

Abstract
DNA double-strand breaks (DSBs) are cytotoxic lesions that threaten genomic integrity. Failure to repair a DSB has deleterious consequences, including genomic instability and cell death. Indeed, misrepair of DSBs can lead to inappropriate end-joining events, which commonly underlie oncogenic transformation due to chromosomal translocations. Typically, cells employ two main mechanisms to repair DSBs: homologous recombination (HR) and classical nonhomologous end joining (C-NHEJ). In addition, alternative error-prone DSB repair pathways, namely alternative end joining (alt-EJ) and single-strand annealing (SSA), have been recently shown to operate in many different conditions and to contribute to genome rearrangements and oncogenic transformation. Here, we review the mechanisms regulating DSB repair pathway choice, together with the potential interconnections between HR and the annealing-dependent error-prone DSB repair pathways.
Mechanisms of DNA DSB Repair
Detection and faithful repair of damaged DNA is essential for genome integrity. Many types of DNA lesions impede replication fork progression, resulting in replication fork collapse and DSB formation with loss of physical continuity of the genome [1]. The repair of DSBs involves four possible mechanisms (Figure 1). The first mechanism is C-NHEJ. In this mechanism, the DSB is repaired by blunt end ligation independently of sequence homology, but requiring many factors such as Ku70/80, DNA-PKcs, and DNA ligase IV (Figure 1A). C-NHEJ can occur throughout the cell cycle but is dominant in G0/G1 and G2 [2,3]. Despite the mutagenicity of C-NHEJ, its fast kinetics has a clear role in protecting genome integrity, notably by suppressing chromosomal translocations, at least for the majority of repair events [4]. Alternatively, the DSB end can be resected, leaving 3′ single-stranded DNA (ssDNA) overhangs. The resected DSB can be repaired by three possible mechanisms: HR, SSA, and alt-EJ. HR predominates in the mid-S and mid-G2 cell cycle phases, where the amount of DNA replication is highest and when the sister template is available [3]. Because HR uses a sister or homologous chromatid for repair, it requires strand invasion mediated by the recombinase RAD51 and the process is typically error-free even though completion of HR often requires error-prone polymerases (Figure 1B) [5]. The resected DSB can also be repaired by mutagenic repair pathways, namely SSA or alt-EJ. SSA mediates end joining between interspersed nucleotide repeats in the genome and involves reannealing of Replication Protein A (RPA)-covered ssDNA by the RAD52 protein. Although this is homology-directed repair, one copy of the repeat and the intervening sequence between the repeats are deleted in the repair product, thus resulting in loss of genetic information (Figure 1C) [5].
Figure 1. Four Approaches to Repair DNA Double-Strand Breaks (DSBs).
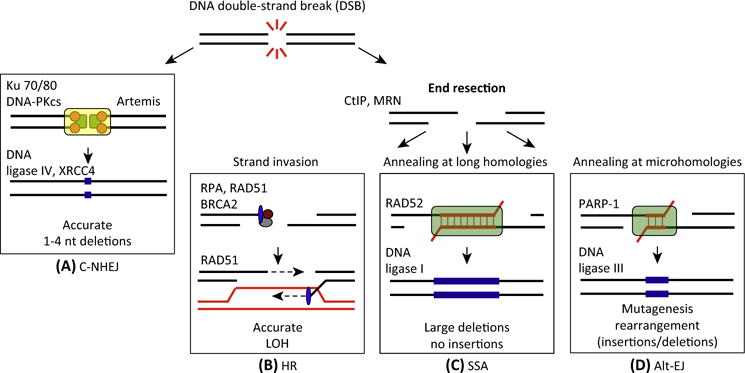
(A–D) The repair of DNA DSBs relies primarily on whether DNA end resection occurs. When resection is blocked, repair through C-NHEJ is favored. However, when DNA resection occurs, three pathways (HR, alt-EJ, and SSA) can compete for the repair of DSBs. Indeed, there are two layers of competition for the repair of DSBs. Initially at the stage of end resection, C-NHEJ competes with the resection-dependent pathways. Secondly, once resection has occurred, HR, alt-EJ, and SSA can compete for the repair. Notably, each of the four repair pathways lead to different genetic outcomes (LOH, deletions, insertions) and the fidelity of the repair mechanism is mentioned for each pathway. Abbreviations: nt, nucleotides; LOH, loss of heterozygosity; C-NHEJ, classical nonhomologous end joining; HR, homologous recombination; alt-EJ, alternative end joining; SSA, single-strand annealing.
In recent years, the notion of an alt-EJ pathway in addition to C-NHEJ has become more evident [6]. The use of alt-EJ for DNA DSB repair has been described in various cellular contexts, but the mechanistic details of this pathway remain unclear. The use of alt-EJ for repairing DSB has harmful consequences on genomic integrity because of its apparent predilection for joining DSBs on different chromosomes, thereby generating chromosomal translocations and mutagenic rearrangements (Figure 1D) [7,8]. Early evidence for alt-EJ came from studies showing that yeast and mammalian cells deficient in C-NHEJ were still able to repair DSBs via end joining [6]. Further evidence for an alternative end-joining pathway arose from the observation that mice deficient in C-NHEJ still exhibited chromosomal translocations and V(D)J recombination [9]. Molecular characterization of this alt-EJ activity in cells lacking C-NHEJ revealed that the XRCC1/DNA ligase III complex and the PARP1 polyribosylating enzyme were involved [10]. Initially, alt-EJ was considered merely a backup repair pathway for C-NHEJ for end joining of chromosomal DSBs in the context of V(D)J recombination because of its error-prone nature and its original detection only in the absence of C-NHEJ [11]. Subsequent studies, however, have demonstrated that alt-EJ might have a more primary role in repairing endogenous chromosomal DSBs, depending on the biological context [6].
Role of End Resection in DSB Repair Choice
Given that three of the pathways diverge at the early step of end resection and have different outcomes (Figure 1), it is likely that end resection dictates pathway choice and repair outcome [12]. The initial phase of end resection, called ‘end clipping’, is carried out by the structure-specific nuclease MRE11 and CtIP. In this phase, a relatively small number of base pairs (i.e., 20 bp in mammalian cells or 100–300 bp in yeast) are processed, making the DNA ends available for alt-EJ [13,14]. In the second phase of end resection called ‘extensive resection’, helicases and exonucleases (i.e., DNA2, BLM, WRN, CtIP, and EXO1) generate long stretches of ssDNA, thereby committing the cells to HR or SSA [12,15].
The cell cycle phase plays a crucial role in the pathway choice decision, and end resection is promoted by cyclin-dependent kinases (CDKs) through the phosphorylation of multiple substrates (Figure 2). S-CDK-mediated phosphorylation of both the budding yeast nucleases Dna2 and Sae2 (CtIP homolog) promotes efficient end resection [16]. The parallel process in mammalian cells involves CDK-dependent CtIP phosphorylation, favoring the CtIP–BRCA1 interaction in the S/G2 phases (Figure 2) [17,18], although the requirement of this interaction for end resection remains controversial [19]. In addition, CDK-dependent phosphorylation of EXO1 promotes end resection, while impairment of EXO1 phosphorylation attenuates resection, cell survival, and HR, but increases NHEJ activity upon DNA damage [20]. Consistent with these results, resection of DSB ends is greatly reduced in noncycling cells, thereby favoring C-NHEJ over resection-dependent repair pathways (HR, alt-EJ, and SSA) [12].
Figure 2. Mechanisms Regulating DNA End Resection in Mammalian Cells and Their Influence on DNA Repair Pathway Choice.

The cell cycle controls the competition between C-NHEJ and resection-dependent repair pathways. Extensive end resection is stimulated in the S/G2 phase of the cell cycle in a manner that depends on CDK activity, which mediates phosphorylation of multiple substrates, such as components of the MRN complex and CtIP. In the G1 phase of the cell cycle, 53BP1 and Rif1 proteins localize to DSBs, inhibit BRCA1 recruitment, block DNA end resection thus promoting C-NHEJ. In the S and G2 phases of the cell cycle, the ATM kinase, which phosphorylates members of the MRN complex, BRCA1, CtIP, or BLM favors the three resection-dependent DSB repair pathways (HR, alt-EJ, or SSA). However, recent evidence also suggests that alt-EJ could occur in G1 [105]. Abbreviations: ATM, Ataxia Telangiectasia Mutated; CDK, cyclin-dependent kinase; DSBs, double-strand breaks; MRN, MRE11, RAD50, and NBS1; C-NHEJ, classical nonhomologous end joining; HR, homologous recombination; alt-EJ, alternative end joining; SSA, single-strand annealing.
Other post-translational modifications (PTMs) have also been implicated in the regulation of end resection. For example, acetylation appears to inhibit end resection in budding yeast, while sumoylation promotes it [21,22]. Some of the relevant substrates have also been identified. These include Sae2, Mre11, Sgs1, and Exo1 in yeast, and BLM and CtIP in human cells [21,23]. A recent study has revealed that RNF111/UBE2M-mediated neddylation acts as an inhibitor of BRCA1 and CtIP-mediated DNA end resection [24]. Additionally, BRCA1, which forms a complex with CtIP and the MRN complex (MRE11, RAD50, and NBS1), and thus plays a crucial role in promoting DSB end resection [25], has been shown to be modified through polyribosylation by PARP1, which has been suggested to play key roles in regulating HR levels [26].
Accessory factors also contribute to repair pathway choice via modulation of end resection. For instance, the balance between BRCA1 and 53BP1 modulates pathway choice by either promoting or preventing end resection. 53BP1 blocks DNA resection by preventing CtIP from accessing DNA ends. Thus, 53BP1 directs repair through C-NHEJ (Figure 2), which is upregulated in BRCA1-null cells [27]. This end protection function of 53BP1 depends upon its phosphorylation by activated Ataxia Telangiectasia Mutated (ATM), which promotes the recruitment of interacting factors Rif1 and PTIP, and subsequent recruitment of downstream nucleases such as Artemis [28–31]. The resection inhibition function of 53BP1 appears to limit mutagenic C-NHEJ over physiological C-NHEJ, which is required for proper immunoglobulin class switch recombination (CSR) [32]. However, when Rif1 or 53BP1 are knocked out, DSBs are extensively resected and not repaired by C-NHEJ, resulting in persistent chromosome breaks and genomic instability [4,33]. More recently, REV7 was also shown to promote C-NHEJ by inhibiting DNA end resection downstream of Rif1 [34,35]. Loss-of-function mutations in factors controlling end resection (such as 53BP1 and REV7) result in reduced C-NHEJ and restore DNA end resection, which then favors HR activity and induces PARP inhibitor (PARPi) resistance of BRCA1-deficient cells.
Factors such as the MRN complex, BRCA1, and ATM function in both C-NHEJ and HR. This link among the MRN complex, ATM, and end resection provides a possible regulatory mechanism for resection-dependent repair pathways (Figure 2). ATM recruits and phosphorylates all members of the MRN complex [36], which subsequently leads to ATM-dependent phosphorylation of other HR components, including BRCA1 [37], CtIP, EXO1, and BLM, and promotes efficient DSB resection [38,39]. It is tempting to speculate that ATM, which is directly involved in HR regulation, might also be a crucial regulator of the end resection-dependent error-prone DSB repair pathways (Figure 2) [6].
While end resection in DSB repair has been extensively described in yeast, less is known about this process in other eukaryotic cells, despite its obvious importance in repair pathway choice and outcome. With the development of new approaches and techniques for the study of end resection in human cells, it is likely that more complex regulatory mechanisms in this system will be uncovered.
Homologous Recombination Control: The RAD51 Hub in Homology-Based Repair Pathway Choice
Once end resection has occurred, the C-NHEJ repair pathway is prevented and any of the three homology-based pathways, that is, SSA, alt-EJ, and HR, can be invoked. Usage of these pathways can be affected by regulation of and competition with the RAD51 presynaptic and postsynaptic steps. We focus on positive and negative regulators of RAD51 function. Upon DNA damage induction, the RPA complex initially competes with RAD51 for ssDNA binding in yeast and mammals [17,40]. However, RPA has also a pro-recombinogenic role once RAD51 is loaded onto ssDNA, since it favors presynaptic nucleofilament formation by eliminating secondary structure formation and by protecting DNA ends from degradation [41]. Moreover, RPA binding to resected DNA prevents spontaneous annealing between microhomologies, suggesting a step of regulation for microhomology-mediated repair [42]. RPA also regulates SSA, as SSA requires RPA binding to ssDNA and is independent of RAD51 in both yeast and humans.
Positive Regulators of HR
Some accessory protein mediators positively regulate RPA displacement, RAD51 nucleofilament formation, and strand exchange activity: the most important are the RAD51 paralogs, BRCA2, and RAD52. BRCA2 is the main mediator of RAD51 nucleofilament formation and strand exchange in mammalian cells, through a series of eight evolutionarily conserved motifs, which are called the BRC repeats. BRCA2 BRC domains promote RAD51 loading onto ssDNA by disrupting self-assembled RAD51 oligomers and favoring one-to-one binding of RAD51 monomers instead. BRCA2 binding stabilizes the ATPase activity of RAD51, thereby supporting the ssDNA binding activity of RAD51. In a second step, the BRCA2 C-terminal domain binds to RAD51 oligomers in the context of the RAD51–ssDNA helix [43], thus promoting nucleofilament growth and participating in strand invasion [44,45]. In yeast, which does not possess BRCA2, an analogous recombinogenic function appears to be carried out by Rad52.
Negative Regulators of HR
Conversely, many factors negatively regulate RAD51 nucleofilament formation to ensure strand exchange regulation, thus avoiding hyper-recombination and crossover (CO)-driven rearrangements. Yeast Srs2 was the first characterized negative regulator of Rad51 function. The Srs2 DNA helicase removes Rad51 from presynaptic filaments through an ATP-hydrolyzing activity coupled to DNA unwinding (Figure 3) [46,47]. In mammalian cells, the helicase and Proliferating Cell Nuclear Antigen (PCNA)-interacting factor, PARI, has been shown to dismantle RAD51 nucleofilaments in a process that requires ATP hydrolysis (Figure 3) [48]. More recently, the polymerase Polυ was shown to act at the presynaptic step as an antirecombinase. Even though Polυ is not able to displace RAD51 already bound to ssDNA, Polυ can still limit the formation of RAD51 nucleofilaments in a mechanism that requires binding to RAD51 and ATP hydrolysis (Figure 3) [49]. Interestingly, Srs2, PARI, and Polυ can also promote error-prone repair mechanisms such as alt-EJ, translesion synthesis (TLS), or SSA [50–53].
Figure 3. Regulation of RAD51-Mediated Recombination.
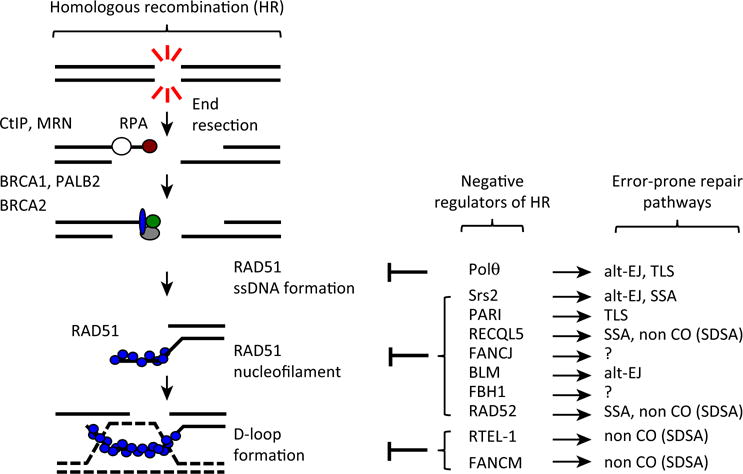
During the HR process, once resection occurs, homology search and DNA-strand invasion generate D loops, a key intermediate for all subpathways of HR. This reaction catalyzed by RAD51 is negatively controlled at different levels. At the presynaptic level, Polυ prevents RAD51 nucleofilament formation. Postsynaptically, many proteins such as yeast Srs2 or human PARI, RECQL5, and BLM are able to dismantle formed RAD51 nucleofilaments. Additionally, HR can also be regulated at the level of strand exchange by RTEL1- or FANCM-mediated D-loop displacement. Concomitantly, players that negatively regulate HR promote DSB repair through alternative error-prone mechanisms such as alt-EJ or SSA. Abbreviations: CO, crossover; SDSA, synthesis-dependent strand annealing; TLS, translesion synthesis; SSA, single-strand annealing; alt-EJ, alternative end joining; DSB, double-strand break; HR, homologous recombination.
Analogous antirecombinogenic activities are shown by several mammalian helicases, including the RecQ helicases. RECQL5 disrupts RAD51 filaments in an ATP-dependent manner and through some BRC-like domains [54,55]. While limiting HR, RECQL5 counteracts the inhibitory effect of RAD51 on RAD52, thus promoting synthesis-dependent strand annealing (SDSA), which limits COs [56]. Deletion of RECQL5 in Fanconi anemia (FA) cells increases genomic instability, suggesting that RECQL5 in the HR-deficient context can induce compensatory repair mechanisms crucial for survival [57]. Another RecQ helicase, BLM, similarly inhibits the early recombinogenic step of displacing RAD51 from ssDNA in an ATP-dependent manner [58]. Two recent studies uncovered a dual role for BLM in DSB repair: BLM can repress alt-EJ in a manner epistatic with 53BP1 and Rif1; conversely, in the absence of 53BP1 or Rif1, BLM can also favor alt-EJ [59,60]. Apart from these two RecQ helicases, the FANCJ and FBH1 helicases also promote disassembly of the RAD51 nucleofilament [58,61]. Even though deletion of FBH1 in chicken DT40 cells causes an elevated rate of sister chromatid exchange (SCE), it remains unclear whether FBH1 can promote error-prone DSB repair [62].
Even after RAD51 nucleofilament formation, HR can be modulated at the level of strand exchange. D-loop displacement by specific DNA helicases during DSB repair prevents CO events. This is favored during mitosis, when imprecise chromatid exchange could cause translocations [63]. The helicase RTEL1 efficiently drives D-loop disassembly through an ATP hydrolysis-dependent mechanism [64]. FANCM can similarly disrupt RAD51 coated D-loops [65]. While RTEL1 and Mph1 (the yeast FANCM homolog) inhibit HR, they also promote SDSA-mediated non-CO product formation [66,67]. Depletion of these factors causes accumulation of RAD51 foci and higher rates of SCE.
After strand exchange, RAD51 is removed from double-stranded DNA (dsDNA), which is pivotal for the downstream steps of strand extension, junction resolution, and chromatin reassembly. ATM, which is known to be a crucial regulator of the presynaptic HR, also regulates the last steps in HR, possibly through RAD54 activation [68]. The HELQ helicase removes RAD51 from dsDNA and promotes strand extension in an ATP-independent manner [69,70]. Altogether, these findings reinforce the centrality of RAD51 in pathway choice control for DSB repair, both in the modulation of non-HR pathways and in the tight control of HR repair steps.
Annealing-Dependent DSB Repair
Connection between HR and alt-EJ
alt-EJ functions in S phase independently of C-NHEJ factors and, until recently, was considered a backup repair pathway for C-NHEJ for joining chromosomal DSBs, particularly during V(D)J recombination [2,71,72]. However, recent advances have shown that the alt-EJ and HR pathways compete for the repair of DSBs [49,73]. While both alt-EJ and HR share a common initial resection mechanism [8], processing of resected ends in HR is catalyzed by the eukaryotic RecA homolog, RAD51, while PARP1 is thought to mediate annealing at microhomologies for alt-EJ [74–76]. In HR, RAD51 forms a filament on ssDNA that drives strand exchange with a homologous template. PARP1 has been proposed to serve as a platform for recruiting alt-EJ repair factors, such as TLS polymerases, to microhomology sequences flanking the break [74–76]. Modulation of RAD51 or PARP1 function through genetic or chemical means affects HR and alt-EJ levels, respectively. For example, the budding yeast Srs2 helicase disrupts the Rad51–ssDNA presynaptic nucleofilament in vitro, and has been implicated in the prevention of undesirable HR [46,77] and promotion of efficient alt-EJ [50]. Interestingly, deletion of Srs2 also reduces SSA frequency [51], suggesting that Srs2 sits at the crossroads of HR and annealing-dependent DSB repair. Chemically inhibiting PARP1 function with PARPi in cells compromised for HR repair confers synthetic lethality [78,79], suggesting that PARP1-mediated alt-EJ compensates for the HR deficiency of these cells. It is to be noted, however, that PARPi likely inhibits other PARP family members and, thus, this synergistic relationship could be a compound effect on alt-EJ. Interestingly, PARP3, another PARP family member, is an important player in the cellular response to DSBs, silencing of which has recently been shown to decrease HR efficiency while enhancing end resection and mutagenic alt-EJ [80]. Also, recent studies indicate a synthetic lethal interaction between PARP1 and PARP2. Indeed, parp1−/− parp2−/− mice display embryonic lethality showing that these proteins have overlapping and nonredundant functions in the maintenance of genomic stability [81].
Recently, a competitive relationship between alt-EJ and HR in human cells has been demonstrated. Polυ (encoded by POLQ), a TLS polymerase that functions in alt-EJ, inhibits HR by binding to RAD51 and inhibiting RAD51 nucleofilament formation in a mechanism that require its ATP hydrolysis and binding to RAD51 [49]. This is consistent with a study in Drosophila showing that spn-A (homolog of human RAD51) mutants were sensitive to ionizing radiation (IR), and that loss of mus308 (homolog of human Polυ) increases spn-A mutant sensitivity to radiation [75]. It is tempting to speculate that by preventing RAD51 assembly on ssDNA, Polυ limits RAD51 toxicity (a possible consequence of RAD51 overexpression) [82]. At the same time, Polυ can perform alt-EJ through direct binding of resected DSBs in a process that depends on an evolutionarily conserved loop domain. This process allows pairing of microhomologies. Thereafter, Polυ can extend each strand from the base-paired region using the opposing overhang as a template (Figure 4) [83,84]. Interestingly, Polυ is upregulated in HR-deficient ovarian and breast cancers, suggesting that alt-EJ can serve as a backup pathway for the repair of DSBs when HR is defective. When depleted of Polυ, HR-deficient cells became hypersensitive to cytotoxic agents and embryos from crossbred HR-deficient and Polυ-deficient mice were not viable, demonstrating a synthetic lethal relationship between HR and Polυ-mediated alt-EJ [49,73]. Accordingly, the generation of small molecule inhibitors of Polυ activity may offer a new potential therapeutic approach for cancers with inactivated HR.
Figure 4. Polυ Regulates the Balance between Homologous Recombination (HR) and Alternative End Joining (alt-EJ).
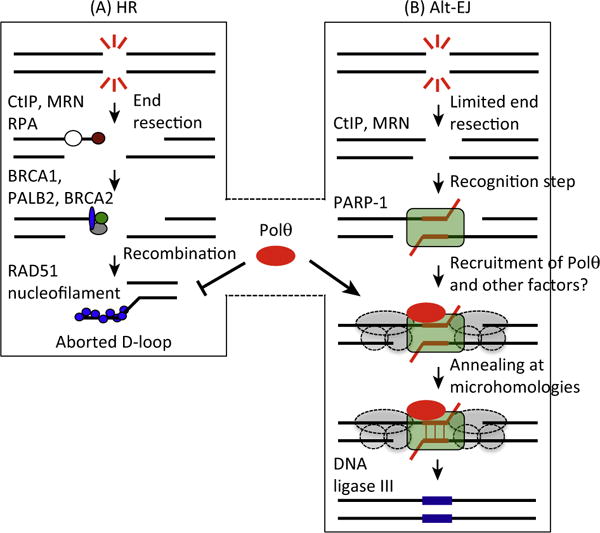
(A) In the case of a HR defect, such as mutations in BRCA1/2 genes, Polυ is overexpressed. Polυ blocks the formation of RAD51–ssDNA nucleofilament and thus RAD51 toxicity in cells deficient in HR. (B) At the same time, Polυ is recruited to DNA damage sites by PARP1 where it performs untemplated error-prone synthesis required for alt-EJ. Polυ recruitment to damage sites might also depend on other alt-EJ factors (gray circles). Abbreviation: ssDNA, single-stranded DNA.
While the alt-EJ factor Polυ inhibits HR, a recent study suggests that many HR factors, including FA proteins, promote alt-EJ [85]. However the function of these proteins in alt-EJ is not completely known. Expression of FANCD2, a key regulator of the FA interstrand crosslink (ICL) repair pathway, directly correlates with Polυ, suggesting common regulatory mechanisms [49]. Moreover, recent studies have shown that FANCD2 directly interacts with CtIP, and is essential for efficient end resection during ICL repair [86,87]. As CtIP is a key node in the regulation of end resection that in turn dictates the balance among HR, alt-EJ, and SSA, it is tempting to speculate that factors interacting with CtIP could directly influence repair pathway choice in a context-dependent manner. Altogether, these findings confirm the notion of multilevel connections between HR and alt-EJ, and demonstrate that some factors can function in several repair pathways, suggesting multiple avenues of regulation.
Connections between HR and SSA: Is RAD52 the Sole Mediator?
As opposed to HR, the SSA pathway is unique in that it does not require a donor sequence; thus, SSA does not entail strand invasion and is independent of RAD51 [88]. Instead, SSA uses the resected ends to anneal exposed complementary sequences to complete repair. The steps of SSA have been elucidated in budding yeast. Rad52, in complex with the homologous Rad59 protein, binds to the exposed repeats flanking the DSB and promotes their annealing. This annealed complex is thought to be stabilized by the Msh2–Msh3 mismatch repair proteins, and the nonhomologous 3′ flaps are cleaved by the Rad1–Rad10 endonuclease in a mechanism dependent on the Saw1 and Slx4 proteins. Studies in mammalian cells have demonstrated the involvement of the mammalian homologs of Rad52 (RAD52) and Rad1–Rad10 (ERCC4–ERCC10) in SSA [17]. However, additional components of this pathway in higher eukaryotic systems remain unknown. For example, there is no clear mammalian Saw1 homolog, although a recent study has proposed that the ICL repair sensor UHRF1 is a functional homolog [89]. It will be interesting to test whether UHRF1 and SLX4 are required for mammalian SSA. Additionally, the absence of yeast Rad52 confers very severe defects and radiosensitization due to its involvement in all HR subpathways, both Rad51-dependent and Rad51-independent [5]. On the contrary, in organisms possessing a BRCA2 homolog (Ustilago maydis, chicken, human, and mice), RAD52 inactivation causes minimal DNA repair defects [90]. These observations raise the question as to whether RAD52 has limited HR function in BRCA2-containing organisms.
Regardless of these differences, both lower and higher eukaryotic cells show competition between HR and SSA for the repair of DSBs. For example, Rad51 prevents Rad52-mediated annealing of complementary ssDNA in yeast [91,92], and RAD51 inhibition in mammalian cells upregulates RAD52-mediated SSA activity [17]. Moreover, even though RAD52 deficiency in mammals had no impact on cell growth and viability in HR-proficient cells, loss of RAD52 function was found to be synthetically lethal with deficiency in some HR factors (BRCA1, PALB2, and BRCA2) [93].
The mechanism by which loss of RAD52 increases genomic instability in HR-deficient cells is unclear, but evidence suggests that RAD52 maintains RAD51-dependent homology-directed gene conversion in BRCA1/2-deficient cells [93]. Whether BRCA-deficient tumors become dependent on SSA, and whether SSA activity is increased by loss of HR, remain under debate and an understanding of SSA usage, particularly in HR-deficient tumors, could uncover novel targets for anticancer therapy. These findings call for another look at RAD52 and its relationship with HR in vertebrates. Indeed, the reduction in the development of T-cell lymphomas in ATM-deficient mice by the RAD52 single knockout indicates a significant in vivo function for mouse RAD52 [94]. Surprisingly, similar results were obtained when ATM-deficient mice were crossbred with POLQ-deficient mice. The double mutant mice displayed a delayed onset of T-cell lymphomas, significantly increasing mouse survival [95]. These observations raise the question of whether POLQ or RAD52 is predominantly used for DNA repair upon a HR deficiency in humans and mice.
Annealing-Dependent DSB Repair Outcomes and Their Mutational Signatures
As the repair of DSBs, via annealing-dependent pathways, introduces insertions and/or deletions (Figure 5), one can predict that the use of such DNA repair mechanisms will leave behind a particular genomic signature, or ‘genomic scar’, distinct from that of HR-mediated repair, which usually preserves genomic integrity. As modern next-generation sequencing technologies can rapidly identify cancer risk alleles of many genes, genome-wide sequencing and loss of heterozygosity (LOH) profiling have detected specific mutation profiles indicative of a HR defect [96–98]. A large proportion of breast and ovarian cancers is attributable to inherited risk conferred by mutations in DNA repair genes, including BRCA1/2, FA genes, or the DNA mismatch repair genes. The utility of this approach lies in the enhanced sensitivity of some HR-deficient tumors to certain drugs, such as PARPi. Indeed, Tutt and colleagues recently showed that copy number aberrations are more prevalent in a subgroup of breast cancers (namely basal-like breast cancers) that respond to platinum-based chemotherapy, thus providing a candidate predictive biomarker for this disease [99].
Figure 5. The Different Outcomes of Annealing-Dependent Error-Prone Double-Strand Break (DSB) Repair.
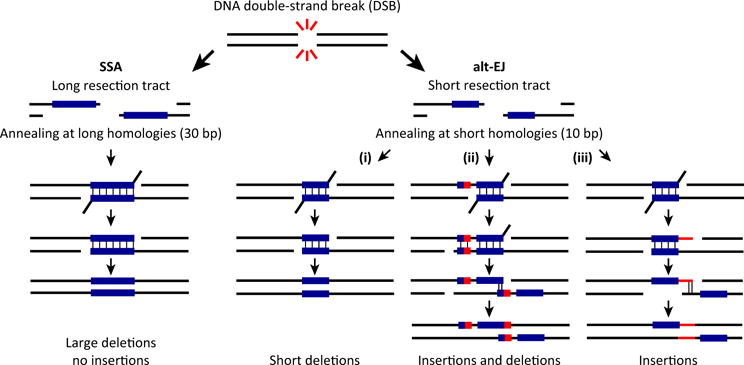
After formation of a DSB, DNA resection results in two single-stranded overhangs and exposure of DNA homology. SSA uses annealing of large DNA sequences of homology, which leads to deletions of large fragments of DNA. By contrast, alt-EJ requires pairing of only small homologous DNA sequences (microhomologies), which can lead to deletions and/or insertions depending on how the repair is orchestrated: (i) When the annealing is stable the overhanging noncomplementary flaps are trimmed by the endonuclease complex and repair is completed by fill-in DNA synthesis and ligation, resulting in deletions of the DNA regions flanking the original break. (ii) Alternatively, translesion polymerases (such as Polυ) may extend the annealed sequences using untemplated error-prone DNA synthesis resulting in realignment at newly created micro-homologous sequences. Then the repair is completed by flap trimming, gap-filling DNA synthesis, and ligation, which results in a deletion plus insertion junction. (iii) Flap trimming followed by untemplated DNA error-prone synthesis can generate new regions of microhomology leading to insertions of DNA sequences. Abbreviations: alt-EJ, alternative end joining; DSB, double-strand break; SSA, single-strand annealing.
Despite our improved understanding of the genetic and molecular features of cancer cells, the association between tumor genomic features (such as HR deficiency) and clinically relevant endpoints (such as disease response and patient outcome) is not well established [100]. This is partly because the current ‘HR-deficient genetic signature’ is not inclusive. For instance, nearly 20% of ovarian serous carcinoma and basal-like breast cancers that are deficient in HR-mediated repair do not have mutations in BRCA1/2 genes, suggesting that not all deleterious HR mutations have been identified. An important step forward would be the identification of other genomic signatures in the absence of known HR biomarkers (such as BRCA status) to more comprehensively define HR deficiency. A clue in this direction is the observation that BRCA1/2 mutant tumors have an obvious mutational spectrum. Indeed, the current HR-deficient genetic signature (or signature 3) associates strongly with elevated numbers of large (longer than 3 bp) insertions and deletions with overlapping microhomology at breakpoint junctionsi. As the mutagenic alt-EJ and SSA pathways play backup roles for HR [49,73,93], and copy number aberrations in HR-deficient basal-like breast cancers promote the use of these error-prone pathways, repair through alt-EJ or SSA could account for the characteristic mutational spectra observed in all HR-deficient tumors.
In alt-EJ, the fill-in synthesis is likely mediated by the Polυ polymerase, which is error-prone and likely produces point mutations, as well as random insertions and deletions (indels) [101]. Indeed, upregulation of budding yeast Polυ appears to generate random deletions or insertions of 20–200 bp [76]. Thus, the use of alt-EJ, which could be indicative of HR deficiency, is likely to leave a mutational signature, composed of indels at sites of microhomology-mediated repair (10 bp) (Figure 5). As HR deficiency also appears to enhance SSA activity, determining the SSA mutational signature would also aid in identifying HR-deficient tumors. Based on the literature and studies in yeast, the predicted mutational signature resulting from upregulated SSA would be large deletions at sites with long homology repeats (30 bp) (Figure 5). Thus, characterizing the mutational signature associated with the use of alt-EJ and SSA may help to expand the current list of HR-deficient genetic signatures, with the ultimate goal of establishing more accurate and comprehensive associations between particular genomic features and relevant clinical endpoints, potentially leading to the identification of novel clinical biomarkers to more confidently direct anticancer treatment choice and/or predict patient outcome.
Concluding Remarks
Here, we have reviewed the current knowledge regarding pathways used for the repair of DSBs through the lens of clinical applications. We have discussed regulation of the four known DSB repair pathways and how error-prone DNA repair pathways can compensate for the loss of HR. Understanding the basic regulation of DNA repair mechanisms has led to advances in synthetic lethality-based drug development. For several decades, significant effort has focused on identifying unique oncoproteins or pathways that could serve as targets for designer therapeutic agents. Despite several notable successes using this approach, first-generation drugs such as cisplatin that nonspecifically damage DNA continue to comprise the backbone of most anticancer therapeutic regimens. As large-scale genomic studies begin to define the mutational status of individual DNA repair pathways in tumors, it may become possible to rationally select DNA damaging agents based on tumor repair pathway mutations, thus allowing available DNA damaging agents to be used in a ‘designer’ manner. Consistent with key roles for DNA repair pathways in preventing oncogenesis, their inactivation can lead to many cancer predispositions (e.g., BRCA1/2 mutations in the early onset of breast/ovarian, prostate, and pancreatic cancers). Indeed, cancer cells often have at least one defective DNA repair pathway [102], and a significant proportion of cancers (especially breast and ovarian cancers) demonstrate defects in DNA repair genes, including many in the HR pathway [103]. This situation is ripe for targeted therapy, which is based on the principle that when tumor cells are deficient in a particular DNA repair pathway, this deficiency is compensated for by the activation of a second pathway. The tumor is thus dependent on this alternative pathway for survival, inhibition of which will specifically kill the malignant cells (synthetic lethality) with little effect on normal cells [102]. The genetic concept of synthetic lethality provides a framework for the design of novel therapeutic approaches to cancer. Already, there are promising indications from clinical trials that this approach could be beneficial when PARPi are given to patients with germline BRCA1 or BRCA2 gene mutations [78,104]. PARP1, inhibition of which is synthetic lethal with a deficiency in HR, is also known to participate in a variety of cellular processes including the repair of DSBs through alt-EJ. Therefore, error-prone DSB repair pathways may be one process that is key for the viability and proliferation of HR-deficient cells. This highlights the importance of studying DSB repair pathways and determining their composition, mechanism, and regulation. Such studies would uncover new targets for drug development and new synthetic lethal opportunities that can be assessed in the clinic (see Outstanding Questions).
Trends.
Of the four known pathways for repairing DNA DSBs, some evolved towards high-fidelity processes (HR and C-NHEJ), while others are intrinsically mutagenic (alt-EJ and SSA).
Some repair pathways are end resection-independent (C-NHEJ), while others are end resection-dependent (HR, alt-EJ, and SSA). End resection likely plays a key role in dictating DNA repair pathway choice.
Homology-based repair pathways (HR, alt-EJ, and SSA) are competitive and mutually regulated around the RAD51 presynaptic and postsynaptic steps of HR.
Error-prone repair pathways can compensate for the loss of HR. Polυ (an alt-EJ polymerase) is upregulated in HR-deficient cancers: loss of the HR and Polυ-mediated alt-EJ pathways is synthetic lethal.
Outstanding Questions.
How does end resection control DNA repair pathway choice for DNA DSBs? What are the crucial players regulating end resection in human cells? How are these players regulated? Are they differentially regulated upon the loss of HR?
How do the annealing-dependent DSB repair pathways (SSA and alt-EJ) compensate for the loss of HR? Which alternative pathway is dominant? What functions of SSA or alt-EJ are crucial for the survival of HR-deficient cells? What triggers the use of SSA versus alt-EJ upon the loss of HR?
What is the relevance of a genomic signature resulting from the use of SSA and/or alt-EJ genomic signature? Would the evaluation of these signatures, in combination with the current HR-deficient genomic signature, allow for a better assessment of HR defects and cancer patient outcome.
Acknowledgments
We would like to thank Jessica C. Liu and Prabha Sarangi for editing the manuscript and helpful discussions. R.C. is a recipient of the Ovarian Cancer Research Fellowship (OCRF). B.R. is a recipient of the Italian Association for Cancer Research (AIRC) Fellowships for Abroad. This work was supported by National Institutes of Health (NIH) grants P50CA168504 and R01HL52725 and by grants from the OCRF and Breast Cancer Research Foundation (BCRF) to A.D.D.
Footnotes
Resources
References
- 1.Aparicio T, et al. DNA double-strand break repair pathway choice and cancer. DNA Repair. 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiruvella KK, et al. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karanam K, et al. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47:320–329. doi: 10.1016/j.molcel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Difilippantonio MJ, et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heyer WD, et al. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- 7.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corneo B, et al. Rag mutations reveal robust alternative end joining. Nature. 2007;449:483–486. doi: 10.1038/nature06168. [DOI] [PubMed] [Google Scholar]
- 10.Wang M, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 12.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 13.Truong LN, et al. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci USA. 2013;110:7720–7725. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sturzenegger A, et al. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem. 2014;289:27314–27326. doi: 10.1074/jbc.M114.578823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, et al. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct Mol Biol. 2011;18:1015–1019. doi: 10.1038/nsmb.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennardo N, et al. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polato F, et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J Exp Med. 2014;211:1027–1036. doi: 10.1084/jem.20131939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomimatsu N, et al. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun. 2014;5:3561. doi: 10.1038/ncomms4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robert T, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cremona CA, et al. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell. 2012;45:422–432. doi: 10.1016/j.molcel.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarangi P, et al. Sumoylation influences DNA break repair partly by increasing the solubility of a conserved end resection protein. PLoS Genet. 2015;11:e1004899. doi: 10.1371/journal.pgen.1004899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jimeno S, et al. Neddylation inhibits CtIP-mediated resection and regulates DNA double strand break repair pathway choice. Nucleic Acids Res. 2015;43:987–999. doi: 10.1093/nar/gku1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badie S, et al. BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. EMBO J. 2015;34:828. doi: 10.15252/embj.201570610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y, et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination mediated DNA repair. Cancer Discov. 2014;4:1430–1447. doi: 10.1158/2159-8290.CD-13-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunting SF, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escribano-Diaz C, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Chapman JR, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Callen E, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–1280. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014;28:2693–2698. doi: 10.1101/gad.252478.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zong D, et al. Ectopic expression of RNF168 and 53BP1 increases mutagenic but not physiological non-homologous end joining. Nucleic Acids Res. 2015;43:4950–4961. doi: 10.1093/nar/gkv336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bothmer A, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu G, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–544. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boersma V, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature. 2015;521:537–540. doi: 10.1038/nature14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 37.Li S, et al. Functionallink of BRCA1 andataxia telangiectasia gene product in DNA damage response. Nature. 2000;406:210–215. doi: 10.1038/35018134. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013;9:e1003277. doi: 10.1371/journal.pgen.1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson SE, et al. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol Cell. 2013;49:657–667. doi: 10.1016/j.molcel.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Haber JE. Role of Saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biol. 2004;2:E21. doi: 10.1371/journal.pbio.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H, et al. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng SK, et al. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:405–412. doi: 10.1038/nsmb.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carreira A, Kowalczykowski SC. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci USA. 2011;108:10448–10453. doi: 10.1073/pnas.1106971108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esashi F, et al. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol. 2007;14:468–474. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- 45.Ayoub N, et al. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol. 2009;19:1075–1085. doi: 10.1016/j.cub.2009.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krejci L, et al. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–309. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- 47.Chiolo I, et al. Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol Cell Biol. 2005;25:5738–5751. doi: 10.1128/MCB.25.13.5738-5751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moldovan GL, et al. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell. 2012;45:75–86. doi: 10.1016/j.molcel.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ceccaldi R, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015;518:258–262. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee K, Lee SE. Saccharomyces cerevisiae Sae2-and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics. 2007;176:2003–2014. doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, et al. Microarray-based genetic screen defines SAW1, a gene required for Rad1/Rad10-dependent processing of recombination intermediates. Mol Cell. 2008;30:325–335. doi: 10.1016/j.molcel.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu AM, McVey M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010;38:5706–5717. doi: 10.1093/nar/gkq379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Connor KW, et al. PARI overexpression promotes genomic instability and pancreatic tumorigenesis. Cancer Res. 2013;73:2529–2539. doi: 10.1158/0008-5472.CAN-12-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwendener S, et al. Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J Biol Chem. 2010;285:15739–15745. doi: 10.1074/jbc.M110.110478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Islam MN, et al. A variant of the breast cancer type 2 susceptibility protein (BRC) repeat is essential for the RECQL5 helicase to interact with RAD51 recombinase for genome stabilization. J Biol Chem. 2012;287:23808–23818. doi: 10.1074/jbc.M112.375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paliwal S, et al. Human RECQ5 helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucleic Acids Res. 2014;42:2380–2390. doi: 10.1093/nar/gkt1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim TM, et al. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015;43:893–903. doi: 10.1093/nar/gku1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sommers JA, et al. FANCJ uses its motor ATPase to destabilize protein–DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem. 2009;284:7505–7517. doi: 10.1074/jbc.M809019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gravel S, et al. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grabarz A, et al. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013;5:21–28. doi: 10.1016/j.celrep.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 61.Simandlova J, et al. FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J Biol Chem. 2013;288:34168–34180. doi: 10.1074/jbc.M113.484493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kohzaki M, et al. Cooperative roles of vertebrate Fbh1 and Blm DNA helicases in avoidance of crossovers during recombination initiated by replication fork collapse. Mol Cell Biol. 2007;27:2812–2820. doi: 10.1128/MCB.02043-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersen SL, Sekelsky J. Meiotic versus mitotic recombination: two different routes for double-strand break repair: the different functions of meiotic versus mitotic DSB repair are reflected in different pathway usage and different outcomes. BioEssays. 2010;32:1058–1066. doi: 10.1002/bies.201000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barber LJ, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135:261–271. doi: 10.1016/j.cell.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gari K, et al. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 66.Stafa A, et al. Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics. 2014;196:1017–1028. doi: 10.1534/genetics.114.162297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adelman CA, Boulton SJ. Metabolism of postsynaptic recombination intermediates. FEBS Lett. 2010;584:3709–3716. doi: 10.1016/j.febslet.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 68.Bakr A, et al. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res. 2015;43:3154–3166. doi: 10.1093/nar/gkv160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moldovan GL, et al. DNA polymerase POLN participates in cross-link repair and homologous recombination. Mol Cell Biol. 2010;30:1088–1096. doi: 10.1128/MCB.01124-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ward JD, et al. Overlapping mechanisms promote post-synaptic RAD-51 filament disassembly during meiotic double-strand break repair. Mol Cell. 2010;37:259–272. doi: 10.1016/j.molcel.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 71.Bentley J, et al. DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 2004;32:5249–5259. doi: 10.1093/nar/gkh842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–454. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mateos-Gomez PA, et al. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–257. doi: 10.1038/nature14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Audebert M, et al. Effect of double-strand break DNA sequence on the PARP-1 NHEJ pathway. Biochem Biophys Res Commun. 2008;369:982–988. doi: 10.1016/j.bbrc.2007.11.132. [DOI] [PubMed] [Google Scholar]
- 75.Chan SH, et al. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet. 2010;6:e1001005. doi: 10.1371/journal.pgen.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Veaute X, et al. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein laments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- 78.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 79.Bryant HE, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 80.Beck C, et al. PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014;42:5616–5632. doi: 10.1093/nar/gku174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Menissier de Murcia J, et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair. 2008;7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kent T, et al. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat Struct Mol Biol. 2015;22:230–237. doi: 10.1038/nsmb.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zahn KE, et al. Human DNA polymerase theta grasps the primer terminus to mediate DNA repair. Nat Struct Mol Biol. 2015;22:304–311. doi: 10.1038/nsmb.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Howard SM, et al. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015;11:e1004943. doi: 10.1371/journal.pgen.1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Unno J, et al. FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Rep. 2014;7:1039–1047. doi: 10.1016/j.celrep.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 87.Murina O, et al. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014;7:1030–1038. doi: 10.1016/j.celrep.2014.03.069. [DOI] [PubMed] [Google Scholar]
- 88.Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997;272:28194–28197. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- 89.Liang CC, et al. UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 2015;10:1947–1956. doi: 10.1016/j.celrep.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kojic M, et al. Compensatory role for Rad52 during recombinational repair in Ustilago maydis. Mol Microbiol. 2008;67:1156–1168. doi: 10.1111/j.1365-2958.2008.06116.x. [DOI] [PubMed] [Google Scholar]
- 91.Ivanov EL, et al. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu Y, et al. Rad51 protein controls Rad52-mediated DNA annealing. J Biol Chem. 2008;283:14883–14892. doi: 10.1074/jbc.M801097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lok BH, et al. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Treuner K, et al. Loss of Rad52 partially rescues tumorigenesis and T-cell maturation in Atm-deficient mice. Oncogene. 2004;23:4655–4661. doi: 10.1038/sj.onc.1207604. [DOI] [PubMed] [Google Scholar]
- 95.Shima N, et al. The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol. 2004;24:10381–10389. doi: 10.1128/MCB.24.23.10381-10389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Popova T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 97.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Watkins J, et al. Genomic complexity profiling reveals that HORMAD1 overexpression contributes to homologous recombination deficiency in triple-negative breast cancers. Cancer Discov. 2015;5:488–505. doi: 10.1158/2159-8290.CD-14-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Helleday T, et al. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yousefzadeh MJ, Wood RD. DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair. 2013;12:1–9. doi: 10.1016/j.dnarep.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24:3799–3808. doi: 10.1200/JCO.2005.05.4171. [DOI] [PubMed] [Google Scholar]
- 103.Bast RC, Jr, et al. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lord CJ, et al. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- 105.Barton O, et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J Cell Biol. 2014;206:877–894. doi: 10.1083/jcb.201401146. [DOI] [PMC free article] [PubMed] [Google Scholar]