

Abstract
An asymmetric decarboxylative Csp3–Csp2 cross-coupling has been achieved via the synergistic merger of photoredox and nickel catalysis. This mild, operationally simple protocol transforms a wide variety of naturally abundant α-amino acids and readily available aryl halides into valuable chiral benzylic amines in high enantiomeric excess, thereby producing motifs found in pharmacologically active agents.
Nickel catalysis has provided numerous pathways to forge carbon–carbon bonds via a variety of well-known coupling protocols (e.g., Negishi, Suzuki–Miyaura, Stille, Kumada, and Hiyama couplings).1 The value of these transformations lies in their capacity to effect useful fragment couplings with broad functional-group tolerance, which enables a highly modular, building-block approach to the construction of complex organic molecules. Beginning in 2005, one of our laboratories has demonstrated that chiral nickel catalysts can be employed to achieve the enantioselective coupling of a broad spectrum of racemic alkyl halides (e.g., α-halocarbonyl compounds, as well as benzylic, allylic, and unactivated alkyl halides) with a wide array of common organometallic nucleophiles (e.g., organozinc, -boron, -silicon, -magnesium, and -zirconium reagents; Scheme 1A).2 A prominent feature of this approach is the capacity to employ racemic Csp3–halide containing starting materials to forge Csp3–Csp3 and Csp3–Csp2 bonds with high levels of stereocontrol in the enantioconvergent fragment-coupling step.
Scheme 1. Asymmetric Decarboxylative Arylation of α-Amino Acids.
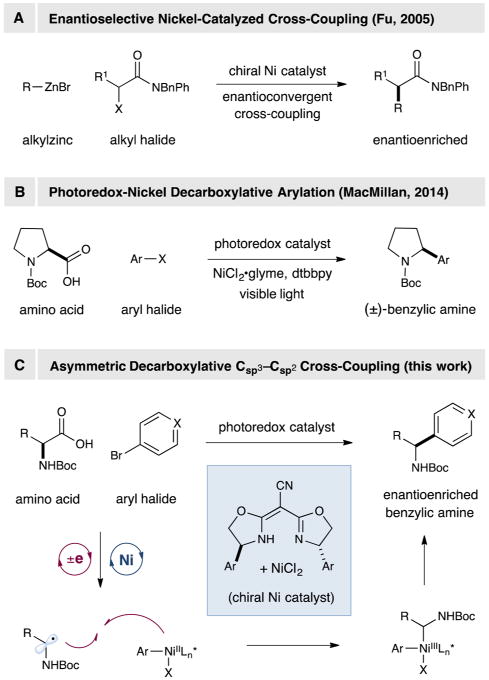
Visible-light-mediated photoredox catalysis has emerged in recent years as a valuable concept in organic synthesis wherein single-electron transfer (SET) events with organic substrates are facilitated.3 This general strategy allows for the development of bond constructions that are often elusive or currently impossible via classical two-electron pathways.4 Recently, one of our laboratories introduced a dual photoredox–nickel catalysis platform that enables carboxylate oxidation–CO2 extrusion as a generic mechanism for nucleophile activation, a development that allows carboxylic acids to be used in lieu of organometallic nucleophiles (e.g., organoboron or organozinc reagents) in nickel-mediated fragment couplings (Scheme 1B).5,6 This strategy has subsequently enabled the catalytic decarboxylative functionalization of a wide range of Csp3 centers with aryl, heteroaryl, and vinyl fragments using abundant, inexpensive, yet structurally complex carboxylic acids (e.g., α-amino acids, carbohydrates, and α-oxy acids).7
In view of the appealing attributes of (i) enantioconvergent nickel-catalyzed cross-coupling and (ii) metallaphotoredox-mediated decarboxylative arylation, we recently decided to pursue the possibility that the two technologies could be merged to enable the enantioselective synthesis of benzylic amines from readily available α-amino acid starting materials. Because benzylic amines are a subunit in a wide array of bioactive compounds, including agrochemicals, natural products, and pharmaceuticals (e.g., rivastigmine for Alzheimer's disease and repaglinide for type II diabetes),8,9 the development of enantioselective methods for the synthesis of this important pharmacophore from inexpensive starting materials remains an important challenge.10 In this communication, we report the successful merger of enantioselective nickel-catalyzed cross-coupling with metallaphotoredox-mediated decarboxylative arylation, specifically, the first example of a one-step synthesis of enantioenriched benzylic amines from a wide array of naturally occurring α-amino acids using light and a synergistic combination of an iridium photocatalyst and a chiral nickel complex.
Design Plan
Based on our previous investigation of the decarboxylative arylation of α-amino acids, we envisioned that an enantioselective variant would be accessible via an analogous mechanism.5 Thus, photocatalyst-mediated oxidation and decarboxylation of an α-amino acid would produce a prochiral α-amino radical (Scheme 1C).5,7,11 Meanwhile, activation of an aryl halide via oxidative addition would lead to a Ni(II)–aryl complex, which would intercept the newly generated α-amino radical. The resulting diorganonickel(III) adduct would then undergo reductive elimination to forge the desired Csp3–Csp2 bond. The presence of an appropriate chiral ligand would induce enantioselectivity.
Results
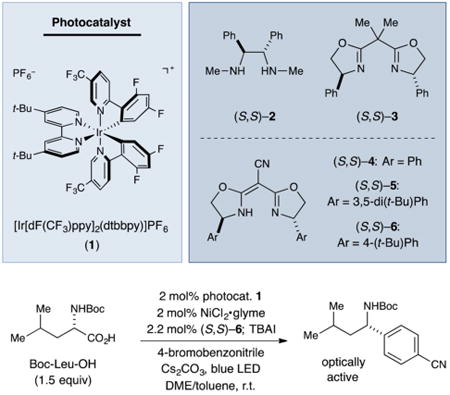
Our investigation into synergistic photoredox- and nickel-catalyzed asymmetric decarboxylative arylation began with the evaluation of several ligand classes for the coupling of N-Boc-leucine with 4-bromobenzonitrile. We were intrigued to determine that chiral 1,2-diamine and bis(oxazoline) ligands, which have found extensive use in related asymmetric nickelcatalyzed cross-coupling reactions that employ organometallic nucleophiles as coupling partners, were ineffective for this enantioselective decarboxylative arylation (Table 1, entries 1 and 2).12,13 In contrast, semicorrin-like ligand 4 delivered the desired product with promising ee, although modest yield (entry 3).14 Whereas the addition of a tert-butyl substituent in the meta positions of ligand 4 was beneficial (ligand 5; entry 4), the incorporation of a tert-butyl substituent in the para positions provided even higher enantioselectivity and yield (ligand 6; entry 5). From a practical point of view, it is worth noting that ligand (S,S)-6 is commercially available and air-stable.15
Table 1. Catalytic Enantioselective Decarboxylative Arylationa.
| |||
|---|---|---|---|
|
| |||
| entry | variation from standard conditions | ee (%) | yield (%)b |
| 1 | (S,S)-2 instead of (S,S)-6 | – | <2 |
| 2 | (S,S)-3 instead of (S,S)-6 | 9 | 28 |
| 3 | (S,S)-4 instead of (S,S)-6 | 77 | 34 |
| 4 | (S,S)-5 instead of (S,S)-6 | 86 | 59 |
| 5 | none | 92 | 69 |
| 6 | no NiCl2·glyme | – | <2 |
| 7 | no (S,S)-6 | – | <2 |
| 8 | no photocat. 1 | – | 0 |
| 9 | no light | – | 0 |
| 10 | (R,R)-6 instead of (S,S)-6 | −91 | 72 |
All data are the average of two experiments.
Yields determined by 1H NMR spectroscopy using 1,3-bis(trifluoromethyl)-5-bromobenzene as an internal standard.
A series of control experiments established that the nickel complex, the chiral ligand, the photocatalyst, and light are all essential (entries 6–9). Consistent with our hypothesis that the reaction proceeds through a radical intermediate (Scheme 1C), the choice of either (S,S)-6 or (R,R)-6 as the chiral ligand leads to the formation of opposite enantiomers of the desired product from enantiopure Boc-l-Leu-OH, with the stereo-chemical outcome determined entirely by the configuration of the ligand (entry 5 versus entry 10).
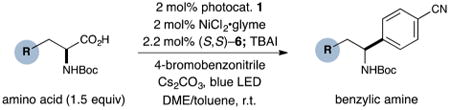
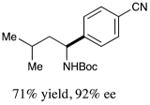
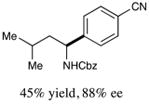
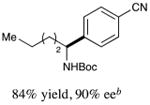
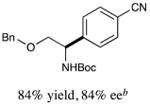
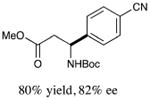
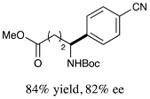
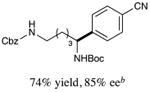
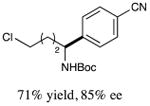
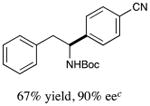
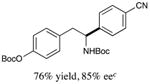
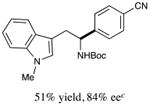
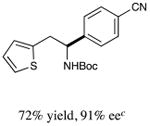
With optimized reaction conditions in hand, we next evaluated the scope of α-amino acids that can be employed in this asymmetric arylation reaction. As illustrated in Table 2, a variety of N-Boc-protected α-amino acids that bear an alkyl side chain (e.g., protected Leu, Ser, Asp, Glu, and Lys) were arylated with good to excellent enantioselectivity (entries 1 and 3–8; 71–84% yield, 82–92% ee).16 Although these conditions were optimized for N-Boc-protected α-amino acids, they can also be applied to an N-Cbz-protected substrate, albeit in more modest yield (entry 2; 45% yield, 88% ee). In the case of α-amino acids that bear a CH2Ar (Ar = aromatic) side chain, ligand 5 proved to be optimal (entries 9–12; 51–76% yield, 84–91% ee). The mild nature of this protocol allows for the presence of a wide range of functional groups, including carbamate, ether, ester, alkyl chloride, carbonate, indole, and thiophene moieties.
Table 2. Scope of the α-Amino Acida.
| |||
|---|---|---|---|
|
| |||
| entry | product | entry | product |
| 1 |
|
2 |
|
| 3 |
|
4 |
|
| 5 |
|
6 |
|
| 7 |
|
8 |
|
|
| |||
| 9 |
|
10 |
|
| 11 |
|
12 |
|
All data are the average of two experiments.
Reaction performed using 5 mol% NiCl2·glyme and 5.5 mol% (S,S)-6.
Reaction performed using 2.2 mol% (S,S)-5, without 10 mol% TBAI (see the Supporting Information).
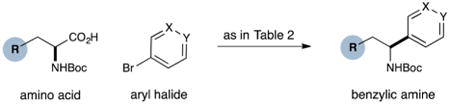
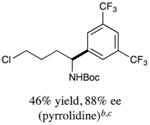
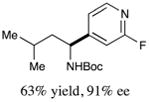
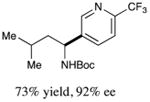
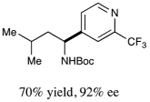
We next turned our attention to determining the scope of aromatic halides that are amenable to cross-coupling under the optimized reaction conditions. As illustrated in Table 3, in the case of α-amino acids that bear an alkyl side chain, a range of aryl halides can be incorporated with good efficiency and high ee, including substituted pyridyl groups, which are commonly found in pharmaceutically relevant compounds (entries 1–8; 46–76% yield, 88–92% ee). Using ligand 5, N-Boc-protected phenylalanine can be cross-coupled with aryl and heteroaryl halides in moderate-to-good yield and with high enantioselectivity (entries 9–12; 47–84% yield, 90–93% ee). When an electron-neutral aryl halide (Ph–I) was employed as a coupling partner with N-Boc-protected phenylalanine, the desired benzylic amine was generated with more modest ee (66%) and comparable yield (64%).
Table 3. Scope of the Aryl Halidea.
| |||
|---|---|---|---|
|
| |||
| entry | product | entry | product |
| 1 |
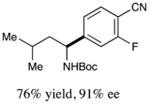
|
2 |
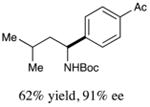
|
| 3 |
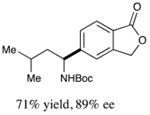
|
4 |
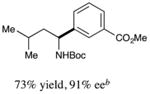
|
| 5 |
|
6 |
|
| 7 |
|
8 |
|
|
| |||
| 9 |
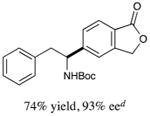
|
10 |
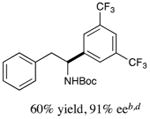
|
| 11 |
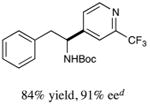
|
12 |
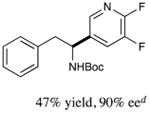
|
All data are the average of two experiments.
Reaction performed using an aryl iodide.
The cross-coupling product was cyclized to form a pyrrolidine.
Reaction performed using 2.2 mol% (S,S)-5, without 10 mol% TBAI (see the Supporting Information).
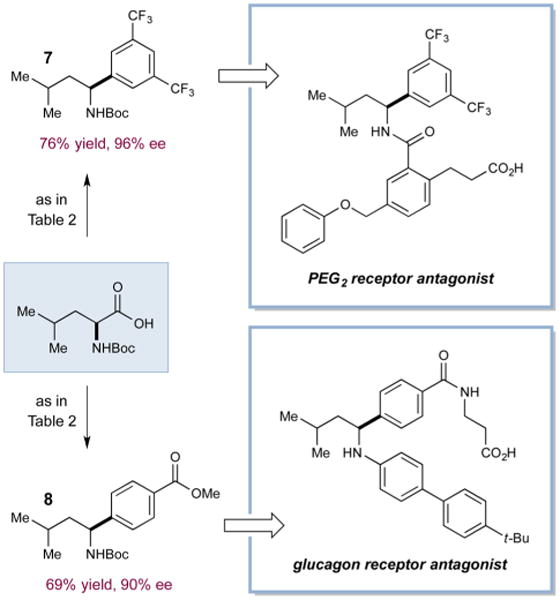
To highlight the utility of this asymmetric cross-coupling reaction, we applied our protocol to the synthesis of the cores of pharmacologically active molecules. Using the conditions outlined in Table 2, N-Boc-leucine was successfully cross-coupled to provide protected benzylic amines 717 and 818 in good yield and excellent enantioselectivity (Scheme 2). Products 7, bearing bis-trifluoromethylphenyl substitution, and 8, bearing a methylbenzoate group, could be advanced to the corresponding bioactive compounds through straightforward synthetic sequences (e.g., amide coupling and Buchwald–Hartwig amination).
Scheme 2. Catalytic Asymmetric Coupling To Produce Pharmacophores.
In conclusion, we have developed an enantioselective decarboxylative Csp3–Csp2 cross-coupling reaction of α-amino acids with aryl halides by interfacing photoredox and nickel catalysis. This enantioconvergent approach provides ready access to enantioenriched benzylic amine products using abundant feedstock α-amino acids as coupling partners in place of traditional organometallic nucleophiles. The generality of this protocol has been illustrated through the cross-coupling of a variety of amino acids and aryl halides. Finally, we have demonstrated the utility of this strategy for the preparation of pharmaceutically relevant compounds.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH (NIGMS R01 GM093213 and R01 GM062871) and by gifts from Merck and Amgen. Z.Z. is grateful for a postdoctoral fellowship from the Shanghai Institute of Organic Chemistry.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b13211.
Crystallographic data (CIF)
Experimental procedures and compound characterization data (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.For a recent review, see: Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299. doi: 10.1038/nature13274.
- 2.For example, see: Fischer C, Fu GC. J Am Chem Soc. 2005;127:4594. doi: 10.1021/ja0506509.Saito B, Fu GC. J Am Chem Soc. 2008;130:6694. doi: 10.1021/ja8013677.Dai X, Strotman NA, Fu GC. J Am Chem Soc. 2008;130:3302. doi: 10.1021/ja8009428.Lou S, Fu GC. J Am Chem Soc. 2010;132:1264. doi: 10.1021/ja909689t.Lou S, Fu GC. J Am Chem Soc. 2010;132:5010. doi: 10.1021/ja1017046. (f) See also: Cordier CJ, Lundgren RJ, Fu GC. J Am Chem Soc. 2013;135:10946. doi: 10.1021/ja4054114.
- 3.For recent reviews, see: Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r.Xie J, Jin H, Xu P, Zhu C. Tetrahedron Lett. 2014;55:36.Schultz DM, Yoon TP. Science. 2014;343:1239176. doi: 10.1126/science.1239176.
- 4.For selected examples, see: Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976.Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756. doi: 10.1021/ja9033582.Hamilton DS, Nicewicz DA. J Am Chem Soc. 2012;134:18577. doi: 10.1021/ja309635w.DiRocco DA, Rovis T. J Am Chem Soc. 2012;134:8094. doi: 10.1021/ja3030164.Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Science. 2013;339:1593. doi: 10.1126/science.1232993.Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR. J Am Chem Soc. 2013;135:17735. doi: 10.1021/ja4100595.
- 5.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.A related dual catalytic transformation has been developed using organotrifluoroborates as the nucleophilic coupling partner: Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647.
- 7.Noble A, McCarver SJ, MacMillan DWC. J Am Chem Soc. 2015;137:624. doi: 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews and leading references, see: Lawrence SA. Amines: Synthesis, Properties and Applications. Cambridge University Press; Cambridge, U.K.: 2004. Nugent TC, editor. Chiral Amine Synthesis: Methods, Developments and Applications. Wiley-VCH; Weinheim, Germany: 2010.
- 9.For recent reviews, see: Lane RM, Darreh-Shori T. J Alzheimer's Dis. 2015;44:1039. doi: 10.3233/JAD-142268.Scott L. J Drugs. 2012;72:249. doi: 10.2165/11207600-000000000-00000.
- 10.For a few recent examples, see: Seel S, Thaler T, Takatsu K, Zhang C, Zipse H, Straub BF, Mayer P, Knochel P. J Am Chem Soc. 2011;133:4774. doi: 10.1021/ja201008e.Chu L, Wang XC, Moore CE, Rheingold AL, Yu JQ. J Am Chem Soc. 2013;135:16344. doi: 10.1021/ja408864c.Zhu Y, Buchwald SL. J Am Chem Soc. 2014;136:4500. doi: 10.1021/ja501560x.
- 11.(a) Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:5257. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chu L, Ohta C, Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:10886. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Noble A, MacMillan DWC. J Am Chem Soc. 2014;136:11602. doi: 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For examples of the use of chiral 1,2-diamine ligands, see: (a) Reference 2b. Wilsily A, Tramutola F, Owston NA, Fu GC. J Am Chem Soc. 2012;134:5794. doi: 10.1021/ja301612y.
- 13.For examples of the use of chiral bis(oxazoline) ligands, see: (a) Reference 2d. Liang Y, Fu GC. J Am Chem Soc. 2015;137:9523. doi: 10.1021/jacs.5b04725.
- 14.(a) Pfaltz A. Acc Chem Res. 1993;26:339. [Google Scholar]; (b) Corey EJ, Wang X. Tetrahedron Lett. 1993;34:4001. [Google Scholar]; (c) Dagorne S, Bellemin-Laponnaz S, Maisse-Francois A. Eur J Inorg Chem. 2007;2007:913. doi: 10.1002/chem.200601112. [DOI] [PubMed] [Google Scholar]; (d) Nolin KA, Ahn RW, Toste FD. J Am Chem Soc. 2005;127:12462. doi: 10.1021/ja050831a. [DOI] [PubMed] [Google Scholar]; (e) Chakrabarti A, Konishi H, Yamaguchi M, Schneider U, Kobayashi S. Angew Chem, Int Ed. 2010;49:1838. doi: 10.1002/anie.200906308. [DOI] [PubMed] [Google Scholar]; (f) Zhang P, Wolf C. Angew Chem, Int Ed. 2013;52:7869. doi: 10.1002/anie.201303551. [DOI] [PubMed] [Google Scholar]
- 15.The TBAI may accelerate the cross-coupling by, for example, increasing the ionic strength of the reaction mixture.
- 16.Under our optimized conditions, N-Boc-protected proline and valine are not suitable coupling partners (<10% yield).
- 17.Asada M, Iwahashi M, Obitsu T, Kinoshita A, Nakai Y, Onoda T, Nagase T, Tanaka M, Yamaura Y, Takizawa H, Yoshikawa K, Sato K, Narita M, Ohuchida S, Nakai H, Toda M. Bioorg Med Chem. 2010;18:1641. doi: 10.1016/j.bmc.2009.12.068. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Conner SE, Zhu G Eli Lilly & Co. U.S Patent 20080300308A1. 2008 Dec 4;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.