

Abstract
Condensins play a key role in the global organization of bacterial chromosomes. In Escherichia coli, the inactivation of its sole condensin MukBEF induces severe growth defects and renders cells hypersusceptible to novobiocin. We report here that this hypersusceptibility can be observed in TolC-deficient cells and is therefore unrelated to multidrug efflux. We further show that mutations in MukE that impair its focal subcellular localization potentiate novobiocin and that the extent of the potentiation correlates with the residual activity of MukE. Finally, both DNA gyrase and topoisomerase IV might partially complement novobiocin susceptibility in a temperature-dependent manner. These data indicate that the observed antibiotic susceptibility resides in both type II DNA topoisomerases and is efflux independent. Furthermore, novobiocin susceptibility is associated with the activity of MukBEF and can be induced by its partial inactivation, which makes the protein a plausible target for inhibition.
INTRODUCTION
Condensins are essential for the global folding of bacterial chromosomes (reviewed in references 1, 2, and 3). These multisubunit complexes act as macromolecular clamps that bridge distant DNA segments and thereby stabilize the looped architecture of the chromosome (4). Escherichia coli encodes a single condensin, MukBEF (5, 6). MukB belongs to the family of the characteristically V-shaped structural maintenance of chromosome (SMC) proteins (7) and is responsible for the ATP-modulated DNA binding and bridging activity of the complex (8). MukBEF forms dynamic clusters at the core of the daughter chromosomes, which can be found in the middle of short cells or at the quarter positions of longer cells (9–11). Such focal localization is essential for cell viability and requires the activity of the regulatory subunit, MukEF (6, 12).
The inactivation of MukBEF leads to severe growth defects, including anucleate cell formation, chromosome decondensation and cutting, a marked decline in colony formation, and an increase in novobiocin susceptibility (13, 14). These phenotypes are alleviated at low temperatures and can be partially complemented by the overexpression of cold shock protein CspE and camphor resistance proteins CrcA and CrcB (15). Partial suppression of MukBEF phenotypes can also be achieved by mutations in topoisomerase I and DNA gyrase that increase the supercoiling of intracellular DNA (16). Such mutations increase the compactness of the chromosome and are expected to compensate for the chromosomal disorganization caused by the deletion of mukB. Besides the aforementioned genetic interaction with DNA gyrase, MukB forms a complex with the ParC subunit of the other type II DNA topoisomerase, topoisomerase IV (topo IV), and modulates its activity (17, 18).
The mechanism of novobiocin susceptibility of MukB-deficient cells remains somewhat enigmatic. Spontaneous suppressors of novobiocin susceptibility of ΔmukB cells have been mapped to GyrB, the ATPase subunit of DNA gyrase (13). However, no increase in susceptibility was found for fluoroquinolones, which, similar to novobiocin, primarily target DNA gyrase (19, 20). This raises the possibility that other modes of novobiocin action might be responsible for the effect. Indeed, novobiocin has been reported to inhibit various ATPases, including topo IV (19) and transmembrane transporters (21). The observed hypersusceptibility might also be explained by pleiotropic effects involving the cell envelope. Compared to fluoroquinolones, novobiocin is a much better substrate for multidrug transporters, which effectively reduce its intracellular concentration (22, 23). Defects in cell integrity or multidrug efflux, therefore, would be expected to selectively affect antibiotic susceptibility.
To address such concerns, we explored the functional interactions between antibiotic susceptibility, MukBEF, and DNA topoisomerases. We found that the enhanced susceptibility of ΔmukB cells to topoisomerase inhibitors remains even upon inactivation of multidrug efflux and is therefore unrelated to drug permeation into the cell. Moreover, both DNA gyrase and topoisomerase IV can partially complement the observed hypersusceptibility of ΔmukB cells. Finally, novobiocin susceptibility correlates with the focal localization of MukE, which links antibiotic susceptibility to the activity of MukBEF and reveals that the protein controls a rate-limiting step in its pathway.
MATERIALS AND METHODS
Plasmids and strains.
MG1655 and BW25113 were used as wild-type E. coli strains, as appropriate. Strains ETBW (BW25113 ΔtolC::kan) (24), OU101 (MG1655 ΔmukB::kan), OU127 (MG1655 ΔmukE::kan), and OU111 (MG1655 mukE::kan lacYA::mukE-gfp-spc) (11) were previously described. OU140 (BW25113 ΔmukB::kan) was constructed by P1vir transduction of the ΔmukB::kan fragment from OU101. Strain OU142 (BW25113 ΔtolC ΔmukB::kan) was constructed by P1vir transduction of the ΔmukB::kan locus from OU101 into GD100 (BW25113 ΔtolC) (24).
Plasmids p15GyrB-R136C and p15ParE-R132C contain genes encoding GyrB and ParE, respectively, with the indicated point mutations, which confer novobiocin resistance to the proteins (19, 25). GyrB was cloned together with the 199-bp upstream fragment, which contains its endogenous promoter (26), and ParE was expressed from the constitutive PMUK promoter. The constructs were assembled using PCR-assisted cloning and inserted into the low-copy-number plasmid p15sp-E02a (11) to replace mukE. pBB03 carries the entire smtA-mukF-mukE-mukB operon under the control of the arabinose-inducible PBAD promoter (27). Due to leakage, MukBEF (but not SmtA) is expressed from this plasmid even in the absence of arabinose, at about 10-fold its endogenous level (27).
Bacteriological techniques.
Bacteria were grown in LB at 200 rpm in the presence of 30 μg/ml kanamycin or 50 μg/ml spectinomycin, when appropriate. Overnight cultures of mukB+ cells were prepared by growing them at 37°C; ΔmukB and ΔmukE cells were grown for 40 h at 23°C (22). MICs were measured using a 2-fold dilution method in 96-well microtiter plates. To this end, 5 × 104 exponentially growing cells (optical density at 600 nm [OD600], 0.6) were inoculated into 0.1 ml of LB supplemented with antibiotics and further incubated with shaking for up to 24 h at 37°C or 30°C, or 48 h at 23°C. Cell growth was evaluated by visual inspection to measure the MIC or by use of the Tecan Spark 10M microplate reader. Fluorescence microscopy was done as previously described (6). The frequency of single-step suppressor mutations was estimated, according to standard protocols (28), by plating 109 exponential OU101 (ΔmukB) cells on LB agar containing 128 μg/ml novobiocin (1/2× the MIC for the parental MG1655 strain) and counting colonies after 24 h of incubation at 37°C.
RESULTS
Antibiotic susceptibility of ΔmukB cells is efflux independent.
Novobiocin is an excellent substrate of efflux pumps, such as the E. coli AcrAB-TolC, and their activity is believed to be the major factor in the resistance of Gram-negative bacteria to this antibiotic (22). To evaluate the contribution of multidrug efflux to the antibiotic susceptibility of MukB-deficient cells, we constructed a set of isogenic strains that harbor ΔmukB and ΔtolC mutations, alone or together. TolC is required for the activity of several multidrug efflux transporters; as a result, ΔtolC cells are hypersusceptible to many antibiotics (29). Regardless of the presence of tolC, ΔmukB cells were able to grow at 37°C when transferred from the permissive room temperature, albeit at a reduced rate (Fig. 1A). Such growth, however, produces cells with severe morphological defects (14), most of which fail to produce colonies (Fig. 1B).
FIG 1.

Fitness defects of MukB-deficient cells. BW25113 (WT), ETBW (ΔtolC), OU140 (ΔmukB), and OU142 (ΔmukB ΔtolC [ΔΔ]) cells were grown at the permissive temperature of 23°C up to an OD600 of 0.6, diluted to OD of 0.05, and further grown at 37°C. (A) Turbidity of the cell culture following temperature upshift. (B) Colony formation (±SD, n = 3) by the cells from panel A before and after growth at 37°C. Note that ΔmukB and ΔmukB ΔtolC cells formed clear colonies by day 2 only. (C and D) MICs for novobiocin (Novo), erythromycin (Ery), norfloxacin (Nor), nalidixic acid (Nal), and rifampin (Rif) for MukB- and TolC-deficient cells at 23°C and (C) and 37°C (D).
In agreement with previous studies (22, 23), the inactivation of TolC had a marked effect on susceptibility to novobiocin, erythromycin, and nalidixic acid but very small, if any, for norfloxacin or rifampin (Table 1 and Fig. 1C and D). The effect of the ΔmukB mutation was similar whether or not tolC was present in the cell. This effect had the same trend but was typically greater at 37°C than at 23°C (Fig. 1C and D). In particular, the inactivation of MukB potentiated novobiocin 25-fold and 128-fold at 23°C and 37°C, respectively, in the wild-type background, whereas the respective values were 13-fold and 21-fold in the absence of TolC. The potentiation of type II topoisomerase inhibitors norfloxacin and nalidixic acid was barely detectable at 23°C, in agreement with previous studies (13), but was obvious, between 2- and 4-fold, at 37°C. In contrast, the potentiation of erythromycin (an inhibitor of translation) and rifampin (an inhibitor of transcription) was about 2-fold and was not affected by the temperature. Since cells are more dependent on MukBEF function at elevated temperatures, these data indicate that the inactivation of MukBEF potentiates antibiotics that target type II DNA topoisomerases but does not have a multidrug effect.
TABLE 1.
MIC (μg/ml) of the indicated antibiotics for MukB- and TolC-deficient cells
| Antibiotic used, by temp | MIC (μg/ml) |
|||
|---|---|---|---|---|
| WT cells | ΔtolC cells | ΔmukB cells | ΔtolC ΔmukB cells | |
| 37°C | ||||
| Novobiocin | 160 | 1 | 1.25 | 0.048 |
| Erythromycin | 100 | 2 | 50 | 1 |
| Norfloxacin | 0.08 | 0.04 | 0.04 | 0.01 |
| Nalidixic acid | 6.4 | 0.8 | 1.6 | 0.25 |
| Rifampin | 10 | 6.4 | 2.0 | 2.5 |
| 23°C | ||||
| Novobiocin | 813 | 4 | 32 | 0.31 |
| Erythromycin | 10 | 2 | 13 | 1 |
| Norfloxacin | 0.032 | 0.02 | 0.04 | 0.01 |
| Nalidixic acid | 6.4 | 0.4 | 6.4 | 0.25 |
| Rifampin | 3.2 | 3.2 | 1.6 | 1.6 |
Notably, deletions of tolC and mukB were additive to each other. This was especially clear at 37°C, when the combined potentiation effect of the two mutations varied between 8- and 16-fold for fluoroquinolones and was >3,000-fold for novobiocin (Fig. 1D). We conclude, therefore, that the mukB-dependent potentiation of antibiotics is not caused by permeation of the cell envelope but arises due to enhanced sensitivity of the intracellular targets of the drugs.
Novobiocin susceptibility resides in type II DNA topoisomerases.
At 37°C, the MIC of novobiocin for ΔmukB ΔtolC cells was 0.05 μg/ml, which is 40-fold lower than its inhibition constant, KD, for DNA gyrase, at 2 μg/ml (3.5 μM) (30). This raises the possibility that the primary drug target might be changing in this strain. Indeed, novobiocin is a competitive inhibitor of DNA gyrase (30, 31), topo IV (19), and, apparently, other cellular ATPases (21). However, we could not raise single-step suppressors of novobiocin susceptibility at 37°C (frequency, <10−9). To evaluate the hypothesis, we instead measured the antibiotic susceptibility of cells that constitutively express novobiocin-resistant versions of DNA gyrase (gyrB-R136C [25]) and topo IV (parE-R132C [19]). These alleles confer about 10-fold drug resistance to each of the enzymes, with only a modest decline in their in vitro activity (20).
At 23°C, the 50% inhibitory concentration (IC50) of novobiocin increased 20-fold when the ΔmukB cells were transformed with a plasmid expressing the novobiocin-resistant allele of gyrB (Fig. 2A). These results are in full accord with those of the previous study, which focused on the suppression of mukB phenotypes at room temperature (13). In contrast, only a small change in novobiocin susceptibility was observed when the cells were transformed with plasmids expressing wild-type gyrB, parE, or the novobiocin-resistant allele of topo IV, parE-R132C (Fig. 2B). Thus, DNA gyrase is indeed the primary target of novobiocin in condensin-deficient cells at room temperature.
FIG 2.

GyrB and ParE complement novobiocin susceptibility of ΔmukB cells. OU101 (ΔmukB) or the parental MG1655 (WT) cells were transformed with a plasmid expressing wild-type or novobiocin-resistant GyrB or ParE, as indicated, and were assessed for growth inhibition by novobiocin at the indicated temperatures. The data were fit to the Hill equation to evaluate the IC50. The error bars indicate the SD (n ≥ 3).
The pattern of complementation changed dramatically at elevated temperatures, when the MIC for novobiocin declined further. The drug-resistant version of DNA gyrase provided only 6-fold and 2-fold increases in the MIC at 30°C and 37°C, respectively (Fig. 2C). Notable were the contributions from the drug-sensitive alleles of gyrB and parE, which reached 2-fold and 5-fold, respectively, at 30°C and 37°C (Fig. 2C and D). The complementation found was unexpected but can be rationalized given the mechanism of inhibition by novobiocin. This drug binds close to the ATPase site of topoisomerases and competitively inhibits their turnover (31). Because of it, mutations in gyrB that confer novobiocin resistance also diminish the activities of the enzymes (25). Apparently, the drug-sensitive allele of topoisomerases is more efficient than the drug-resistant one at low concentrations of the antibiotic.
Novobiocin susceptibility correlates with focal formation.
In principle, any DNA binding protein can have a secondary and often fortuitous function as a transcriptional regulator. Thus, the observed potentiation of antibiotics might be associated with such nonspecific activity of MukB, the DNA binding subunit of the complex, rather than the holoenzyme. To assess this notion, we explored novobiocin potentiation in MukE-deficient cells. We found that the MIC of novobiocin for the ΔmukE OU127 cells, 1 μg/ml, was indistinguishable from that for the ΔmukB OU140 cells. This firmly associates novobiocin susceptibility with the activity of the holoenzyme. To determine whether or not this association is quantitative, we took advantage of the previously constructed series of MukE point mutants (6). These loss-of-function mutants were defective in focal subcellular localization but varied in their ability to be recruited to the endogenous MukBEF clusters, indicating that they retained their partial activity (6) (Fig. 3A). Here, we measured the novobiocin susceptibility of cells in which these mutants replace the endogenous MukE.
FIG 3.
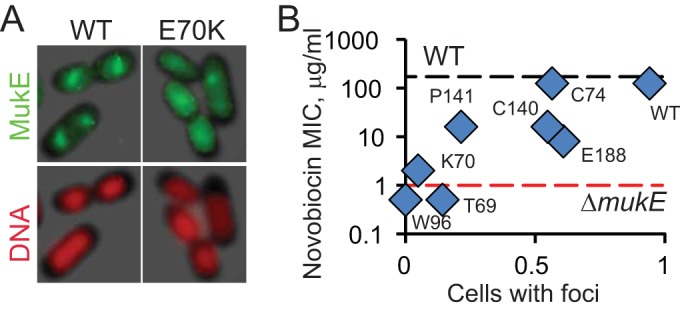
Correlation between novobiocin susceptibility and focal subcellular localization of MukE. (A) Green fluorescent protein (GFP)-tagged MukE forms foci in the middle of the nucleoids of live cells. Random mutagenesis of MukE revealed nine loss-of-function point mutations, all of which were unable to form foci in the absence of endogenous MukE but formed foci with reduced frequency in the presence of the native MukE (OU110 strains; lacY::mukE-GFP*), as shown for one of the mutants (6). (B) Frequency of focal formation by point MukE mutants plotted against their novobiocin susceptibility, as measured for the MukE-deficient OU111 strains (ΔmukE::kan lacY::mukE-GFP*).
In Fig. 3B, the measured MICs for novobiocin are plotted against the previously determined frequencies of focal formation. These frequencies represent the extent of the residual activity in the complex and, therefore, can be viewed as a mimic of its partial inhibition. The point mukE mutants displayed intermediate levels of novobiocin potentiation, which clearly correlated with the residual activity of MukBEF. With MICs on a logarithmic scale, the Pearson correlation coefficient was estimated to be 0.84. This correlation reveals a quantitative link between antibiotic susceptibility and the activity of condensin. As discussed below, this feature qualifies MukBEF as a rate-limiting enzyme in its pathway and makes it a plausible target for drug discovery. However, the MIC of novobiocin did not change when MG1655 cells were transformed with pBB03 (data not shown), which overproduces MukBEF by about 10-fold (see Materials and Methods). Thus, the endogenous level of MukBEF exceeds the cellular demand for it.
DISCUSSION
The inactivation of condensins in E. coli causes a dramatic increase in susceptibility to novobiocin, an inhibitor of DNA topoisomerases. This increase in novobiocin susceptibility is comparable to that of the efflux-deficient cells but has a different mechanism and acts in synergy with efflux. Intriguingly, the effect of such inactivation on fluoroquinolones is much smaller, suggesting that perhaps a secondary target of novobiocin is sensitized in ΔmukB cells. We show here that this is indeed the case, and that both DNA gyrase and topo IV serve as targets of novobiocin in the absence of MukB. The result clearly establishes both these enzymes as members of the MukBEF pathway and expands the conclusions of the previous study, which revealed DNA gyrase as a partial suppressor of the ΔmukB phenotype (13).
This finding offers insight into the mechanism of potentiation. It is unlikely to be due to the homeostasis of DNA supercoiling, since DNA gyrase and topo IV pull it in opposite directions (32). In contrast, the two topoisomerases act in concert in support of DNA replication, and can partially compensate for each other in the removal of topological links (33). It is conceivable, therefore, that the inactivation of condensins places an additional burden on DNA replication and increases the demand on topoisomerase activity. The strong temperature dependence of the effect is also consistent with this view, since the cellular demand for DNA topoisomerases increases at elevated temperatures, when the rates of DNA and RNA synthesis increase.
The reason why potentiation is much stronger for novobiocin than for fluoroquinolones remains obscure, given that they inhibit the same enzyme. The main distinction between these drug classes is in the mechanism of inhibition. Novobiocin competitively inhibits ATP turnover and all related activities of the enzymes (19, 25). Fluoroquinolones, in contrast, trap the DNA cleavage intermediate and convert it to a poison (34–36). As a result, fluoroquinolones effectively inhibit bacterial growth at concentrations well below their KD. The same becomes true for novobiocin in ΔmukB cells (Fig. 1D). Apparently, a decline in the topoisomerase activity generates a toxic product in the absence of MukBEF.
Novobiocin susceptibility is triggered by the inactivation of MukE or MukB. Hence, it depends on the activity of the entire complex, not only its DNA binding subunit MukB; therefore, it cannot be explained by the disruption of fortuitous protein-DNA interactions. It might be notable in this respect that the affected activity was the focal subcellular localization of MukBEF, which further underscores its importance for operation of the complex.
Importantly, novobiocin susceptibility scales proportionally with partial inactivation of the complex (Fig. 3). This reveals that the amount of strain on DNA topoisomerases and the overall decline in cell fitness vary depending on the extent of MukBEF inactivation. This, in turn, implicates MukBEF as a rate-limiting enzyme in its pathway. This makes it a promising target for external regulation. Indeed, metabolic pathways usually have a limited number of rate-limiting steps, in which the amount or activity of the enzyme controls the flux through the entire pathway (37). Such enzymes frequently serve in homeostatic regulation. They are also good targets for inhibition by small molecules, since even partial inactivation of the enzyme would have adverse effects on cell fitness.
ACKNOWLEDGMENTS
This work was supported in parts by grant 1049755 from the National Science Foundation and the award for project no. HR14-042 from the Oklahoma Center for Advancement of Science and Technology to V.V.R. and the award HDTRA1-14-1-0019 from the Department of the Defense, Defense Threat Reduction Agency, to H.I.Z.
The content of the information in this article does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Kleine Borgmann LA, Graumann PL. 2014. Structural maintenance of chromosome complex in bacteria. J Mol Microbiol Biotechnol 24:384–395. doi: 10.1159/000368931. [DOI] [PubMed] [Google Scholar]
- 2.Reyes-Lamothe R, Nicolas E, Sherratt DJ. 2012. Chromosome replication and segregation in bacteria. Annu Rev Genet 46:121–143. doi: 10.1146/annurev-genet-110711-155421. [DOI] [PubMed] [Google Scholar]
- 3.Rybenkov VV, Herrera V, Petrushenko ZM, Zhao H. 2014. MukBEF, a chromosomal organizer. J Mol Microbiol Biotechnol 24:371–383. doi: 10.4014/jmb.1309.09072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui Y, Petrushenko ZM, Rybenkov VV. 2008. MukB acts as a macromolecular clamp in DNA condensation. Nat Struct Mol Biol 15:411–418. doi: 10.1038/nsmb.1410. [DOI] [PubMed] [Google Scholar]
- 5.Hiraga S, Ichinose C, Onogi T, Niki H, Yamazoe M. 2000. Bidirectional migration of SeqA-bound hemimethylated DNA clusters and pairing of oriC copies in Escherichia coli. Genes Cells 5:327–341. doi: 10.1046/j.1365-2443.2000.00334.x. [DOI] [PubMed] [Google Scholar]
- 6.She W, Mordukhova E, Zhao H, Petrushenko ZM, Rybenkov VV. 2013. Mutational analysis of MukE reveals its role in focal subcellular localization of MukBEF. Mol Microbiol 87:539–552. doi: 10.1111/mmi.12112. [DOI] [PubMed] [Google Scholar]
- 7.Melby TE, Ciampaglio CN, Briscoe G, Erickson HP. 1998. The symmetrical structure of structural maintenance of chromosomes (SMC) and MukB proteins: long, antiparallel coiled coils, folded at a flexible hinge. J Cell Biol 142:1595–1604. doi: 10.1083/jcb.142.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petrushenko ZM, Lai CH, Rybenkov VV. 2006. Antagonistic interactions of kleisins and DNA with bacterial condensin MukB. J Biol Chem 281:34208–34217. doi: 10.1074/jbc.M606723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badrinarayanan A, Reyes-Lamothe R, Uphoff S, Leake MC, Sherratt DJ. 2012. In vivo architecture and action of bacterial structural maintenance of chromosome proteins. Science 338:528–531. doi: 10.1126/science.1227126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohsumi K, Yamazoe M, Hiraga S. 2001. Different localization of SeqA-bound nascent DNA clusters and MukF-MukE-MukB complex in Escherichia coli cells. Mol Microbiol 40:835–845. doi: 10.1046/j.1365-2958.2001.02447.x. [DOI] [PubMed] [Google Scholar]
- 11.She W, Wang Q, Mordukhova EA, Rybenkov VV. 2007. MukEF is required for stable association of MukB with the chromosome. J Bacteriol 189:7062–7068. doi: 10.1128/JB.00770-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin HC, Lim JH, Woo JS, Oh BH. 2009. Focal localization of MukBEF condensin on the chromosome requires the flexible linker region of MukF. FEBS J 276:5101–5110. doi: 10.1111/j.1742-4658.2009.07206.x. [DOI] [PubMed] [Google Scholar]
- 13.Adachi S, Hiraga S. 2003. Mutants suppressing novobiocin hypersensitivity of a mukB null mutation. J Bacteriol 185:3690–3695. doi: 10.1128/JB.185.13.3690-3695.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niki H, Jaffe A, Imamura R, Ogura T, Hiraga S. 1991. The new gene mukB codes for a 177 kd protein with coiled-coil domains involved in chromosome partitioning of E. coli. EMBO J 10:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu KH, Liu E, Dean K, Gingras M, DeGraff W, Trun NJ. 1996. Overproduction of three genes leads to camphor resistance and chromosome condensation in Escherichia coli. Genetics 143:1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawitzke JA, Austin S. 2000. Suppression of chromosome segregation defects of Escherichia coli muk mutants by mutations in topoisomerase I. Proc Natl Acad Sci U S A 97:1671–1676. doi: 10.1073/pnas.030528397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayama R, Marians KJ. 2010. Physical and functional interaction between the condensin MukB and the decatenase topoisomerase IV in Escherichia coli. Proc Natl Acad Sci U S A 107:18826–18831. doi: 10.1073/pnas.1008140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Stewart NK, Berger AJ, Vos S, Schoeffler AJ, Berger JM, Chait BT, Oakley MG. 2010. Escherichia coli condensin MukB stimulates topoisomerase IV activity by a direct physical interaction. Proc Natl Acad Sci U S A 107:18832–18837. doi: 10.1073/pnas.1008678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy CD, Cozzarelli NR. 2003. Alteration of Escherichia coli topoisomerase IV to novobiocin resistance. Antimicrob Agents Chemother 47:941–947. doi: 10.1128/AAC.47.3.941-947.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khodursky AB, Zechiedrich EL, Cozzarelli NR. 1995. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc Natl Acad Sci U S A 92:11801–11805. doi: 10.1073/pnas.92.25.11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan P, You G. 2009. Novobiocin is a potent inhibitor for human organic anion transporters. Drug Metab Dispos 37:1203–1210. doi: 10.1124/dmd.109.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tikhonova EB, Wang Q, Zgurskaya HI. 2002. Chimeric analysis of the multicomponent multidrug efflux transporters from Gram-negative bacteria. J Bacteriol 184:6499–6507. doi: 10.1128/JB.184.23.6499-6507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang S, Clayton SR, Zechiedrich EL. 2003. Relative contributions of the AcrAB, MdfA and NorE efflux pumps to quinolone resistance in Escherichia coli. J Antimicrob Chemother 51:545–556. doi: 10.1093/jac/dkg126. [DOI] [PubMed] [Google Scholar]
- 24.Dhamdhere G, Zgurskaya HI. 2010. Metabolic shutdown in Escherichia coli cells lacking the outer membrane channel TolC. Mol Microbiol 77:743–754. doi: 10.1111/j.1365-2958.2010.07245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Contreras A, Maxwell A. 1992. gyrB mutations which confer coumarin resistance also affect DNA supercoiling and ATP hydrolysis by Escherichia coli DNA gyrase. Mol Microbiol 6:1617–1624. doi: 10.1111/j.1365-2958.1992.tb00886.x. [DOI] [PubMed] [Google Scholar]
- 26.Menzel R, Gellert M. 1987. Modulation of transcription by DNA supercoiling: a deletion analysis of the Escherichia coli gyrA and gyrB promoters. Proc Natl Acad Sci U S A 84:4185–4189. doi: 10.1073/pnas.84.12.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Q, Mordukhova EA, Edwards AL, Rybenkov VV. 2006. Chromosome condensation in the absence of the non-SMC subunits of MukBEF. J Bacteriol 188:4431–4441. doi: 10.1128/JB.00313-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice LB, Bonomo RA. 1996. Genetic and biochemical mechanisms of bacterial resistance to antimicrobial agents, p 450–497. In Lorian V. (ed), Antibiotics in laboratory medicine, 4th ed Williams & Wilkins, Baltimore, MD. [Google Scholar]
- 29.Zgurskaya HI, Nikaido H. 2000. Multidrug resistance mechanisms: drug efflux across two membranes. Mol Microbiol 37:219–225. doi: 10.1046/j.1365-2958.2000.01926.x. [DOI] [PubMed] [Google Scholar]
- 30.Sugino A, Higgins NP, Brown PO, Peebles CL, Cozzarelli NR. 1978. Energy coupling in DNA gyrase and the mechanism of action of novobiocin. Proc Natl Acad Sci U S A 75:4838–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis RJ, Singh OMP, Smith CV, Skarzynski T, Maxwell A, Wonacott AJ, Wigley DB. 1996. The nature of the inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J 15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 32.Zechiedrich EL, Khodursky AB, Bachellier S, Schneider R, Chen D, Lilley DM, Cozzarelli NR. 2000. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J Biol Chem 275:8103–8113. doi: 10.1074/jbc.275.11.8103. [DOI] [PubMed] [Google Scholar]
- 33.Khodursky AB, Peter BJ, Schmid MB, DeRisi J, Botstein D, Brown PO, Cozzarelli NR. 2000. Analysis of topoisomerase function in bacterial replication fork movement: use of DNA microarrays. Proc Natl Acad Sci U S A 97:9419–9424. doi: 10.1073/pnas.97.17.9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewis CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, Gwynn MN. 2010. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466:935–940. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 35.Hiasa H, Yousef DO, Marians KJ. 1996. DNA strand cleavage is required for replication fork arrest by a frozen topoisomerase-quinolone-DNA ternary complex. J Biol Chem 271:26424–26429. doi: 10.1074/jbc.271.42.26424. [DOI] [PubMed] [Google Scholar]
- 36.Kreuzer KN, Cozzarelli NR. 1979. Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth. J Bacteriol 140:424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kacser H. 1995. Recent developments beyond metabolic control analysis. Biochem Soc Trans 23:387–391. doi: 10.1042/bst0230387. [DOI] [PubMed] [Google Scholar]