8C since it was the most active, and compared its activity to P307.
FIG 2.
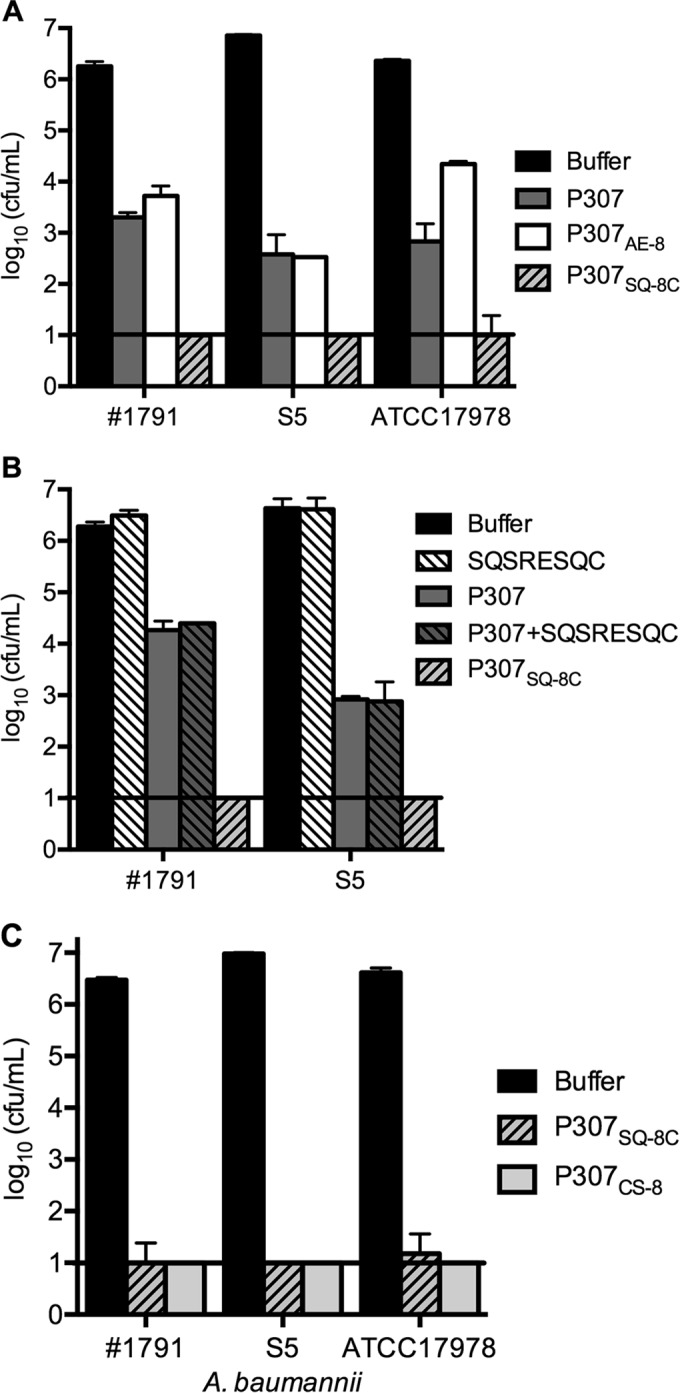
Comparison of in vitro bactericidal activities of peptides. Bacteria were treated with 50 μg/ml of each peptide for 2 h at 22 to 25°C. Serial dilutions were plated for CFU counting. (A) The bactericidal activities of P307 and its variants P307AE-8 and P307SQ-8C against A. baumannii strains 1791, S5, and ATCC 17978. (B) The activities of P307, P307SQ-8C, and equimolar SQSRESQC peptide with or without P307 against strains 1791 and S5. (C) The activities of P307SQ-8C and a scramble variant P307CS-8 against strains 1791, S5, and ATCC 17978. The error bars show the standard deviations, and the black horizontal lines mark the limit of detection.
Bactericidal activities of P307 and P307SQ-8C.
The effects of pH and NaCl on the in vitro activities of P307 and P307SQ-8C were investigated. A. baumannii strain 1791 was treated with 50 μg/ml of either peptide in the presence of various pH and salt conditions. Using two buffer systems (sodium phosphate and Tris-HCl), we compared pH 6.8, 7.5, 8.0, and 8.8 and found that the peptides were more active in Tris-HCl buffer and that higher pH values elicited better killing (see Fig. S1A in the supplemental material). Thus, we elected to continue our in vitro experiments with 50 mM Tris-HCl (pH 7.5) to approximate physiological pH. The activities of both peptides were inversely proportional to the concentration of NaCl (see Fig. S1B in the supplemental material). When titration and killing kinetics were investigated, we found that the activities of both peptides were concentration-dependent (see Fig. S1C in the supplemental material). P307SQ-8C acted faster than P307, resulting in ∼3.2-log decrease after 5 min, with a continued reduction in bacterial count up to ∼5 logs 120 min after P307SQ-8C addition (see Fig. S1D in the supplemental material). There was no difference in the activities of both peptides either at room temperature (22 to 25°C) or 37°C (data not shown). From these in vitro characterization experiments, we chose our optimal experimental conditions to be 50 μg/ml peptides in 50 mM Tris-HCl (pH 7.5) for 2 h at 22 to 25°C, unless otherwise indicated.
We also examined the in vitro bactericidal spectra of P307 and P307SQ-8C. Among the bacteria tested, A. baumannii strains were consistently the most sensitive to the peptides, showing an average of 2.7- and 6.2-log decrease with P307 and P307SQ-8C, respectively. Bacillus anthracis, Pseudomonas aeruginosa, and Staphylococcus aureus were moderately sensitive to P307 and P307SQ-8C, with ∼1.3- and 2.9-log average decreases, respectively. Both Escherichia coli and Klebsiella pneumoniae were resistant to the peptides under these experimental conditions (Fig. 3). However, at pH 8.8, both were sensitive to 50 μg/ml of the peptides (see Fig. S2 in the supplemental material).
FIG 3.

In vitro bactericidal spectra of P307 and P307SQ-8C. Different bacterial species (A. baumannii, B. anthracis, E. coli, P. aeruginosa, S. aureus, and K. pneumoniae) were treated with 50 μg/ml P307 or P307SQ-8C in 50 mM Tris-HCl (pH 7.5) for 2 h at 22 to 25°C to investigate the specificity of the peptides. Serial dilutions were plated for CFU counting. The error bars show the standard deviations, and the black horizontal line marks the limit of detection.
To examine the activity of each peptide against A. baumannii at different growth phases, we compared the sensitivities of strain 1791 at log phase, at stationary phase, and in biofilms. The log-phase organisms were slightly more sensitive to P307 than at stationary phase (an ∼3.7-log decrease versus a 2.4-log decrease) (Fig. 4A). However, no such difference was observed for P307SQ-8C, with both growth phases being equally sensitive to the maximal killing effect of the P307SQ-8C (>5-log decrease). Biofilm-associated A. baumannii was more resistant (Fig. 4B). Biofilms were treated with 250 μg/ml P307 or P307SQ-8C for 2 or 24 h. After 2 h, ∼3- and 4-log decreases in CFU/ml were observed with P307 and P307SQ-8C, respectively. After 24 h, an additional ∼1.3-log decrease was observed with P307, while there was no further decrease with P307SQ-8C.
FIG 4.
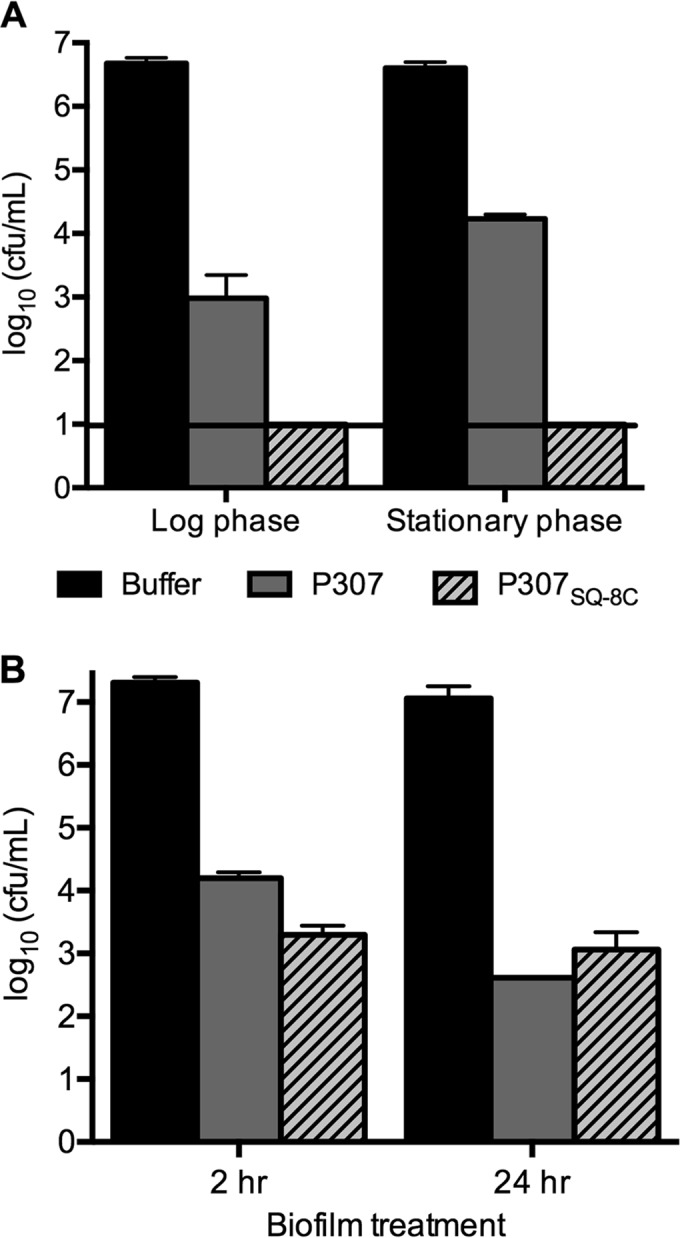
Bactericidal activities of P307 and P307SQ-8C against A. baumannii strain 1791 at log phase, at late stationary phase, and in biofilm. (A) Bacteria in log phase and late stationary phase were treated with 50 μg/ml P307 or P307SQ-8C for 2 h at 22 to 25°C. (B) A. baumannii biofilms in PVC catheters were treated with 250 μg/ml of P307 or P307SQ-8C for 2 or 24 h at 22 to 25°C. Serial dilutions were plated for CFU counting. The error bars show the standard deviations, and the black horizontal line marks the limit of detection.
MIC and synergy.
In order to compare the efficiency of the peptides with clinically used antibiotics, we performed MIC assays on four A. baumannii strains: 1791, 1798, S5, and ATCC 17978. The strains displayed various degrees of sensitivity to levofloxacin, ceftazidime, and P307, while the MICs of polymyxin B and P307SQ-8C were more consistent (Table 2). P307SQ-8C had a lower MIC than P307, which was in accordance with in vitro killing activity (Fig. 2 and 3).
TABLE 2.
MIC comparison for peptides and antibiotics examined in this study
| A. baumannii strain | MIC (μg/ml and μM)a |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Levofloxacin |
Ceftazidime |
Polymyxin B |
P307 |
P307SQ-8C |
||||||
| μg/ml | μM | μg/ml | μM | μg/ml | μM | μg/ml | μM | μg/ml | μM | |
| 1791 | 6 | 16.6 | 128 | 234 | 0.25 | 0.19 | 750 | 221 | 62.5 | 14.5 |
| 1798 | 32 | 88.6 | >1,024 | >1,873 | 0.25 | 0.19 | 1,000 | 294 | 62.5 | 14.5 |
| S5 | 6 | 16.6 | 64 | 117 | 0.25 | 0.19 | 2,000 | 588 | 125 | 29 |
| ATCC 17978 | 0.075 | 0.21 | 6 | 11 | 0.25 | 0.19 | 750 | 221 | 125 | 29 |
Numbers in boldface indicate resistance to the antibiotics.
We also examined the synergistic effects between each peptide and levofloxacin, ceftazidime, or polymyxin B by the checkerboard method (isobologram). No synergy was observed between either peptide and levofloxacin or ceftazidime (data not shown). However, polymyxin B acted synergistically with both P307 and P307SQ-8C (FICIs = 0.125).
Mechanism of action.
Since P307SQ-8C contained a C-terminal cysteine, we investigated the importance of disulfide bond formation for the high activity of P307SQ-8C. SDS-PAGE analysis showed that a portion of P307SQ-8C (∼5 to 10%) ran at twice the theoretical molecular mass of this peptide (data not shown), suggesting a role for disulfide bond formation. To examine this, the C-terminal cysteine residue of P307SQ-8C (the only cysteine in this peptide) was changed to alanine. The resulting alanine variant, P307SQ-8A, ran at the expected size for a peptide monomer (4.3 kDa) with no observable band at the dimer location by SDS-PAGE (data not shown). The difference in bactericidal activities of P307SQ-8A and P307SQ-8C were compared within the limit of detection by lowering the peptide concentration from 50 to 10 μg/ml. Furthermore, TCEP (0.1 and 1 mM) was added to specifically reduce disulfide bonds. Without TCEP, P307SQ-8C was more active than P307SQ-8A (an ∼5-log decrease versus an ∼2.7-log decrease) (see Fig. S3 in the supplemental material). The addition of TCEP did not affect P307SQ-8A, whereas the activity of P307SQ-8C was reduced by ∼2 logs. However, P307SQ-8A was still more active than P307, suggesting that although disulfide bond formation was important, it was not the only contributing factor for the superior activity of P307SQ-8C.
Next, due to the presence of several cationic residues on the peptides (net charge of +7), we investigated their effects on two of the negatively charged components of the bacteria: membrane and DNA. TEM was used to visualize the effects of P307SQ-8C on A. baumannii membrane. The samples were prepared by treating A. baumannii strain 1791 with buffer (control) or 300 μg/ml P307SQ-8C for 5 min or 2 h. A comparison of the TEM images revealed that treatment with P307SQ-8C led to (i) changes in intracellular density, (ii) an appearance of “intact bacterial ghosts,” and (iii) occasional disruption of the cell wall and cytoplasmic membrane (Fig. 5). The affinity of P307 for DNA was examined by electrophoretic mobility shift assay. There was no shift in molecular weight at all ratios of P307 to DNA tested, whereas a positive-control peptide known to bind DNA caused a clear shift in electrophoretic mobility (see Fig. S4 in the supplemental material).
FIG 5.
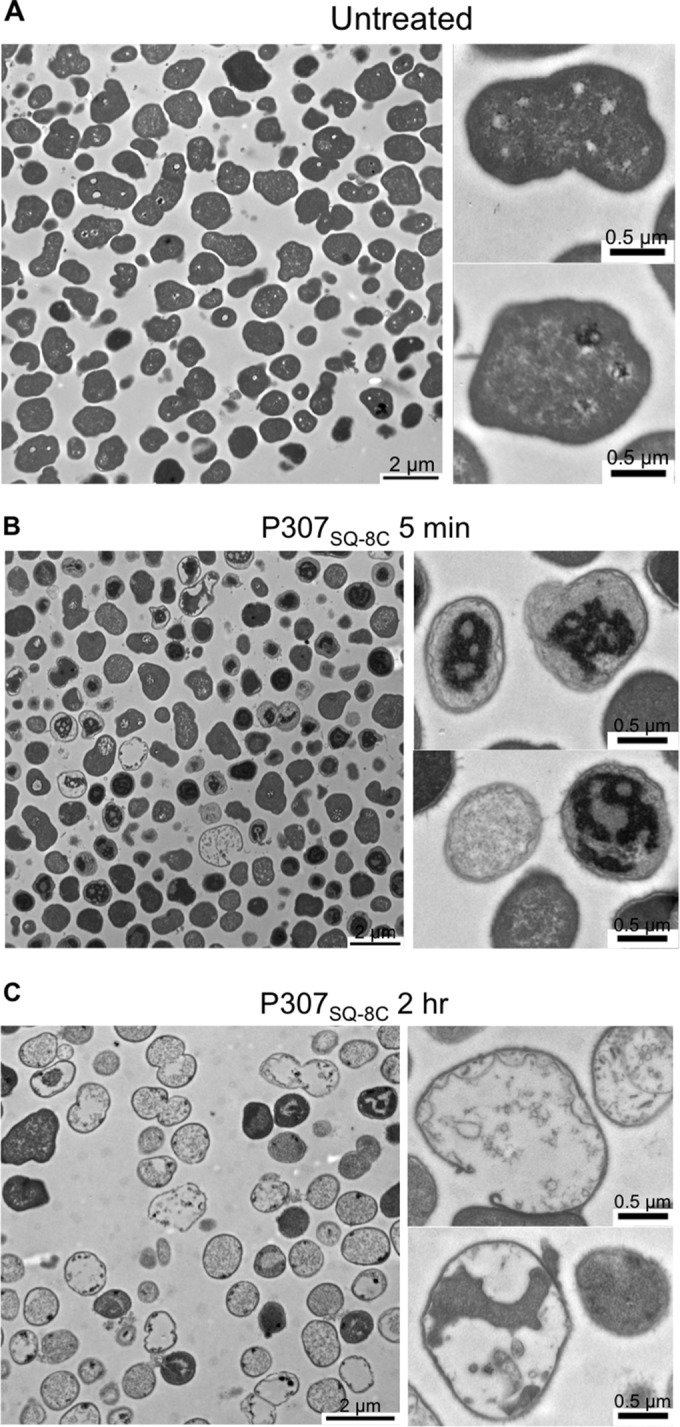
Representative TEM images of A. baumannii strain 1791: untreated control (A) and treatment with 300 μg/ml of P307SQ-8C for 5 min (B) or for 2 h (C). Magnifications: ×2,600 (left panels; scale bar, 2 μm) and ×5,000 (right panels [top and bottom]; scale bar, 0.5 μm).
Since our data suggested a killing mechanism by cytoplasmic membrane destabilization, we measured bacterial membrane disruption by P307 and P307SQ-8C using the SYTOX green uptake assay. Both peptides permeabilized the membranes of A. baumannii strain 1791 at pH 7.5 and K. pneumoniae strain ATCC 700603 at pH 8.8, giving rise to an increase in fluorescent signals of SYTOX green dye as it bound to intracellular DNA (see Fig. S5 in the supplemental material). In contrast, the signal increase was minimal for K. pneumoniae ATCC 700603 at pH 7.5. The results were consistent with the difference in sensitivities of the bacterial species at different pH (Fig. 3 and see Fig. S2 in the supplemental material).
Cytotoxicity.
Since membrane-acting antimicrobial peptides have been shown to be active against mammalian membranes (9), the cytotoxicity of the peptides were evaluated using human RBCs and B cells. In contrast to the 1% Triton X-100 positive control, 345 μg/ml P307SQ-8C did not lyse RBCs but killed A. baumannii strain 1791 (∼4.8 logs) in PBS (see Fig. S6A in the supplemental material). Similarly, the survival of B cells was not significantly affected by P307SQ-8C at 172.5, 345, and 517.5 μg/ml (see Fig. S6B in the supplemental material).
Endotoxin release upon peptide treatment.
Even though the peptides did not display cytotoxicity against human RBCs, toxicity could possibly arise from the bacterial endotoxin released upon peptide treatment. Hence, we measured the amount of endotoxin released by A. baumannii strain 1791 after 2 h of incubation with peptides using an endpoint chromogenic LAL assay (Lonza). We found that the peptides generated endotoxin unit concentrations similar to that of the 100% ethanol positive control. Polymyxin B released more endotoxin at 0.5 μg/ml than at 2 μg/ml (see Fig. S6C in the supplemental material), but less than the peptides. This apparent reduced release was likely due to the known interaction of polymyxin B with lipid A (7), the active component for the LAL assay. The peptides did not interfere with endotoxin detection (data not shown).
Bactericidal activities in plasma and its components.
In order to choose a suitable in vivo model, we examined the activities of the peptides in human blood plasma. Both P307 and P307SQ-8C (100 μg/ml) were inactive in 100% plasma. The activities in Tris buffer were also interfered to various degrees by the addition of monovalent and divalent cations, as well as human serum albumin, at physiological concentrations (see Fig. S6D in the supplemental material).
In vivo mouse skin model.
Since blood plasma interfered with the activities of both peptides, and the skin is a common route of infection by A. baumannii, we decided to investigate the in vivo activity of P307SQ-8C using a mouse skin infection model. Skin abrasions were induced on the shaved backs of the mice by tape-stripping, and the irritated skin was infected with 106 CFU of A. baumannii strain 1791 for 16 h. The infected area was then treated with 2 μg of polymyxin B or 200 μg of P307SQ-8C for 2 h; the infected skin was then excised and processed for bacterial counts. Both treatments were found to significantly reduce the bacterial load (∼2 logs) in the infected skin using this single dose (P < 0.02, ordinary one-way ANOVA) (Fig. 6).
FIG 6.
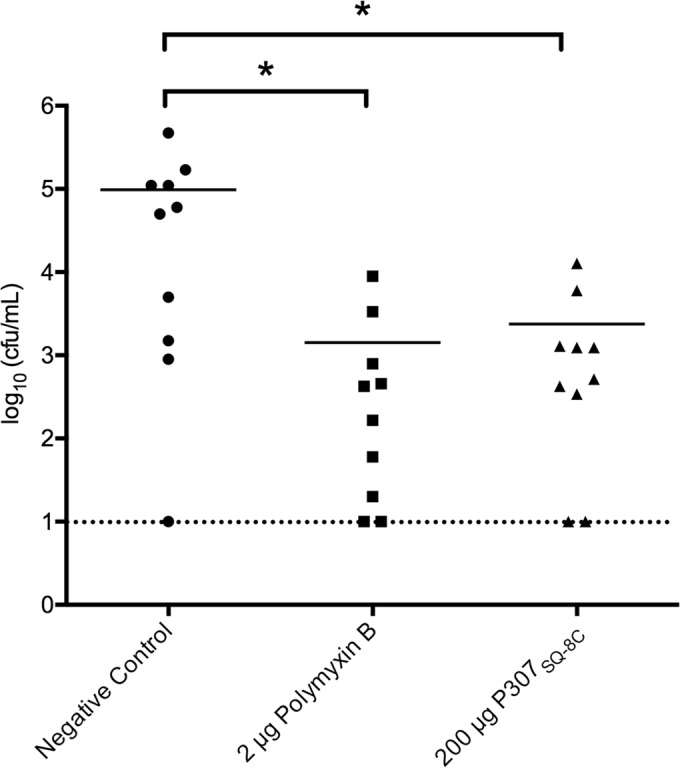
In vivo activity of P307SQ-8C versus that of polymyxin B on the skin of tape-stripped mice infected with A. baumannii strain 1791. Topical treatments included no treatment (negative control), 2 μg of polymyxin B (1.5 nmol), and 200 μg of P307SQ-8C (46.5 nmol) for 2 h. The bars show the mean values, and each dot represents one mouse. Statistically significant differences were noted for the comparison pairs—(i) negative control and polymyxin B (P = 0.0148) and (ii) negative control and P307SQ-8C (P = 0.0157)—with an ∼2-log decrease in A. baumannii in each treatment (ordinary one-way ANOVA, multiple comparisons, uncorrected Fisher LSD test). The dotted line marks the limit of detection.
DISCUSSION
Due to the notable antibiotic resistance mechanisms of A. baumannii (1), membrane-acting drugs such as polymyxin B and colistin, with toxic side effects and rising resistance, are now the last-resort drugs (34). To address the A. baumannii threat, several groups have evaluated the use of bacteriophage endolysins to kill multidrug-resistant A. baumannii (15, 17, 18). It has been hypothesized that the innate bactericidal activities of some of these lysins stem from the permeabilization of the bacterial outer membrane by the highly positively charged C-terminal portions (15, 18). We showed here that P307, a C-terminal-based peptide from PlyF307 (18), by itself, had high in vitro bactericidal activity (Fig. 2A). Furthermore, we demonstrated that P307 could be further engineered for log-fold-increased activity. Compared to P307, or to P307AE-8 with the eight amino acids (AEMELFLK) constituting the full C-terminal part of native PlyF307 (Fig. 1), the modified peptide P307SQ-8C with polar amino acids (SQSRESQC) at the C terminus was 10 to 15 times more active (Fig. 3 and Table 2). Comparison of P307SQ-8C to the scrambled P307CS-8 revealed that the six neutral and two charged polar amino acids could be added in an alternate order for similar bactericidal activity (Fig. 2C). Thus, while both AEMELFLK and SQSRESQC complete the length of the alpha helix to correspond to the adjacent helix in the hairpin structure (Fig. 1), it is the characteristics of the amino acids in the SQSRESQC sequence (not the structure) that dictate the increase in activity.
Both P307 and P307SQ-8C displayed high in vitro bactericidal activities. Not only were they effective against numerous clinical isolates of A. baumannii (Fig. 3) but they also killed bacteria in biofilms on the surface of catheters (Fig. 4B). Since the success of A. baumannii as an opportunistic pathogen is attributed to its improved survival through biofilm formation and persistence in hospital environments (35), the ability of the peptides to kill biofilm-associated bacteria could be harnessed to disinfect hospital environments, as well as to eliminate A. baumannii from the lumens of catheters in situ. While the in vivo utility of the peptides might be limited due to the diminished activity in plasma, the peptides could still be useful on abiotic surfaces such as coatings for ventilators and catheters.
To further increase the potency of the peptides, we analyzed the mechanism of action by various in vitro characterizations. The peptides were more active at higher pH but did not tolerate salinity. E. coli and K. pneumoniae were resistant to the peptides at pH 7.5 but were susceptible at pH 8.8 (Fig. 3 and see Fig. S2 in the supplemental material). Since the charges on the peptide were not expected to vary as the pH changed from 7.5 to 8.8, we reasoned that the changes likely occurred on the bacterial membrane. At higher pH, the bacterial membrane can lose its cations, allowing the positively charged peptides to establish ionic interactions to disrupt the membrane integrity. Based on these observations and data, we postulate the following mechanism of action: P307SQ-8C traverses the outer membrane and establishes ionic interactions with the bacterial cytoplasmic membrane to gain entry into the cell. In the process, the peptide disrupts both outer and inner membranes (as suggested by endotoxin release assay and as shown by TEM images and SYTOX green uptake assay) killing the cell. Assay with TCEP and P307SQ-8A also suggested that the bactericidal activity was higher (by ∼2 logs) when P307SQ-8C was dimerized. With this knowledge base, we are currently exploring additional engineering strategies to further improve both PlyF307 and P307SQ-8C.
Nosocomial A. baumannii infections of deep wounds, burns, and bone or bone marrow are highly prevalent, and the isolates are often multidrug resistant (36). Since one of the common infection routes of A. baumannii is through damaged skin, we utilized an already established skin infection model (30) for our in vivo characterization of P307SQ-8C. The backs of the mice were shaved and tape stripped to create skin irritations, which were then inoculated with 106 CFU of A. baumannii strain 1791. After 16 h of infection, mice were topically treated with a single dose of 46.5 nmol of P307SQ-8C or 1.5 nmol of polymyxin B. Both treatments significantly lowered the bacterial burden (an ∼2-log decrease) in the skin (Fig. 6).
In conclusion, we engineered the positively charged C-terminal peptide P307 from the A. baumannii phage lysin PlyF307 for improved bactericidal activity. The peptides exhibited high in vitro activities and thus could potentially be utilized to control A. baumannii on abiotic surfaces. The target of the peptides appeared to be the bacterial membrane and the interaction likely occurred through electrostatic forces. Despite the membrane target, the peptides did not lyse or kill human RBCs and B cells. Although body fluids and cations interfered with the activities of the peptides, P307SQ-8C significantly decreased the bacterial cell count in a murine skin infection by A. baumannii. We are currently investigating different designs to improve the activities of the peptides, as well as other phage lysins, against A. baumannii.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a grant from Contrafect Corporation.
We thank Michael Wittekind and Raymond Schuch for useful discussions and suggestions during the course of these studies. We thank Clara Eastby, Assaf Raz, Ravenne Reid, and Gabriella Balaa for their contributions to the project. The Proteomics Resource Center and the Electron Microscopy Resource Center at The Rockefeller University are acknowledged for providing expert technical assistance.
ADDENDUM IN PROOF
After submission of our manuscript, a paper was published (H. Yang, M. Wang, J. Yu, and H. Wei, Front Microbiol 6:1471, 2015, http://dx.doi.org/10.3389/fmicb.2015.01471) in which the authors showed that the antibacterial activity of Acinetobacter lysin OBPgp279 was enhanced by the addition of the first eight amino acids (KWKLFKKI) of the cecropin A peptide to the N terminus. However, direct comparisons of the modified lysin termed PlyA to the native OBPgp279 lysin were not shown. PlyA was also found to be inactive in serum and other media.
Funding Statement
Mya Thandar also received stipend support from The David Rockefeller Graduate Program at The Rockefeller University.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02972-15.
REFERENCES
- 1.Potron A, Poirel L, Nordmann P. 2015. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int J Antimicrob Agents 45:568–585. doi: 10.1016/j.ijantimicag.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Zheng W, Yuan S, Li L. 2013. Analysis of hospital departmental distribution and antibiotic susceptibility of Acinetobacter isolated from sputum samples. Am J Infect Control 41:e73–76. doi: 10.1016/j.ajic.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Bonnin RA, Nordmann P, Poirel L. 2013. Screening and deciphering antibiotic resistance in Acinetobacter baumannii: a state of the art. Expert Rev Anti Infect Ther 11:571–583. doi: 10.1586/eri.13.38. [DOI] [PubMed] [Google Scholar]
- 4.Poirel L, Bonnin RA, Nordmann P. 2011. Genetic basis of antibiotic resistance in pathogenic Acinetobacter species. IUBMB Life 63:1061–1067. doi: 10.1002/iub.532. [DOI] [PubMed] [Google Scholar]
- 5.Gayoso CM, Mateos J, Méndez JA, Fernández-Puente P, Rumbo C, Tomás M, Martínez de Ilarduya O, Bou G. 2014. Molecular mechanisms involved in the response to desiccation stress and persistence in Acinetobacter baumannii. J Proteome Res 13:460–476. doi: 10.1021/pr400603f. [DOI] [PubMed] [Google Scholar]
- 6.Fishbain J, Peleg AY. 2010. Treatment of Acinetobacter infections. Clin Infect Dis 51:79–84. doi: 10.1086/653120. [DOI] [PubMed] [Google Scholar]
- 7.Kanazawa K, Sato Y, Ohki K, Okimura K, Uchida Y, Shindo M, Sakura N. 2009. Contribution of each amino acid residue in polymyxin B3 to antimicrobial and lipopolysaccharide binding activity. Chem Pharm Bull 57:240–244. doi: 10.1248/cpb.57.240. [DOI] [PubMed] [Google Scholar]
- 8.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 9.Boohaker RJ, Lee MW, Vishnubhotla P, Perez JM, Khaled AR. 2012. The use of therapeutic peptides to target and to kill cancer cells. Curr Med Chem 19:3794–3804. doi: 10.2174/092986712801661004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Justo JA, Bosso JA. 2015. Adverse reactions associated with systemic polymyxin therapy. Pharmacotherapy:35:28–33. doi: 10.1002/phar.1493. [DOI] [PubMed] [Google Scholar]
- 11.Ko KS, Suh JY, Kwon KT, Jung SI, Park KH, Kang CI, Chung DR, Peck KR, Song JH. 2007. High rates of resistance to colistin and polymyxin B in subgroups of Acinetobacter baumannii isolates from Korea. J Antimicrob Chemother 60:1163–1167. doi: 10.1093/jac/dkm305. [DOI] [PubMed] [Google Scholar]
- 12.Yele AB, Thawal ND, Sahu PK, Chopade BA. 2012. Novel lytic bacteriophage AB7-IBB1 of Acinetobacter baumannii: isolation, characterization and its effect on biofilm. Arch Virol 157:1441–1450. doi: 10.1007/s00705-012-1320-0. [DOI] [PubMed] [Google Scholar]
- 13.Popova AV, Zhilenkov EL, Myakinina VP, Krasilnikova VM, Volozhantsev NV. 2012. Isolation and characterization of wide host range lytic bacteriophage AP22 infecting Acinetobacter baumannii. FEMS Microbiol Lett 332:40–46. doi: 10.1111/j.1574-6968.2012.02573.x. [DOI] [PubMed] [Google Scholar]
- 14.Jin J, Li ZJ, Wang SW, Wang SM, Huang DH, Li YH, Ma YY, Wang J, Liu F, Chen XD, Li GX, Wang XT, Wang ZQ, Zhao GQ. 2012. Isolation and characterization of ZZ1, a novel lytic phage that infects Acinetobacter baumannii clinical isolates. BMC Microbiol 12:156. doi: 10.1186/1471-2180-12-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai MJ, Lin NT, Hu A, Soo PC, Chen LK, Chen LH, Chang KC. 2011. Antibacterial activity of Acinetobacter baumannii phage ϕAB2 endolysin (LysAB2) against both Gram-positive and Gram-negative bacteria. Appl Microbiol Biotechnol 90:529–539. doi: 10.1007/s00253-011-3104-y. [DOI] [PubMed] [Google Scholar]
- 16.Lim JA, Shin H, Kang DH, Ryu S. 2012. Characterization of endolysin from a Salmonella Typhimurium-infecting bacteriophage SPN1S. Res Microbiol 163:233–241. doi: 10.1016/j.resmic.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Walmagh M, Boczkowska B, Grymonprez B, Briers Y, Drulis-Kawa Z, Lavigne R. 2013. Characterization of five novel endolysins from Gram-negative infecting bacteriophages. Appl Microbiol Biotechnol 97:4369–4375. doi: 10.1007/s00253-012-4294-7. [DOI] [PubMed] [Google Scholar]
- 18.Lood R, Winer BY, Pelzek AJ, Diez-Martinez R, Thandar M, Euler CW, Schuch R, Fischetti VA. 2015. Novel phage lysin capable of killing the multidrug-resistant Gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob Agents Chemother 59:1983–1991. doi: 10.1128/AAC.04641-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischetti VA. 2008. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol 11:393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briers Y, Walmagh M, Van Puyenbroeck V, Cornelissen A, Cenens W, Aertsen A, Oliveira H, Azeredo J, Verween G, Pirnay JP, Miller S, Volckaert G, Lavigne R. 2014. Engineered endolysin-based “artilysins” to combat multidrug-resistant Gram-negative pathogens. mBio 5:e01379–14. doi: 10.1128/mBio.01379-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukacik P, Barnard TJ, Keller PW, Chaturvedi KS, Seddiki N, Fairman JW, Noinaj N, Kirby TL, Henderson JP, Steven AC, Hinnebusch BJ, Buchanan SK. 2012. Structural engineering of a phage lysin that targets Gram-negative pathogens. Proc Natl Acad Sci U S A 109:9857–9862. doi: 10.1073/pnas.1203472109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukacik P, Barnard TJ, Buchanan SK. 2012. Using a bacteriocin structure to engineer a phage lysin that targets Yersinia pestis. Biochem Soc Trans 40:1503–1506. doi: 10.1042/BST20120209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wellings DA, Atherton E. 1997. Standard Fmoc protocols. Methods Enzymol 289:44–67. doi: 10.1016/S0076-6879(97)89043-X. [DOI] [PubMed] [Google Scholar]
- 24.Knorr R, Trzeciak A, Bannwarth W, Gillessen D. 1989. New coupling reagents in peptide chemistry. Tetrahedron Lett 30:1927–1930. doi: 10.1016/S0040-4039(00)99616-3. [DOI] [Google Scholar]
- 25.Clinical Laboratory and Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—8th ed. CLSI document M07-A9, vol 32, no 2 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 26.Djurkovic S, Loeffler JM, Fischetti VA. 2005. Synergistic killing of Streptococcus pneumoniae with the bacteriophage lytic enzyme Cpl-1 and penicillin or gentamicin depends on the level of penicillin resistance. Antimicrob Agents Chemother 49:1225–1228. doi: 10.1128/AAC.49.3.1225-1228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park CB, Kim HS, Kim SC. 1998. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun 244:253–257. doi: 10.1006/bbrc.1998.8159. [DOI] [PubMed] [Google Scholar]
- 28.Chen HL, Su PY, Chang YS, Wu SY, Liao YD, Yu HM, Lauderdale TL, Chang K, Shih C. 2013. Identification of a novel antimicrobial peptide from human hepatitis B virus core protein arginine-rich domain (ARD). PLoS Pathog 9:e1003425. doi: 10.1371/journal.ppat.1003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saugar JM, Alarcón T, López-Hernández S, López-Brea M, Andreu D, Rivas L. 2002. Activities of polymyxin B and cecropin A-melittin peptide CA(1-8)M(1-18) against a multiresistant strain of Acinetobacter baumannii. Antimicrob Agents Chemother 46:875–878. doi: 10.1128/AAC.46.3.875-878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pastagia M, Euler C, Chahales P, Fuentes-Duculan J, Krueger JG, Fischetti VA. 2011. A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob Agents Chemother 55:738–744. doi: 10.1128/AAC.00890-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy A, Yang J, Zhang Y. 2012. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 40:W471–W477. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484. doi: 10.1128/AAC.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roca I, Espinal P, Vila-Farrés X, Vila J. 2012. The Acinetobacter baumannii oxymoron: commensal hospital dweller turned pan-drug-resistant menace. Front Microbiol 3:148. doi: 10.3389/fmicb.2012.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dijkshoorn L, Nemec A, Seifert H. 2007. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5:939–951. doi: 10.1038/nrmicro1789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.