

Calcineurin (CaN) is protein phosphatase with characteristic calcium and calmodulin dependent activation through its regulatory subunits1. Activated CaN dephosphorylates downstream transcription factor NFAT which leads to its nuclear translocation and transcriptional activation1. CaN-NFAT signaling axis is initially discovered as an essential pathway for T-cell activation1 but has now been implicated in broad range of cellular processes and cell types ranging from fungus to plants. In heart, CaN mediated signaling is recognized as a common intracellular pathway leading to cardiac hypertrophy and pathological remodeling triggered by a plethora of pathological stressors2. In this issue, Zhu et al adds a new piece of evidence3 to a significant body of literature4 and further demonstrates that genetic or pharmacological attenuation of CaN signaling can have a significant ameliorative effect on the pathogenesis of cardiac hypertrophy and dysfunction in response to varies of stresses.
Like many stress-induced signaling, CaN pathway is also tightly controlled by a cohort of endogenous negative regulators in cells5. The prototypic Regulator-of-Calcinurin-1 (RCAN1, also known as ADAPT78; CSP1; DSC1; DSCR1; MCIP1; RCN1 and Calcipressin1) is transcriptionally controlled by NFAT and serves as a key negative feed-back regulator for CaN signaling by direct binding and inhibiting its phosphatase activities. In addition to RCAN1, a number of other negative regulators for CaN-NFAT signaling have been identified, including Cain/Cabin1 (calcineurin inhibitor 1), RCAN2 (also known as CSP2; DSCR1L1; MCIP2; RCN2; ZAKI-4; ZAKI4), RCAN3 (also known as DSCR1L2; MCIP3; RCN3; hRCN3), FHL2 (four-and-a-half LIM domain protein 2), CHP1 and CHP2 (calcineurin B homologous protein 1, also known as CHP1; SLC9A1BP; Sid470p; p22; p24). Most of them function through direct interaction with CaN as a scaffold. However, specific inhibitory function for CaN has also been identified for Muscle-specific RING figure 1 (MuRF1) as an E3 ubiquitin ligase through targeted CaN degradation6 and Plasma Membrane Calcium ATPase (PMCA) as membrane calcium pump7. Different from RCAN1, many of these endogenous inhibitors are not necessarily bona fide negative feedback regulators for CaN signaling as they are not directly induced by CaN-NFAT mediated transcription upon stimulation, but nevertheless modulate CaN signaling under different extracellular stimuli. Indeed, like RCAN1, manipulating many of these endogenous CaN inhibitors can have a significant impact on cardiac hypertrophy and pathological remodeling.
Figure 1.
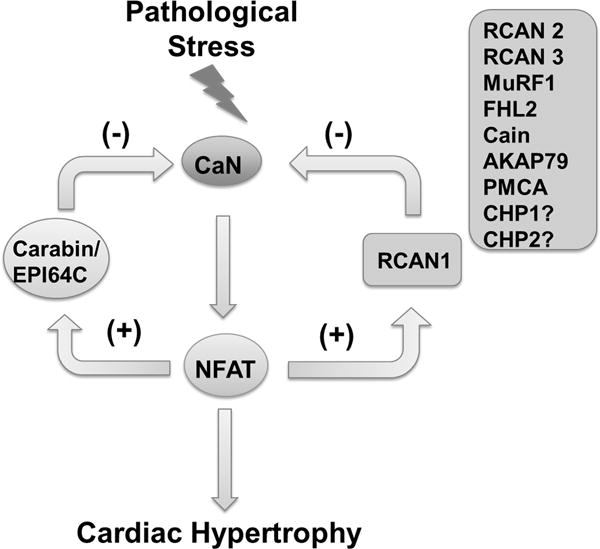
Illustration of negative regulators of CaN-NFAT signaling pathway in cardiac hypertrophy. ? indicates genes with no known evidence for their impact on hypertrophy.
In 2007, Pan et al identified yet another negative feedback regulator for CaN, termed Carabin or EPI64C which fulfills the criteria of both negative inhibitory function to CaN and induction by CaN mediated signaling following T-cell receptor induction8. In addition to its inhibitory effect on CaN activity, Carabin/EPI64C is also reported to have additional inhibitory role for Ras mediated MAP kinase activation through an intrinsic Ras GTPase-activating protein (GAP) activity8. In a recent report by Bisserier et al using both genetic knockout mouse model and AAV mediated cardiac targeted gene transfer, Carabin/EPI64C is shown to be both necessary and sufficient to attenuate pressure-overload induced cardiac hypertrophy and pathological remodeling9, thus adding yet another negative regulator of CaN into the player list in the cardiac hypertrophy regulatory network. In this issue, Zhu et al further advance this notion that Carabin/EPI64C is a critical regulator of cardiac hypertrophy by offering several important new lines of evidence3. First, these investigators generated cardiac specific knockout and cardiomyocyte-specific transgenic mouse models to demonstrate in vivo that Carabin/EPI64C mediated regulation of cardiac hypertrophy and pathological remodeling is a cardiomyocyte cell-autonomous process. Second, the underlying mechanism appears to involve direct interaction and inhibition of CaN signaling rather than Ras-MAP Kinase pathway as originally reported. Lastly, Carabin/EPI64C mediated hypertrophy regulation is conserved across different species and its expression exerts cardiac protection against pressure-overload induced cardiac hypertrophy and dysfunction in both mice and non-human primates. These findings further demonstrate the translational potential of Carabin/EPI64C as a therapeutic target for pathological hypertrophy in stressed human heart.
As an endogenous feed-back regulator for CaN, Carabin/EPI64C is both a downstream target of CaN/NFAT mediated transcriptional induction and an upstream negative inhibitor for CaN signaling8. Since CaN/NFAT mediated signaling is a common pathway significantly elevated in diseased heart, we should expect to observe an induced expression of Carabin/EPI64C in stressed hypertrophic myocardium as observed for RCNA1. Yet, both reports by Bisserier and Zhu showed a significantly reduced expression of Carabin/EPI64C in the pathologically stressed heart and human failing heart3, 9. Therefore, the loss of Carabin/EPI64C (but not RCAN1) mediated negative feedback may represent an interesting new mechanism underlying the hyperactivity of CaN in cardiac hypertrophy and pathological remodeling. It is not clear why Carabin/EPI64C expression is downregulated in the diseased heart and whether restoring its expression in established hypertrophic heart is able to reverse the pathogenic progression. Understanding the uncoupling mechanism between CaN and Carabin/EPI64C, and testing the therapeutic effect of restoring Carabin/EPI64C expression in established hypertrophy may uncover a truly translational path to treat cardiac hypertrophy and pathological remodeling. It is important to note that although pharmacological inhibition of CaN has proved to be effective in clinic to suppress immune response and are widely used for organ transplant and other immune disorders, they have not been demonstrated efficacious to treat heart failure or hypertrophic cardiomyopathy in human. Considering the hypertensive effect of cyclosporine (a pharmacological inhibitor of CaN) at systemic level10, cautions must be taken to translate these insights learnt from tissue-specific and precision genetic manipulations to clinical applications. Clearly, there are a lot more sciences still waiting to be done.
Supplementary Material
Acknowledgments
Sources of Funding:
This work is supported in part by grants from NIH (HL103205, HL098954, HL108186, HL114437) to YW.
Footnotes
DISCLOSURE:
None.
References
- 1.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in t-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 2.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu X, Fang J, Gong J, Guo JH, Zhao GN, Ji YX, Liu HY, Wei X, Li H. Cardiac specific epi64c blunts pressure overload-induced cardiac hypertrophhy. Hypertension. 2016 doi: 10.1161/HYPERTENSIONAHA.115.07042. In Press. [DOI] [PubMed] [Google Scholar]
- 4.Fiedler B, Wollert KC. Interference of antihypertrophic molecules and signaling pathways with the ca2+-calcineurin-nfat cascade in cardiac myocytes. Cardiovasc Res. 2004;63:450–457. doi: 10.1016/j.cardiores.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Kingsbury TJ, Cunningham KW. A conserved family of calcineurin regulators. Genes & Development. 2000;14:1595–1604. [PMC free article] [PubMed] [Google Scholar]
- 6.Maejima Y, Usui S, Zhai P, Takamura M, Kaneko S, Zablocki D, Yokota M, Isobe M, Sadoshima J. Muscle-specific ring finger 1 negatively regulates pathological cardiac hypertrophy through downregulation of calcineurin A. Circulation Heart Failure. 2014;7:479–490. doi: 10.1161/CIRCHEARTFAILURE.113.000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu X, Chang B, Blair NS, Sargent M, York AJ, Robbins J, Shull GE, Molkentin JD. Plasma membrane ca2+-atpase isoform 4 antagonizes cardiac hypertrophy in association with calcineurin inhibition in rodents. J Clin Invest. 2009;119:976–985. doi: 10.1172/JCI36693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan F, Sun L, Kardian DB, Whartenby KA, Pardoll DM, Liu JO. Feedback inhibition of calcineurin and ras by a dual inhibitory protein carabin. Nature. 2007;445:433–436. doi: 10.1038/nature05476. [DOI] [PubMed] [Google Scholar]
- 9.Bisserier M, Berthouze-Duquesnes M, Breckler M, et al. Carabin protects against cardiac hypertrophy by blocking calcineurin, ras, and ca2+/calmodulin-dependent protein kinase ii signaling. Circulation. 2015;131:390–400. doi: 10.1161/CIRCULATIONAHA.114.010686. [DOI] [PubMed] [Google Scholar]
- 10.El-Gowelli HM, El-Mas MM. Central modulation of cyclosporine-induced hypertension. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:351–361. doi: 10.1007/s00210-014-1074-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.